A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia

 , and
, and 
Abstract
:Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organisation Global Hepatitis Report. 2017. Available online: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 27 April 2018).
- McLuckie, A.; Barrs, V.; Lindsay, S.; Aghazadeh, M.; Sangster, C.; Beatty, J. Molecular diagnosis of felis catus gammaherpesvirus 1 (FcaGHV1) infection in cats of known retrovirus status with and without lymphoma. Viruses 2018, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from rna-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, X.L.; Li, W.; Zhu, Y.; Ge, X.Y.; Zhang, L.B.; Zhang, Y.Z.; Bock, C.T.; Shi, Z.L. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in china. Virol. J. 2017, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F.F. Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 2017, 22, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nie, F.Y.; Lin, X.D.; Hao, Z.Y.; Chen, X.N.; Wang, Z.X.; Wang, M.R.; Wu, J.; Wang, H.W.; Zhao, G.; Ma, R.Z.; et al. Extensive diversity and evolution of hepadnaviruses in bats in China. Virology 2018, 514, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Geipel, A.; Konig, A.; Corman, V.M.; van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D.; et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef] [PubMed]
- ICTV. Family—Hepadnaviridae. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 445–455. [Google Scholar]
- Lisco, A.; Vanpouille, C.; Margolis, L. Coinfecting viruses as determinants of hiv disease. Curr. HIV/AIDS Rep. 2009, 6, 5–12. [Google Scholar] [CrossRef] [PubMed]
- McGovern, B.H. The epidemiology, natural history and prevention of hepatitis B: Implications of hiv coinfection. Antivir. Ther. 2007, 12, H3–H13. [Google Scholar] [PubMed]
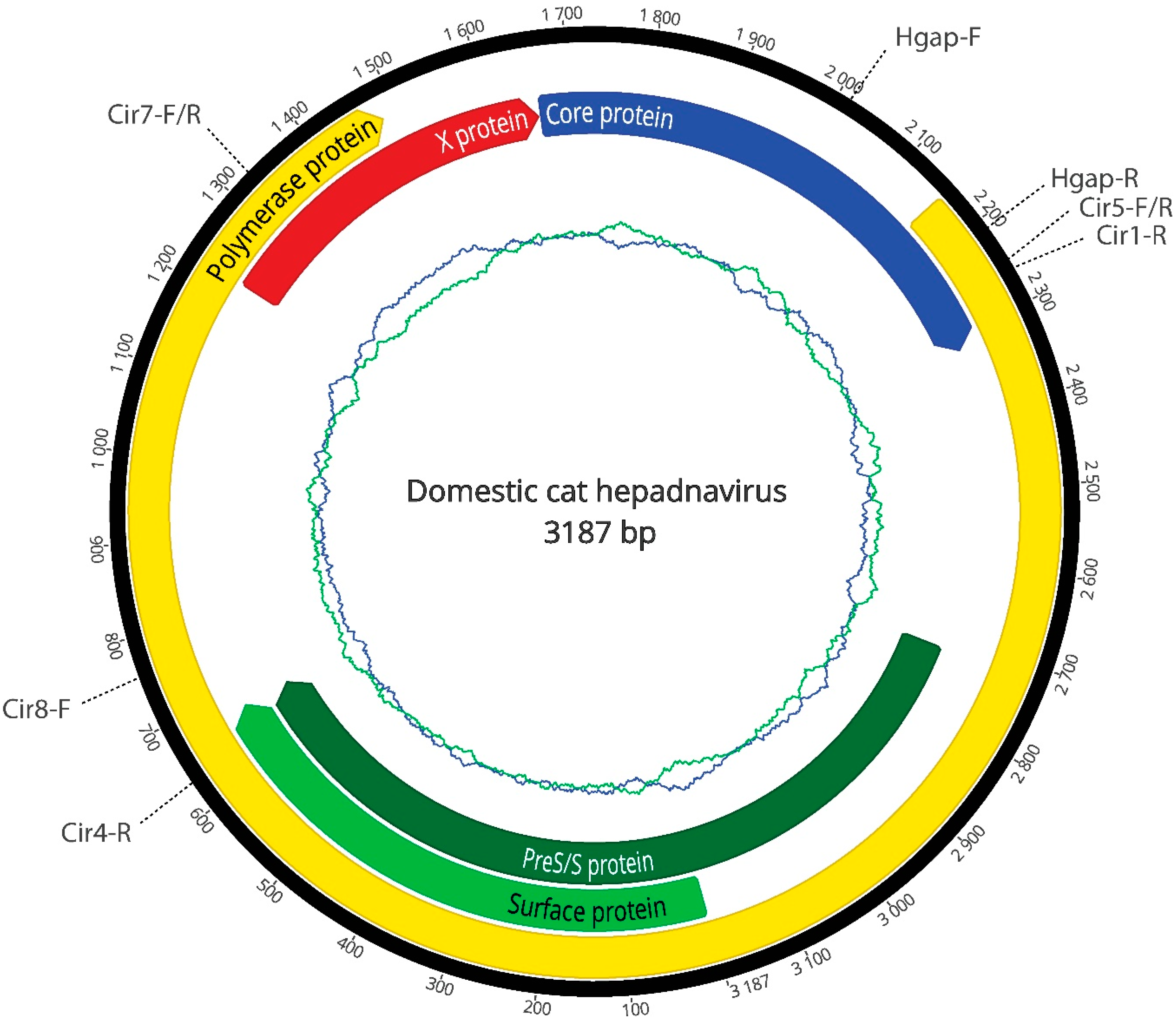
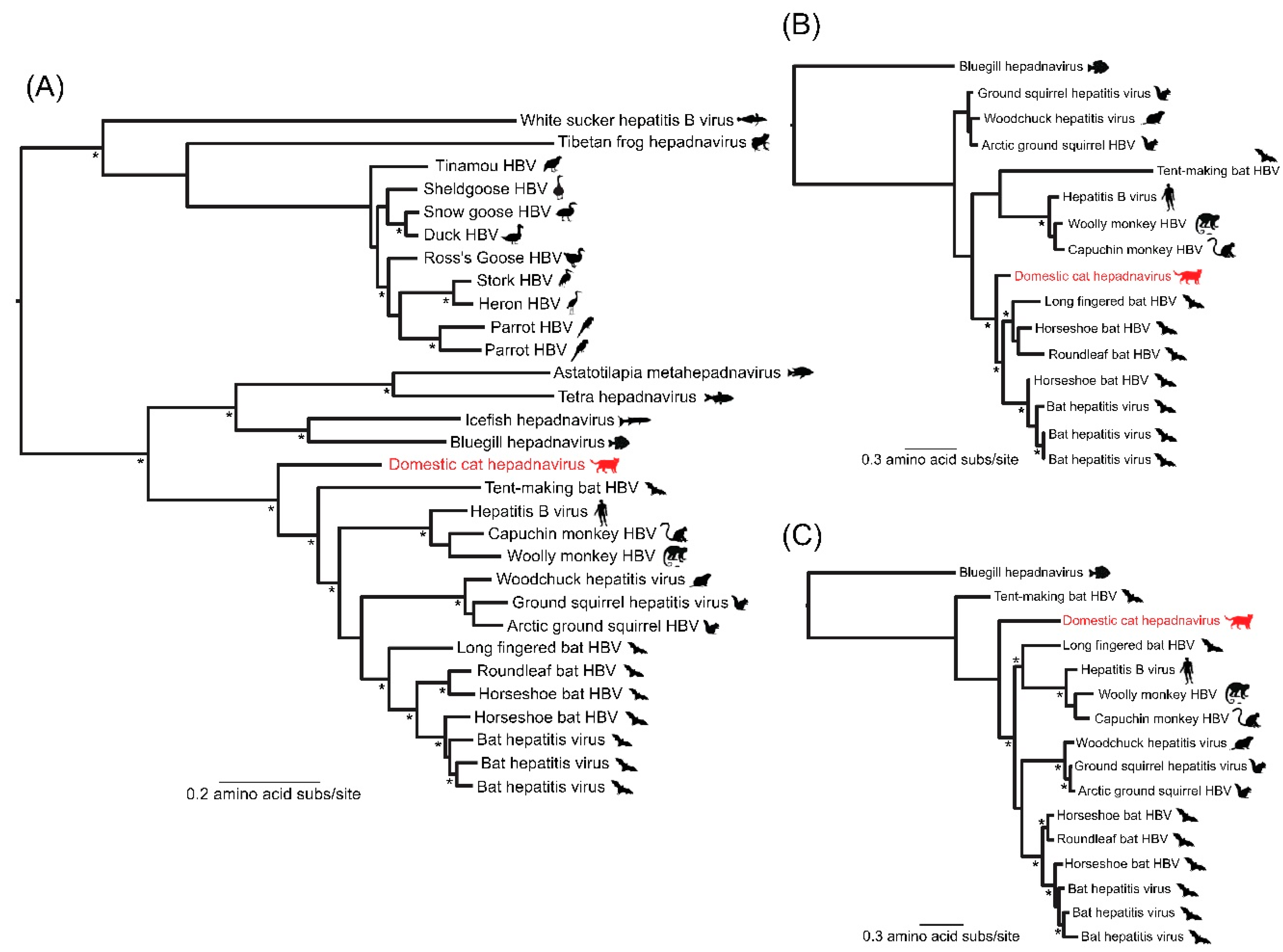

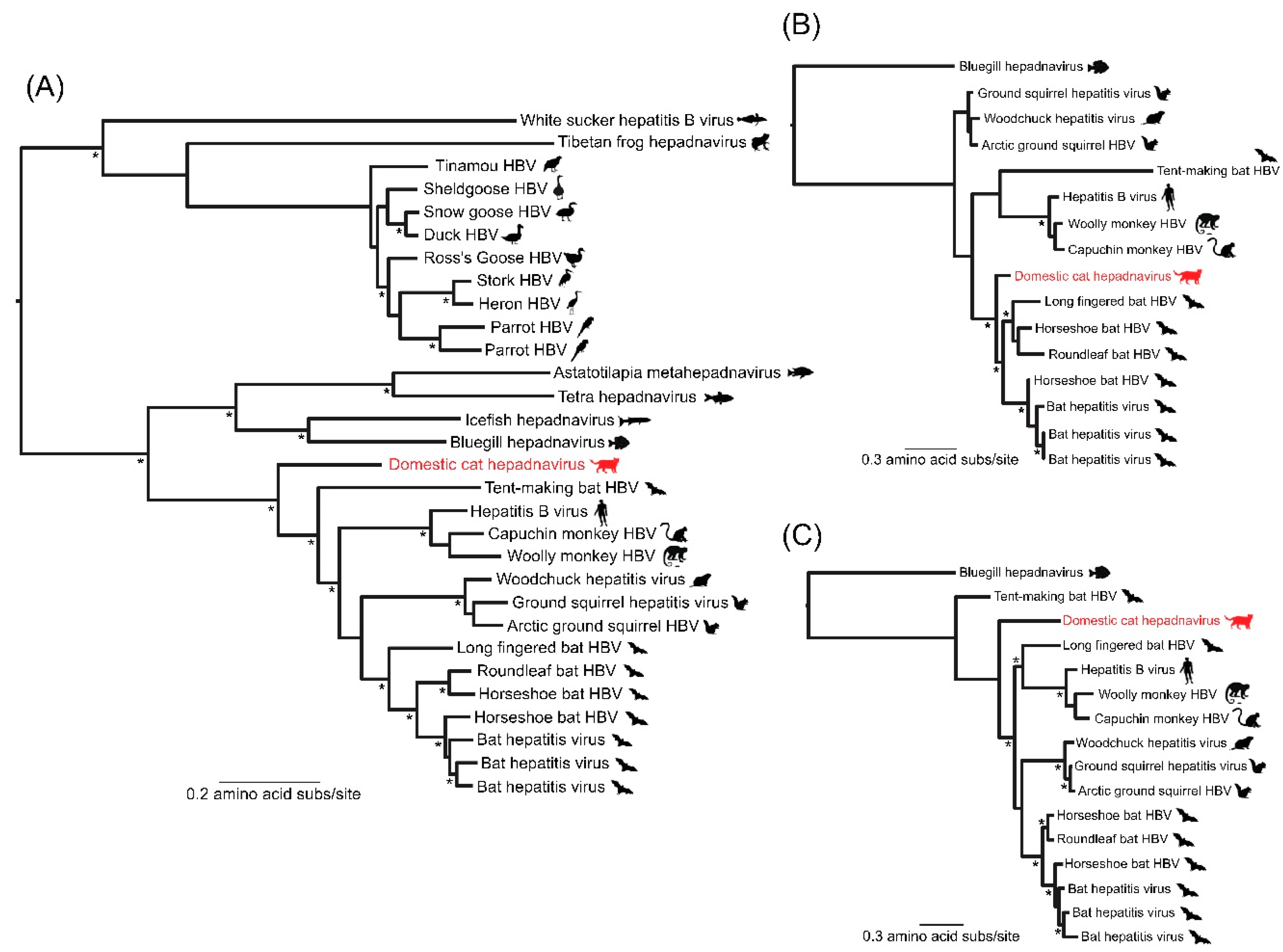
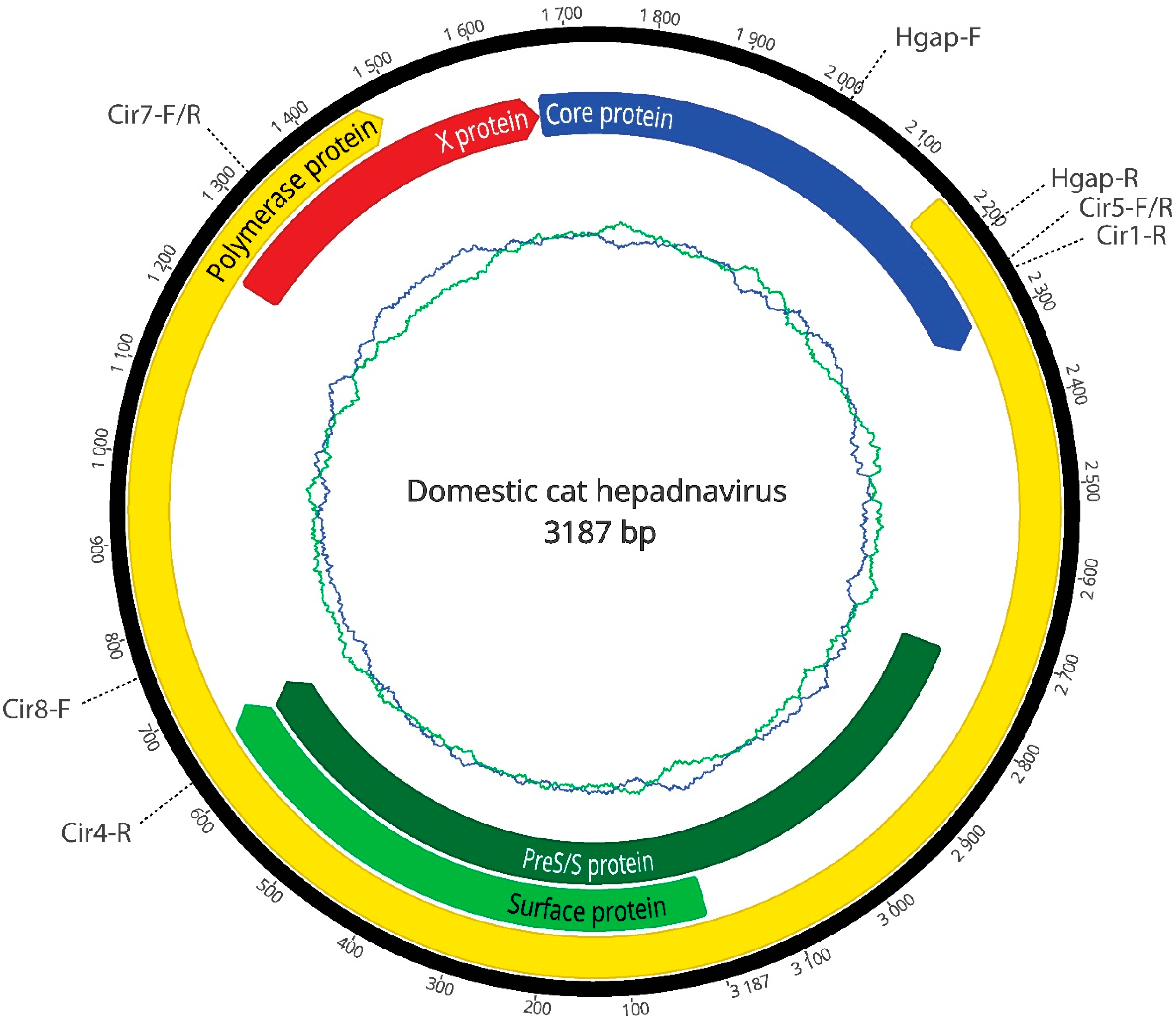{kind=link}
{kind=link}
| Oligonucleotides | Sequence | Size (bp) | Tm (°C) |
|---|---|---|---|
| Cir5-F | 5′-TTGGCACCTGGATTCGCA-3′ | 1400 | 57 |
| Cir4-R | 5′-AGATGTTCCACACTCTTAGCC-3′ | ||
| Cir8-F | 5′-TTGGCACCTGGATTCGCA-3′ | 900 | 58 |
| Cir7-R | 5′-CGTAGACGAAGGACACGTC-3′ | ||
| Cir7-F | 5′-CCATCGATTTACACACTTCCCA-3′ | 950 | 57 |
| Cir5-R | 5′-TGCGAATCCAGGTGCCAA-3′ | ||
| Cir1-R | 5′-ATAACCGTATGCTCCGGAAG-3′ | 1000 | 55 |
| Hgap-F | 5′-GTGCTCTGATAACCGTATGCTC-3′ | 230 | 55 |
| Hgap-R | 5′-CTAGAATGGCTACATGGGGTTAG-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghazadeh, M.; Shi, M.; Barrs, V.R.; McLuckie, A.J.; Lindsay, S.A.; Jameson, B.; Hampson, B.; Holmes, E.C.; Beatty, J.A. A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia. Viruses 2018, 10, 269. https://doi.org/10.3390/v10050269
Aghazadeh M, Shi M, Barrs VR, McLuckie AJ, Lindsay SA, Jameson B, Hampson B, Holmes EC, Beatty JA. A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia. Viruses. 2018; 10(5):269. https://doi.org/10.3390/v10050269
Chicago/Turabian StyleAghazadeh, Mahdis, Mang Shi, Vanessa R. Barrs, Alicia J. McLuckie, Scott A. Lindsay, Barbara Jameson, Bronte Hampson, Edward C. Holmes, and Julia A. Beatty. 2018. "A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia" Viruses 10, no. 5: 269. https://doi.org/10.3390/v10050269
APA StyleAghazadeh, M., Shi, M., Barrs, V. R., McLuckie, A. J., Lindsay, S. A., Jameson, B., Hampson, B., Holmes, E. C., & Beatty, J. A. (2018). A Novel Hepadnavirus Identified in an Immunocompromised Domestic Cat in Australia. Viruses, 10(5), 269. https://doi.org/10.3390/v10050269