A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages



Abstract
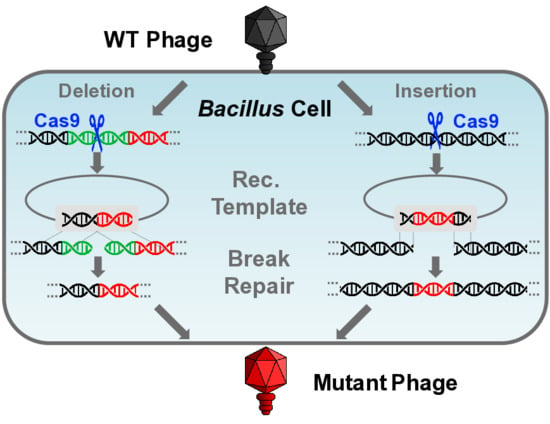
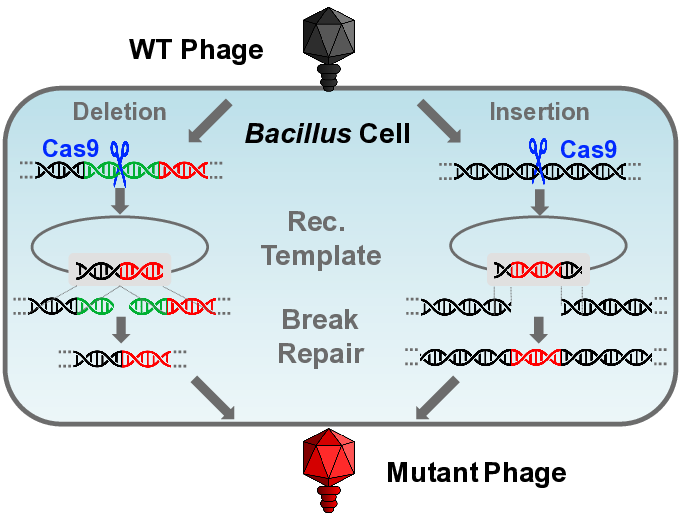
:
1. Introduction
2. Materials and Methods
2.1. CutSPR
2.2. Media and Solutions
2.3. Cloning
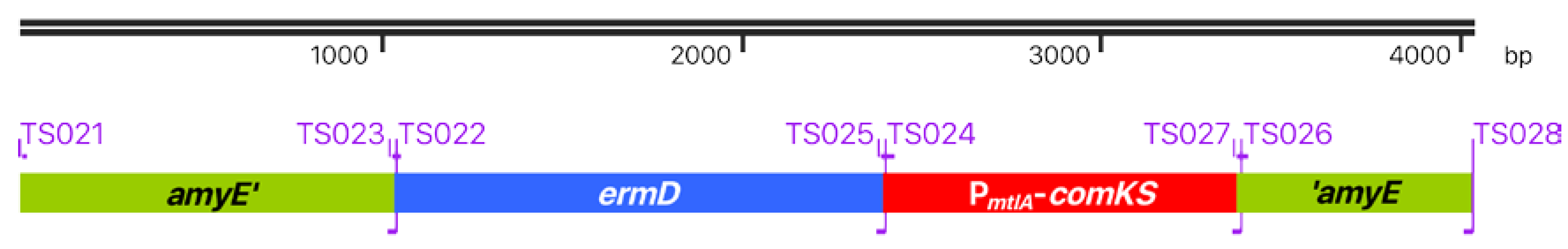
2.4. Primer Design for the Super-Competence Cassette
2.5. Cloning of the sgRNA Targeting Genes Goe1_c00180 and Goe1_c00030
2.6. Recombination Cassette Cloning
2.7. Construction of Mutagenesis Vectors
2.8. Plaque Assay and Analysis
2.9. Specific Plaque Assay
2.10. Phage Propagation in Liquid Culture
2.11. Transmission Electron Microscopy
2.12. Pairwise Sequence Alignment
3. Results
3.1. CutSPR
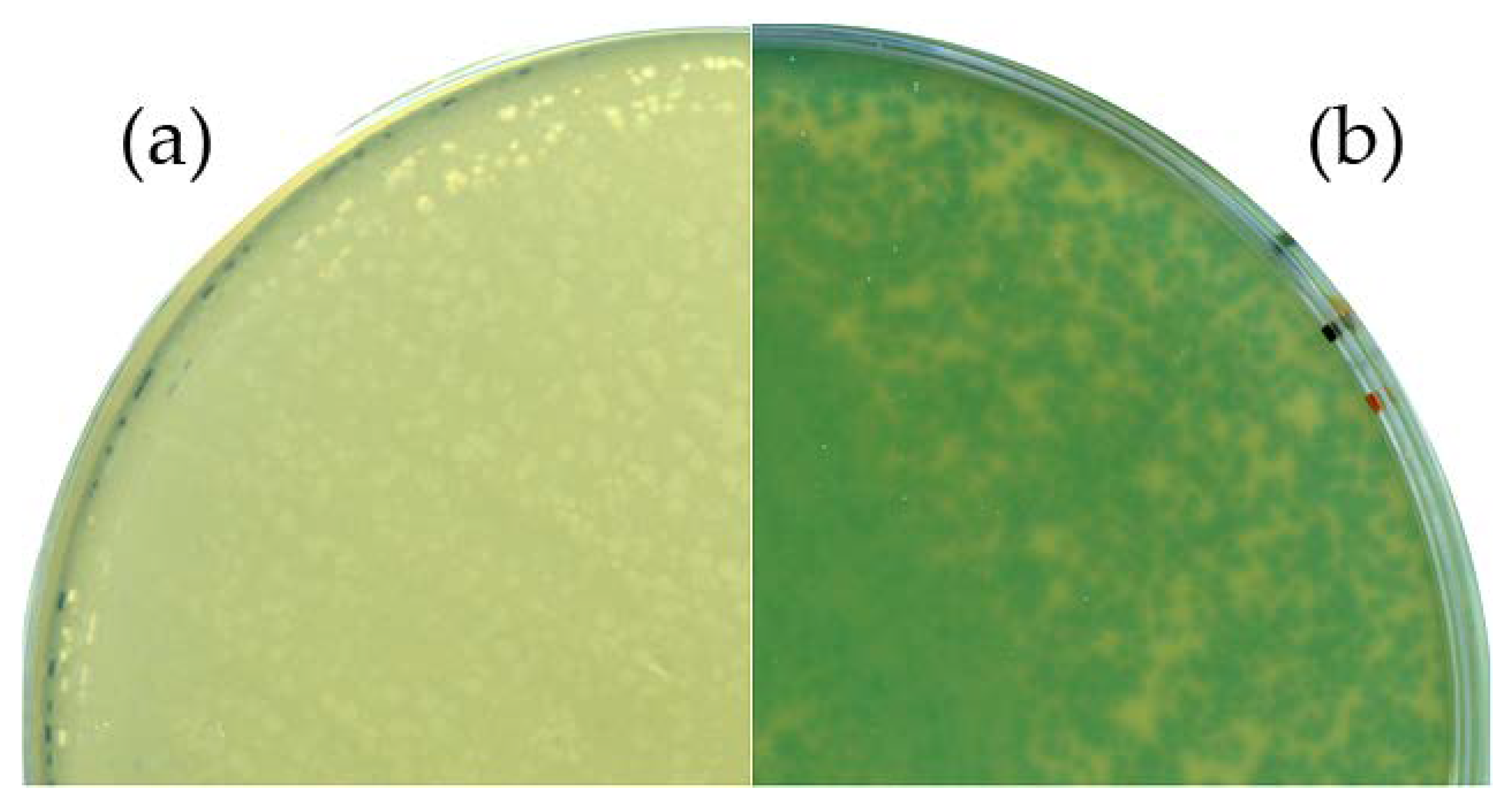
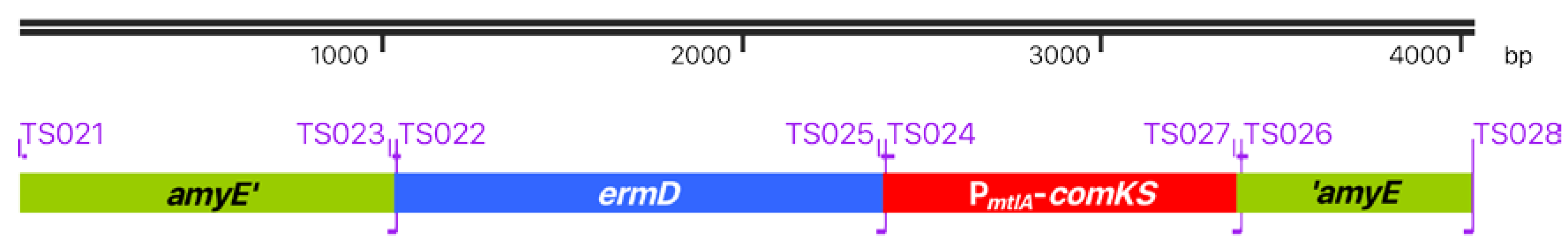
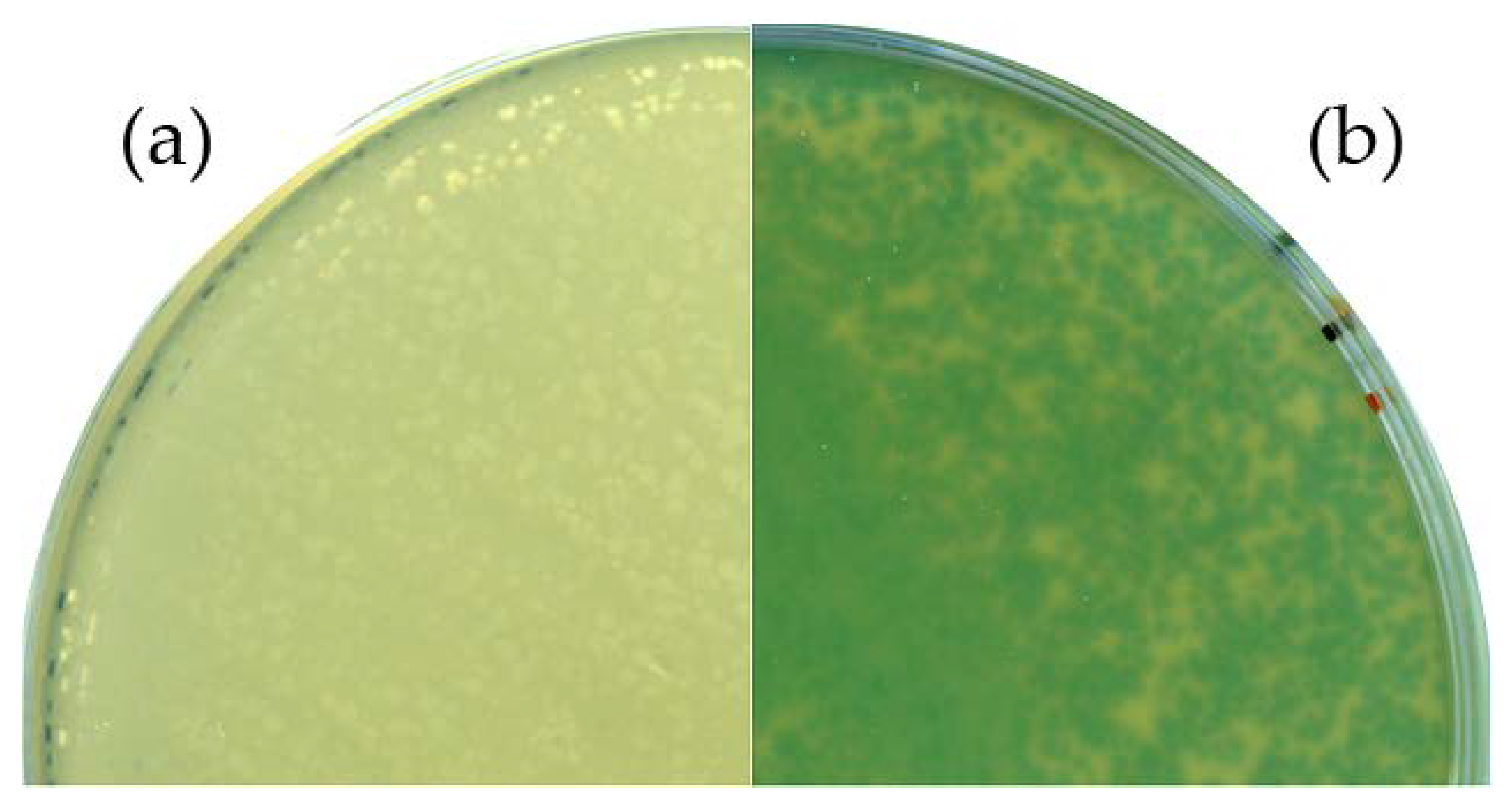
3.2. B. Subtilis TS01
3.3. Genome Modification of vB_BsuP-Goe1
3.3.1. Gene Deletion
3.3.2. Gene Insertion
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Khairnar, K.; Raut, M.P.; Chandekar, R.H.; Sanmukh, S.G.; Paunikar, W.N. Novel bacteriophage therapy for controlling metallo-beta-lactamase producing Pseudomonas aeruginosa infection in catfish. BMC Vet. Res. 2013, 9, 264. [Google Scholar] [CrossRef]
- Ho, Y.-H.; Tseng, C.-C.; Wang, L.-S.; Chen, Y.-T.; Ho, G.-J.; Lin, T.-Y.; Wang, L.-Y.; Chen, L.-K. Application of Bacteriophage-containing Aerosol against Nosocomial Transmission of Carbapenem-Resistant Acinetobacter baumannii in an Intensive Care Unit. PLoS ONE 2016, 11, e0168380. [Google Scholar] [CrossRef]
- WHO. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/ (accessed on 16 November 2017).
- Szaleniec, J.; Górski, A.; Szaleniec, M.; Międzybrodzki, R.; Weber-Dąbrowska, B.; Stręk, P.; Składzień, J. Can phage therapy solve the problem of recalcitrant chronic rhinosinusitis? Future Microbiol. 2017, 12, 1427–1442. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M.; Pajunen, M.; Kiljunen, S. Biotechnological challenges of phage therapy. Biotechnol. Lett. 2007, 29, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A bioinformatic tool for comparative bacteriophage genomics. BMC Bioinform. 2011, 12, 395. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Murugan, K.; Babu, K.; Sundaresan, R.; Rajan, R.; Sashital, D.G. The Revolution Continues: Newly Discovered Systems Expand the CRISPR-Cas Toolkit. Mol. Cell 2017, 68, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Terns, M.P.; Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.-È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Wu, X.; Tang, W.-C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M. Anthrax. In Facts on File, 1st ed.; Alcano, I.E., Ed.; Facts on File News Services: New York, NY, USA, 2003; ISBN 9780791073025. [Google Scholar]
- Schoeni, J.L.; Wong, A.C.L. Bacillus cereus food poisoning and its toxins. J. Food Prot. 2005, 68, 636–648. [Google Scholar] [PubMed]
- Schallmey, M.; Singh, A.; Ward, O.P. Developments in the use of Bacillus species for industrial production. Can. J. Microbiol. 2004, 50, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Altenbuchner, J. Editing of the Bacillus subtilis Genome by the CRISPR-Cas9 System. Appl. Environ. Microbiol. 2016, 82, 5421–5427. [Google Scholar] [CrossRef] [PubMed]
- Willms, I.M.; Hertel, R. Phage vB_BsuP-Goe1: The smallest identified lytic phage of Bacillus subtilis. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Westers, H.; Dorenbos, R.; van Dijl, J.M.; Kabel, J.; Flanagan, T.; Devine, K.M.; Jude, F.; Seror, S.J.; Beekman, A.C.; Darmon, E.; et al. Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol. Biol. Evol. 2003, 20, 2076–2090. [Google Scholar] [CrossRef] [PubMed]
- Spizizen, J. Transformation of biochemically deficient strains of bacillus subtilis by deoxyribonucleate. Proc. Natl. Acad. Sci. USA 1958, 44, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Jensen, G.L.; Burnett, S.H.; Breakwell, D.P. Correction: Genomic comparison of 93 Bacillus phages reveals 12 clusters, 14 singletons and remarkable diversity. BMC Genom. 2014, 15, 1184. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- SantaLucia, J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Bertani, G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951, 62, 293–300. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; ISBN 0879695773. [Google Scholar]
- Wenzel, M.; Altenbuchner, J. Development of a markerless gene deletion system for Bacillus subtilis based on the mannose phosphoenolpyruvate-dependent phosphotransferase system. Microbiology 2015, 161, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Rahmer, R.; Morabbi Heravi, K.; Altenbuchner, J. Construction of a Super-Competent Bacillus subtilis 168 Using the PmtlA-comKS Inducible Cassette. Front. Microbiol. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Willms, I.; Hoppert, M.; Hertel, R. Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3. Viruses 2017, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Valentine, R.C.; Shapiro, B.M.; Stadtman, E.R. Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 1968, 7, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Hoppert, M.; Holzenburg, A. Electron Microscopy in Microbiology (Royal Microscopical Society Microscopy Handbooks 43); BIOS Scientific Publishers: Oxford, UK, 1998; ISBN 1859960162. [Google Scholar]
- Rachinger, M.; Volland, S.; Meinhardt, F.; Daniel, R.; Liesegang, H. First Insights into the Completely Annotated Genome Sequence of Bacillus licheniformis Strain 9945A. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.P.; Lingohr, E.J.; Ceyssens, P.-J.; Kropinski, A.M. Bacillus Phage phi29, Complete Genome. Available online: http://www.ncbi.nlm.nih.gov/nuccore/194186868 (accessed on 3 January 2018).
- Reilly, B.E.; Nelson, R.A.; Anderson, D.L. Morphogenesis of bacteriophage phi 29 of Bacillus subtilis: Mapping and functional analysis of the head fiber gene. J. Virol. 1977, 24, 363–377. [Google Scholar] [PubMed]
- Salas, M.; Vásquez, C.; Méndez, E.; Viñuela, E. Head fibers of bacteriophage phi 29. Virology 1972, 50, 180–188. [Google Scholar] [CrossRef]
- Hirata, H.; Fukazawa, T.; Negoro, S.; Okada, H. Structure of a beta-galactosidase gene of Bacillus stearothermophilus. J. Bacteriol. 1986, 166, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Rachinger, M.; Bauch, M.; Strittmatter, A.; Bongaerts, J.; Evers, S.; Maurer, K.-H.; Daniel, R.; Liebl, W.; Liesegang, H.; Ehrenreich, A. Size unlimited markerless deletions by a transconjugative plasmid-system in Bacillus licheniformis. J. Biotechnol. 2013, 167, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Weller, G.R.; Kysela, B.; Roy, R.; Tonkin, L.M.; Scanlan, E.; Della, M.; Devine, S.K.; Day, J.P.; Wilkinson, A.; D’Adda di Fagagna, F.; et al. Identification of a DNA nonhomologous end-joining complex in bacteria. Science 2002, 297, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Charusanti, P.; Zhang, L.; Weber, T.; Lee, S.Y. CRISPR-Cas9 Based Engineering of Actinomycetal Genomes. ACS Synth. Biol. 2015, 4, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Liu, F.; Gu, P.; Jin, H.; Chang, Y.; Wang, Q.; Liang, Q.; Qi, Q. A CRISPR-Cas9 Assisted Non-Homologous End-Joining Strategy for One-step Engineering of Bacterial Genome. Sci. Rep. 2016, 6, 37895. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Bikard, D. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016, 44, 4243–4251. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, C.; Weidinger, L.A.F.; Dörrbecker, B.; Hübner, S.; Stülke, J.; Commichau, F.M. SPINE: A method for the rapid detection and analysis of protein–protein interactions in vivo. Proteomics 2007, 7, 4032–4035. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | ∆6 | REG19 | TS01 |
|---|---|---|---|
| 1 | 0 | 11 | 120 |
| 2 | 0 | 64 | 714 |
| 3 | 4 | 111 | 445 |
| 4 | no data | no data | 599 |
| average | 133 | 6200 | 42,633 |
| Modification | Verified Plaques | Correct Mutants | Efficiency |
|---|---|---|---|
| Goe1 Δc00180 | 36 | 15 | 40% |
| Goe1 Δc00030 | 40 | 4 | 10% |
| Goe1 Δc00180::bgaB | 40 | 2 | 5% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilling, T.; Dietrich, S.; Hoppert, M.; Hertel, R. A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses 2018, 10, 241. https://doi.org/10.3390/v10050241
Schilling T, Dietrich S, Hoppert M, Hertel R. A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses. 2018; 10(5):241. https://doi.org/10.3390/v10050241
Chicago/Turabian StyleSchilling, Tobias, Sascha Dietrich, Michael Hoppert, and Robert Hertel. 2018. "A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages" Viruses 10, no. 5: 241. https://doi.org/10.3390/v10050241
APA StyleSchilling, T., Dietrich, S., Hoppert, M., & Hertel, R. (2018). A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses, 10(5), 241. https://doi.org/10.3390/v10050241