Paradoxical Effect of Chloroquine Treatment in Enhancing Chikungunya Virus Infection

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Prophylactic Treatment in a Preclinical NHP Model
2.1.1. Animals
2.1.2. Viral Stock
2.1.3. Animal Treatment and Infection
2.1.4. In Vitro Infection of Primary Macrophages and Fibroblast Cells
2.1.5. Plasma Viral RNA Extraction and Quantification
2.1.6. Virion-Based Ig ELISA
2.1.7. Cytokine and ELISPOT Assays
2.2. Curative Treatment from the CuraChik Clinical Trial
Biochemical Analysis of Human Samples
2.3. Statistical Analysis
3. Results
3.1. Pre-Clinical Studies
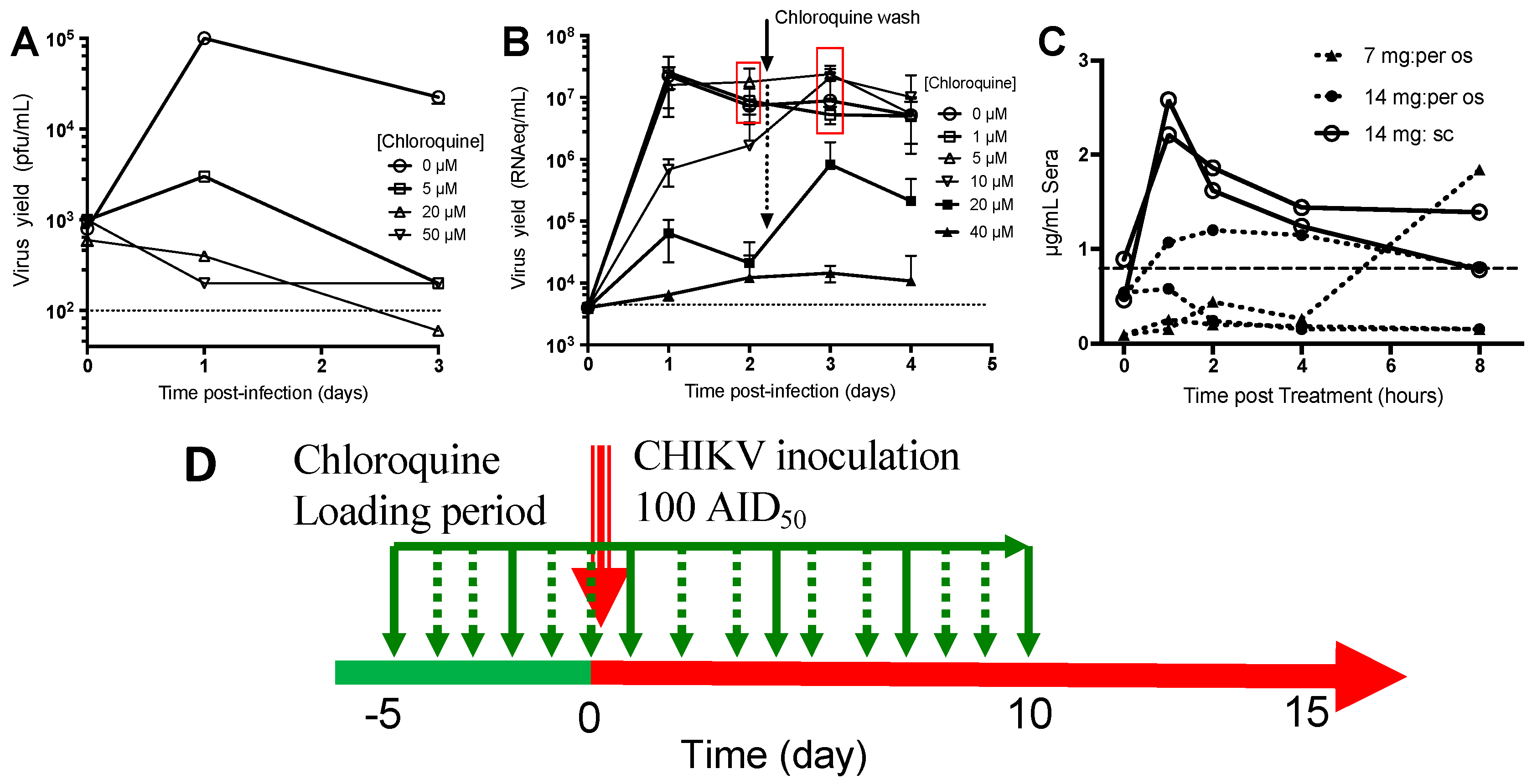
3.1.1. Chloroquine Inhibits CHIKV Replication in Monocyte-Derived Macrophages and Fibroblasts
3.1.2. Pharmacokinetics of Chloroquine in Macaque
3.1.3. Chloroquine Treatment Exacerbates Acute Chikungunya Fever in Macaques
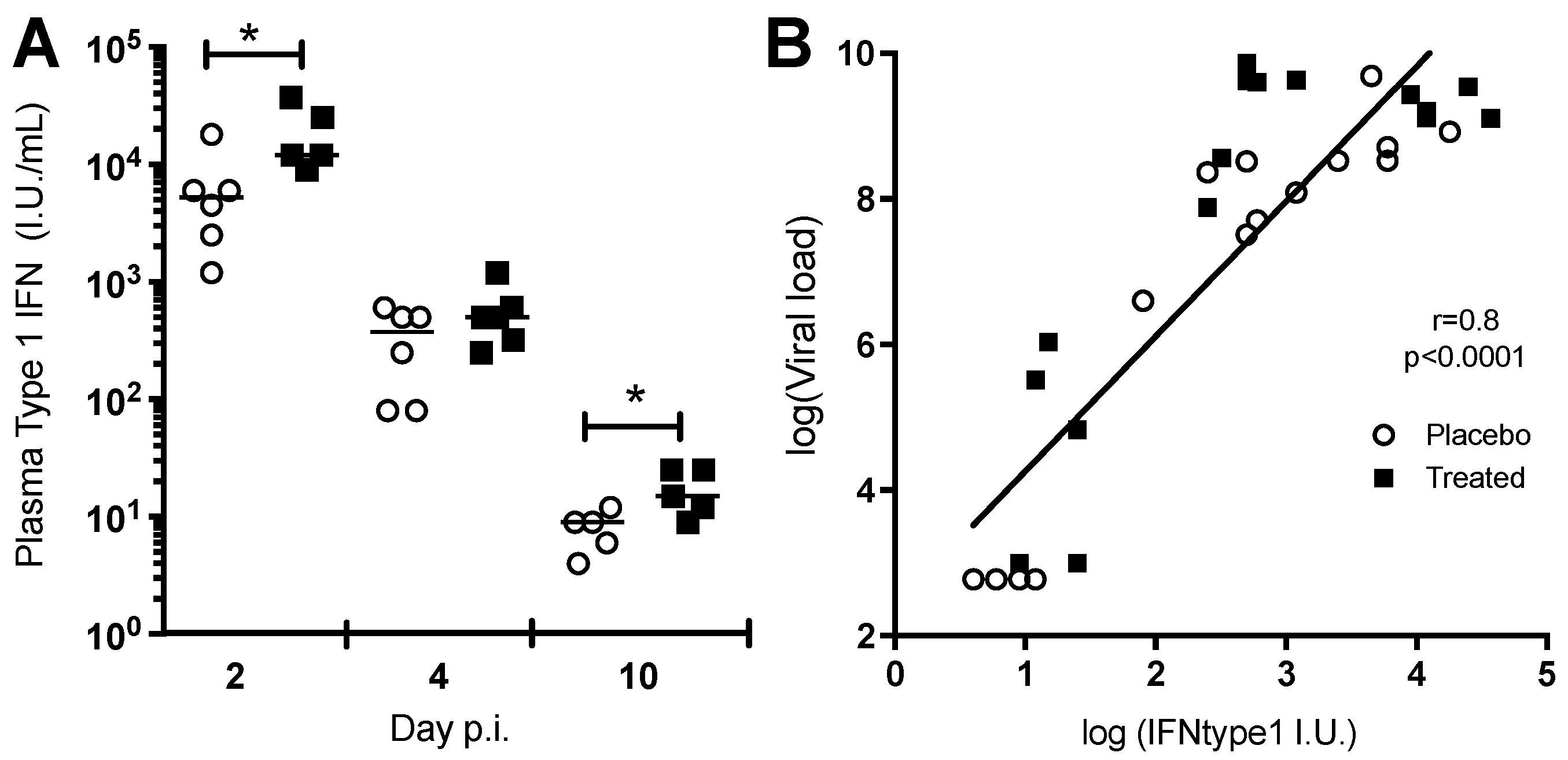
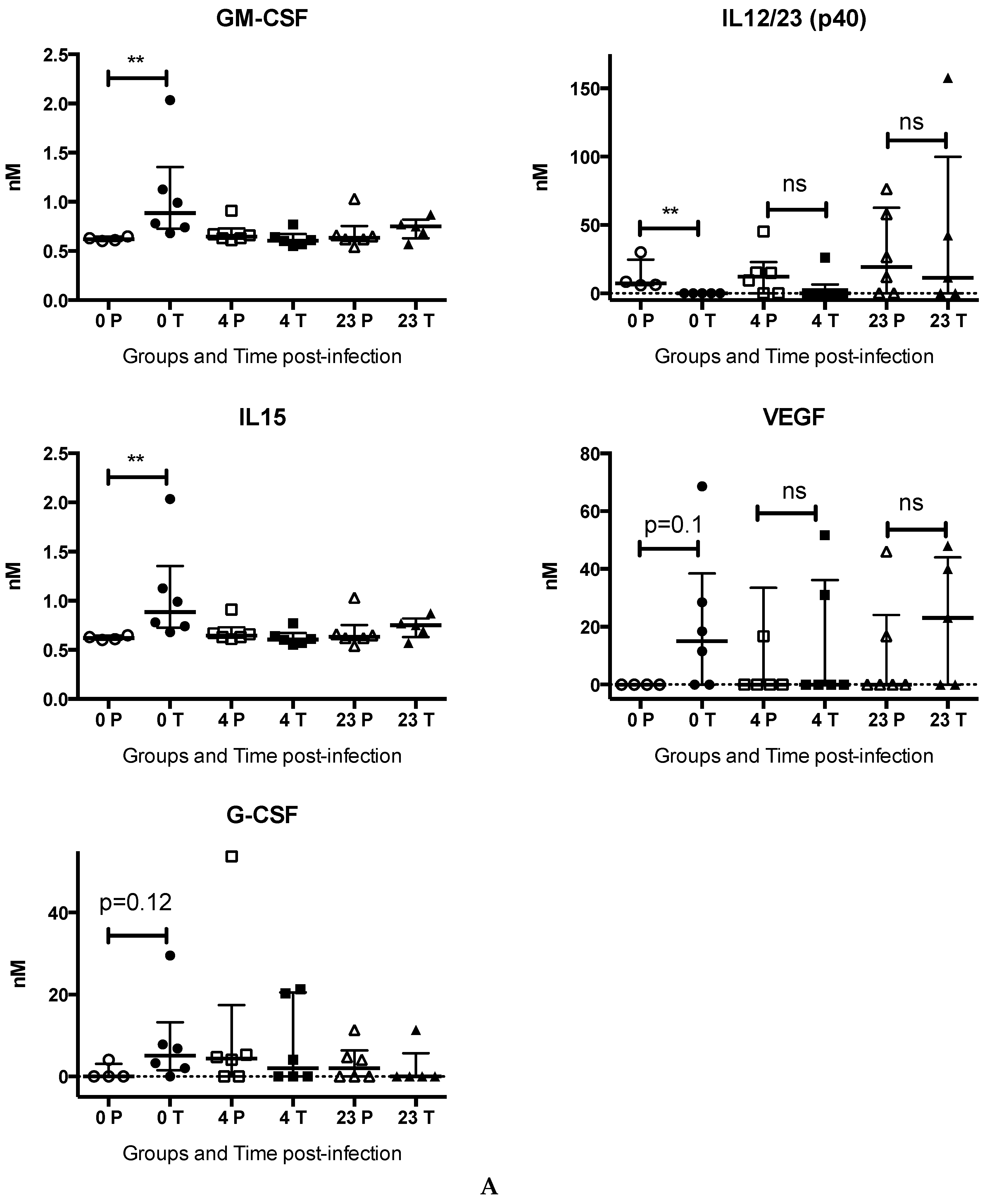
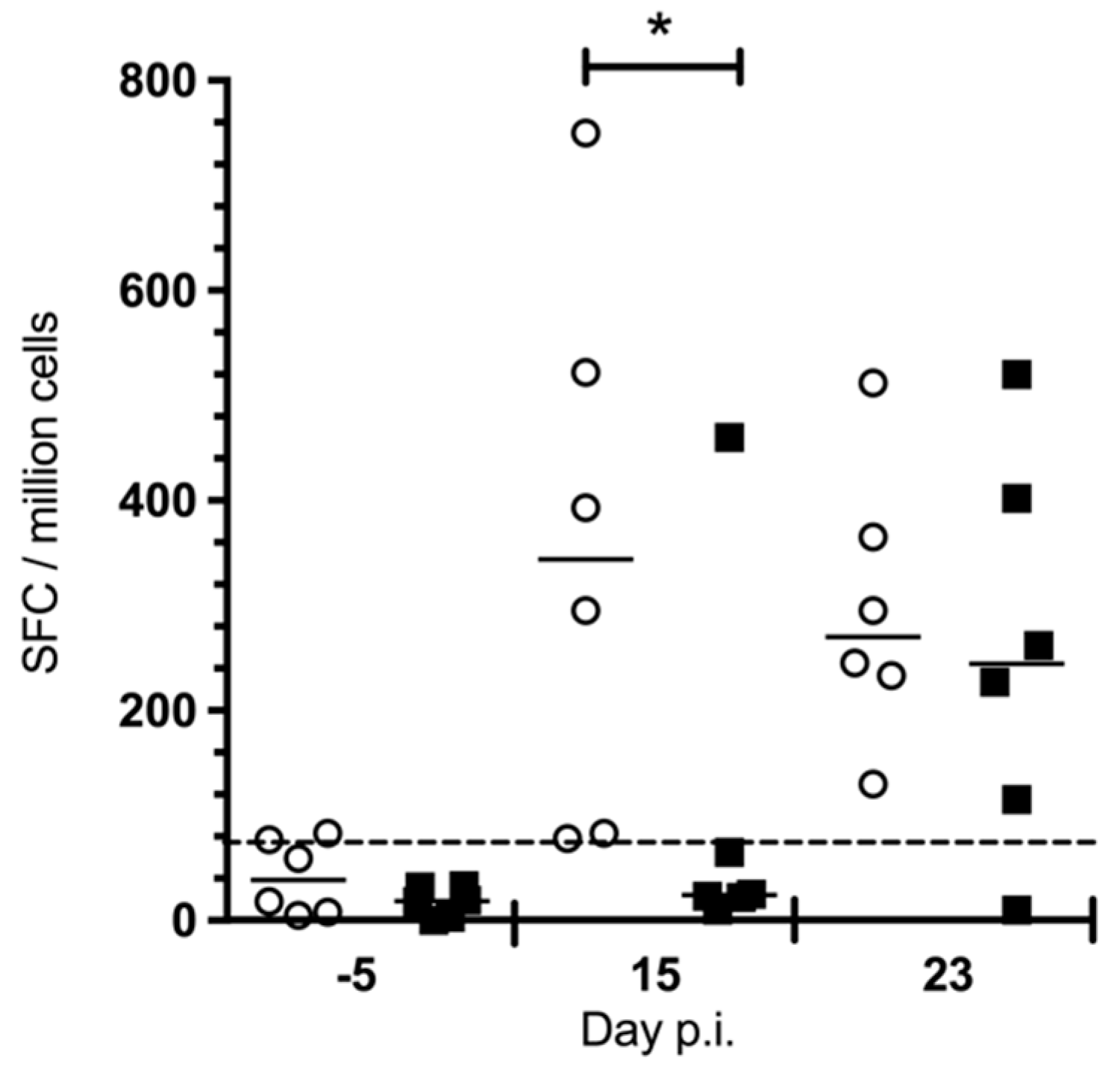
3.1.4. Chloroquine Skews the Immune Response to CHIKV
3.2 Clinical Trial
Immunological Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mason, P.J.; Haddow, A.J. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–53: An additional note on Chikungunya virus isolations and serum antibodies. Trans. R. Soc. Trop. Med. Hyg. 1957, 51, 238–240. [Google Scholar] [CrossRef]
- Dupuis-Maguiraga, L.; Noret, M.; Brun, S.; Le Grand, R.; Gras, G.; Roques, P. Chikungunya disease: Infection-associated markers from the acute to the chronic phase of arbovirus-induced arthralgia. PLoS Negl. Trop. Dis. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Courderc, T.; Ng, L.F.; Hallengärd, D.; Powers, A.; Lecuit, M.; Esteban, M.; Merits, A.; Roques, P.; Liljeström, P. Therapeutics and vaccines against chikungunya virus. Vector Borne Zoonotic Dis. 2015, 15, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Bettadapura, J.; Herrero, L.J.; Taylor, A.; Mahalingam, S. Approaches to the treatment of disease induced by chikungunya virus. Indian J. Med. Res. 2013, 138, 762–765. [Google Scholar] [PubMed]
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Cassell, S.; Edwards, J.; Brown, D.T. Effects of lysosomotropic weak bases on infection of BHK-21 cells by Sindbis virus. J. Virol. 1984, 52, 857–864. [Google Scholar] [PubMed]
- Coombs, K.; Mann, E.; Edwards, J.; Brown, D.T. Effects of chloroquine and cytochalasin B on the infection of cells by Sindbis virus and vesicular stomatitis virus. J. Virol. 1981, 37, 1060–1065. [Google Scholar] [PubMed]
- Helenius, A.; Marsh, M.; White, J. Inhibition of Semliki forest virus penetration by lysosomotropic weak bases. J. Gen. Virol. 1982, 58, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Inglot, A.D. Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs. J. Gen. Virol. 1969, 4, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Shimizu, Y.; Yamamoto, S.; Homma, M.; Ishida, N. Effect of chloroquine on the growth of animal viruses. Arch. Die Gesamte Virusforsch. 1972, 36, 93–104. [Google Scholar] [CrossRef]
- De Lamballerie, X.; Boisson, V.; Reynier, J.C.; Enault, S.; Charrel, R.N.; Flahault, A.; Roques, P.; Le Grand, R. On chikungunya acute infection and chloroquine treatment. Vector Borne Zoonotic Dis. 2008, 8, 837–839. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Santhosh, S.R.; Tiwari, M.; Lakshmana Rao, P.V.; Parida, M. Assessment of in vitro prophylactic and therapeutic efficacy of chloroquine against Chikungunya virus in vero cells. J. Med. Virol. 2010, 82, 817–824. [Google Scholar] [CrossRef] [PubMed]
- De Lamballerie, X.; Ninove, L.; Charrel, R.N. Antiviral treatment of chikungunya virus infection. Infect. Disord. Drug Targets 2009, 9, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Thiberville, S.D.; Boisson, V.; Gaudart, J.; Simon, F.; Flahault, A.; de Lamballerie, X. Chikungunya Fever: A clinical and virological investigation of outpatients on reunion island, South-west Indian ocean. PLoS Negl. Trop. Dis. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Spouge, J.L. Statistical analysis of sparse infection data and its implications for retroviral treatment trials in primates. Proc. Natl. Acad. Sci. USA 1992, 89, 7581–7585. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.M.; Mallet, M.N.; Raoult, D. Correlation between serum doxycycline concentrations and serologic evolution in patients with Coxiella burnetii endocarditis. J. Infect. Dis. 2003, 188, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Malleret, B.; Maneglier, B.; Karlsson, I.; Lebon, P.; Nascimbeni, M.; Perié, L.; Brochard, P.; Delache, B.; Calvo, J.; Andrieu, T.; et al. Primary infection with simian immunodeficiency virus: Plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood 2008, 112, 4598–4608. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Kaldma, K.; Sikut, R.; Culina, S.; Romain, G.; Tuomela, M.; Adojaan, M.; Männik, A.; Toots, U.; Kivisild, T.; et al. Persistent immune responses induced by a human immunodeficiency virus DNA vaccine delivered in association with electroporation in the skin of nonhuman primates. Hum. Gene Ther. 2009, 20, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Zeger, S.L.; Liang, K.Y.; Albert, P.S. Models for longitudinal data: A generalized estimating equation approach. Biometrics 1988, 44, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, J.J.; Jaffar Bandjee, M.C.; Trotot, P.K.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef] [PubMed]
- Mzayek, F.; Deng, H.; Mather, F.J.; Wasilevich, E.C.; Liu, H.; Hadi, C.M.; Chansolme, D.H.; Murphy, H.A.; Melek, B.H.; Tenaglia, A.N.; et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin. Trials 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Couderc, T.; Khandoudi, N.; Grandadam, M.; Visse, C.; Gangneux, N.; Bagot, S.; Prost, J.F.; Lecuit, M. Prophylaxis and therapy for Chikungunya virus infection. J. Infect. Dis. 2009, 200, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Jaffar-Bandjee, M.C.; Das, T.; Hoarau, J.J.; Krejbich Trotot, P.; Denizot, M.; Ribera, A.; Roques, P.; Gasque, P. Chikungunya virus takes centre stage in virally induced arthritis: Possible cellular and molecular mechanisms to pathogenesis. Microbes Infect. 2009, 11, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.-W.; Simarmata, D.; Chow, A.; Her, Z.; Teng, T.-S.; Ong, E.K.S.; Renia, L.; Leo, Y.-S.; Ng, L.F.P. Early appearance of neutralizing IgG3-antibodies is associated with Chikungunya Virus clearance and long-term clinical protection. J. Infect. Dis. 2012, in press. [Google Scholar]
- Teng, T.S.; Kam, Y.W.; Lee, B.; Hapuarachchi, H.C.; Wimal, A.; Ng, L.C.; Ng, L.F. A systematic meta-analysis of immune signatures in patients with acute chikungunya virus infection. J. Infect. Dis. 2015, 211, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Rainsford, K.D.; Parke, A.L.; Clifford-Rashotte, M.; Kean, W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015, 23, 231–269. [Google Scholar] [CrossRef] [PubMed]
- Soumahoro, M.-K.; Boëlle, P.-Y.; Gauzere, B.A.; Atsou, K.; Pelat, C.; Lambert, B.; La Ruche, G.; Gastellu-Etchegorry, M.; Renault, P.; Sarazin, M.; et al. The chikungunya epidemic on la réunion Island in 2005–2006: A cost-of-illness study. PLoS Negl. Trop. Dis. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Brighton, S.W. Chloroquine phosphate treatment of chronic Chikungunya arthritis. An open pilot study. S. Afr. Med. J. 1984, 66, 217–218. [Google Scholar] [PubMed]
- Savarino, A.; Cauda, R.; Cassone, A. On the use of chloroquine for chikungunya. Lancet Infect. Dis. 2007, 7. [Google Scholar] [CrossRef]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Guerrero-Plata, A.; Gilfoy, F.D.; Garofalo, R.P.; Mason, P.W. Differential activation of human monocyte-derived and plasmacytoid dendritic cells by West Nile virus generated in different host cells. J. Virol. 2007, 81, 13640–13648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [PubMed]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.; Wong, S.C.; Kwek, D.J.; Tolou, H.; Lin, R.T.; Tambyah, P.A.; Rénia, L.; et al. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar] [CrossRef] [PubMed]
- Roques, P.; Gras, G.; Labadie, K.; Larcher, T.; Cherel, Y.; Suhrbier, A.; Grand, R.L. Chikungunya virus infection involved monocytes and during chronic phase of the disease persisted in tissue macrophages. In Proceedings of the 45th Annual Scientific Meeting of the European Society for Clinical Investigation, Crete, Greece, 13–16 April 2011. [Google Scholar]
- Maheshwari, R.K.; Srikantan, V.; Bhartiya, D. Chloroquine enhances replication of Semliki Forest virus and encephalomyocarditis virus in mice. J. Virol. 1991, 65, 992–995. [Google Scholar] [PubMed]
- Seth, P.; Mani, H.; Singh, A.K.; Banaudha, K.K.; Madhavan, S.; Sidhu, G.S.; Gaddipati, J.P.; Vogel, S.N.; Maheshwari, R.K. Acceleration of viral replication and up-regulation of cytokine levels by antimalarials: implications in malaria-endemic areas. Am. J. Trop. Med. Hyg. 1999, 61, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.M.; Stecher, V.J.; Danis, E.; Pruden, D.J.; LaBrie, T. Alteration of interleukin-1 activity and the acute phase response in adjuvant arthritic rats treated with disease modifying antirheumatic drugs. Agents Actions 1988, 25, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.J.; Russell, A.S. Re-evaluation of antimalarials in treating rheumatic diseases: Re-appreciation and insights into new mechanisms of action. Curr. Opin. Rheumatol. 2011, 23, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Werneke, S.W.; Schilte, C.; Rohatgi, A.; Monte, K.J.; Michault, A.; Arenzana-Seisdedos, F.; Vanlandingham, D.L.; Higgs, S.; Fontanet, A.; Albert, M.L.; et al. ISG15 is critical in the control of Chikungunya Virus infection independent of UbE1L mediated conjugation. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, H.K.; Unanue, E.R. Decrease in macrophage antigen catabolism caused by ammonia and chloroquine is associated with inhibition of antigen presentation to T cells. Proc. Natl. Acad. Sci. USA 1982, 79, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.T.; Flavell, R.A.; Liljeström, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Belizaire, R.; Unanue, E.R. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc. Natl. Acad. Sci. USA 2009, 106, 17463–17468. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I.; Kang, H.I. Mechanism of action of antimalarial drugs: Inhibition of antigen processing and presentation. Lupus 1993, 2 (Suppl. 1), S9–S12. [Google Scholar] [PubMed]
- Accapezzato, D.; Visco, V.; Francavilla, V.; Molette, C.; Donato, T.; Paroli, M.; Mondelli, M.U.; Doria, M.; Torrisi, M.R.; Barnaba, V. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 2005, 202, 817–828. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
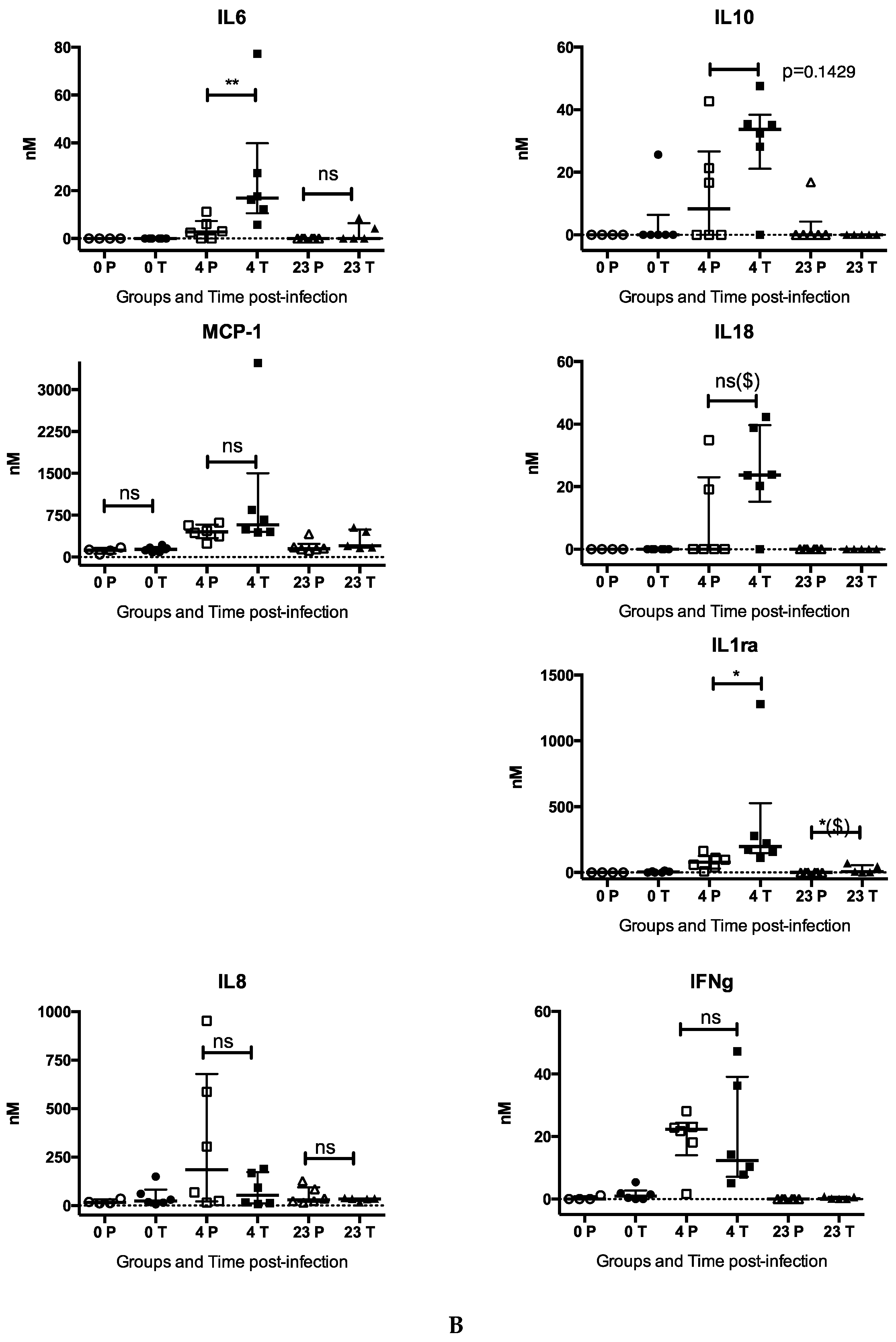{kind=link}
{kind=link}
{kind=link}
| Ig Class | Group | ≤450 | 4000 | 37,000 | ≥110,000 | p Value (Chi2 Test) |
|---|---|---|---|---|---|---|
| IgM titer | Placebo | 0 $ | 2 | 4 | 0 | |
| Chloroquine | 1 | 5 | 0 | 0 | 0.0432 | |
| IgG titer | Placebo | 0 | 0 | 4 | 2 | |
| Chloroquine | 1 | 0 | 5 | 0 | 0.2111 |
| D300 | Placebo Group n = 27 | Chloroquine Group n = 19 | p-Value |
|---|---|---|---|
| Recovery N/T $ (%) | 21/26 (80.8) | 12/17 (70.6) | 0.48 * |
| Presence of Arthralgia N/T (%) | 6/26 (23.1) | 8/15 (53.3) | 0.08 * |
| N Joint involved (SD, mean-max) | 1.5 (3.55, 0–12) | 3.38 (4.6, 0–13) | 0.038 ** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roques, P.; Thiberville, S.-D.; Dupuis-Maguiraga, L.; Lum, F.-M.; Labadie, K.; Martinon, F.; Gras, G.; Lebon, P.; Ng, L.F.P.; De Lamballerie, X.; et al. Paradoxical Effect of Chloroquine Treatment in Enhancing Chikungunya Virus Infection. Viruses 2018, 10, 268. https://doi.org/10.3390/v10050268
Roques P, Thiberville S-D, Dupuis-Maguiraga L, Lum F-M, Labadie K, Martinon F, Gras G, Lebon P, Ng LFP, De Lamballerie X, et al. Paradoxical Effect of Chloroquine Treatment in Enhancing Chikungunya Virus Infection. Viruses. 2018; 10(5):268. https://doi.org/10.3390/v10050268
Chicago/Turabian StyleRoques, Pierre, Simon-Djamel Thiberville, Laurence Dupuis-Maguiraga, Fok-Moon Lum, Karine Labadie, Frédéric Martinon, Gabriel Gras, Pierre Lebon, Lisa F. P. Ng, Xavier De Lamballerie, and et al. 2018. "Paradoxical Effect of Chloroquine Treatment in Enhancing Chikungunya Virus Infection" Viruses 10, no. 5: 268. https://doi.org/10.3390/v10050268
APA StyleRoques, P., Thiberville, S.-D., Dupuis-Maguiraga, L., Lum, F.-M., Labadie, K., Martinon, F., Gras, G., Lebon, P., Ng, L. F. P., De Lamballerie, X., & Le Grand, R. (2018). Paradoxical Effect of Chloroquine Treatment in Enhancing Chikungunya Virus Infection. Viruses, 10(5), 268. https://doi.org/10.3390/v10050268