Concepts in Light Microscopy of Viruses

 , and
, and
Abstract
1. Introduction
2. Probes and Labeling Strategies for Imaging of Viruses
2.1. Labeling of Chemically Fixed Specimens with Single Molecule Sensitivity
2.2. Live Cell Imaging of Viruses Labeled with Organic Fluorophores and Quantum-Dots
2.3. Transgenic Approaches for Live Cell Imaging of Viruses
3. Diffraction Limited Microscopy
3.1. Confocal Microscopy
3.2. Multi-Photon Imaging
3.3. Fluorescence Resonance Energy Transfer
3.4. TIRF Microscopy
3.5. Selective Plane Illumination Microscopy
3.6. Expansion Microscopy
4. Super-Resolution Microscopy
4.1. Super-Resolution Imaging
4.2. Image Scanning Microscopy
5. Image Processing
5.1. Deconvolution
5.2. Software Based Super-Resolution
5.3. Data Analysis
6. Emerging Techniques
6.1. Correlative Light and Electron Microscopy
6.2. Digital Holographic Microscopy
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CARS | Coherent Anti-Stokes Raman Spectroscopy |
| CLEM | Correlative Light and Electron Microscopy |
| CLSM | Confocal Laser Scanning Microscope |
| DHM | Digital Holographic Microscope |
| dSTORM | Direct Stochastic Optical Reconstruction Microscopy |
| EM | Electron Microscope |
| ExM | Expansion Microscopy |
| FRAP | Fluorescence Recovery after Photobleaching |
| FLIM | Fluorescence-Lifetime Imaging Microscopy |
| FRET | Fluorescence/Förster Resonance Energy Transfer |
| iSIM | Instant Structured Illumination Microscopy |
| ISM | Image Scanning Microscopy |
| OPFOS | Orthogonal Plane Fluorescence Optical Sectioning |
| PAINT | Points Accumulation for Imaging in Nanoscale Topography |
| PALM | Photo-Activated Localization Microscopy |
| QE | Quantum Efficiency |
| SDCM | Spinning-Disk Confocal Microscope |
| SIM | Structured Illumination Microscopy |
| SNR | Signal-to-Noise Ratio |
| SPIM | Selective Plane Illumination Microscopy |
| STED | Stimulated Emission Depletion |
| STORM | Stochastic Optical Reconstruction Microscopy |
| TIRF | Total Internal Reflection Fluorescence |
| AAV | Adeno-Associated Virus |
| AdV | Adenovirus |
| HBV | Hepatitis B Virus |
| HCV | Hepatitis C Virus |
| HIV | Human Immune-Deficiency Virus |
| HPV | Human Papilloma Virus |
| HSV | Herpes Simplex Virus |
| IAV | Influenza A Virus |
| RSV | Respiratory Syncytial Virus |
| SV | Simian Virus |
| TMV | Tobacco Mosaic Virus |
| VACV | Vaccinia Virus |
| bDNA | Branched DNA |
| ExM | Expansion Microscopy |
| (e)GFP | (Enhanced) Green Fluorescent Protein |
| (F)ISH | (Fluorescence) In Situ Hybridization |
| FUCCI | Fluorescent Ubiquitination-based Cell Cycle Indicator |
| IF | Immunofluorescence |
| Q-Dots | Quantum Dots |
| SiR | Silicon-Rhodamine |
| smFISH | Single Molecule FISH |
| TRITC | Tetramethylrhodamine |
| CLI | Command-Line Interface |
| GUI | Graphical User Interface |
| ML | Machine Learning |
| SOFI | Super-resolution Optical Fluctuation Imaging |
| SRRF | Super-Resolution Radial Fluctuations |
References
- Helenius, A.; Kartenbeck, J.; Simons, K.; Fries, E. On the entry of Semliki forest virus into BHK-21 cells. J. Cell Biol. 1980, 84, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Matlin, K.S.; Reggio, H.; Helenius, A.; Simons, K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 1981, 91, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Garoff, H.; Helenius, A. How an animal virus gets into and out of its host cell. Sci. Am. 1982, 246, 58–66. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Chemistry the Nobel Prize in Chemistry 1925. Available online: https://www.nobelprize.org/nobel_prizes/chemistry/laureates/1925/index.html (accessed on 3 September 2017).
- The Nobel Prize in Physics the Nobel Prize in Physics 1953. Available online: https://www.nobelprize.org/nobel_prizes/physics/laureates/1953/ (accessed on 3 September 2017).
- The Nobel Prize in Physics the Nobel Prize in Physics 1971. Available online: https://www.nobelprize.org/nobel_prizes/physics/laureates/1971/ (accessed on 3 September 2017).
- The Nobel Prize in Physics the Nobel Prize in Physics 1986. Available online: https://www.nobelprize.org/nobel_prizes/physics/laureates/1986/ (accessed on 3 September 2017).
- The Nobel Prize in Chemistry the Nobel Prize in Chemistry 2014. Available online: https://www.nobelprize.org/nobel_prizes/chemistry/laureates/2014/ (accessed on 26 May 2017).
- The Nobel Prize in Chemistry the Nobel Prize in Chemistry 2017. Available online: https://www.nobelprize.org/nobel_prizes/chemistry/laureates/2017/ (accessed on 6 November 2017).
- Lane, N. The unseen world: Reflections on Leeuwenhoek (1677) “Concerning little animals”. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. Mikrosk. Anat. 1873, 9, 413–418. [Google Scholar] [CrossRef]
- Kausche, G.A.; Pfankuch, E.; Ruska, H. Die Sichtbarmachung von pflanzlichem Virus im Übermikroskop. Naturwissenschaften 1939, 27, 292–299. [Google Scholar] [CrossRef]
- Franklin, R.E. Structure of tobacco mosaic virus. Nature 1955, 175, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C.; Olson, A.J.; Schutt, C.E.; Winkler, F.K.; Bricogne, G. Tomato bushy stunt virus at 2.9 A resolution. Nature 1978, 276, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-H.; Burckhardt, C.; Yakimovich, A.; Greber, U. Imaging, Tracking and Computational Analyses of Virus Entry and Egress with the Cytoskeleton. Viruses 2018, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Korobchevskaya, K.; Lagerholm, B.; Colin-York, H.; Fritzsche, M. Exploring the potential of airyscan microscopy for live cell imaging. Photonics 2017, 4, 41. [Google Scholar] [CrossRef]
- Hanne, J.; Göttfert, F.; Schimer, J.; Anders-Össwein, M.; Konvalinka, J.; Engelhardt, J.; Müller, B.; Hell, S.W.; Kräusslich, H.-G. Stimulated Emission Depletion Nanoscopy Reveals Time-Course of Human Immunodeficiency Virus Proteolytic Maturation. ACS Nano 2016, 10, 8215–8222. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, B.; Zhuang, X. Virus trafficking-learning from single-virus tracking. Nat. Rev. Microbiol. 2007, 5, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Nägerl, U.V.; Willig, K.I.; Hein, B.; Hell, S.W.; Bonhoeffer, T. Live-cell imaging of dendritic spines by STED microscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 18982–18987. [Google Scholar] [CrossRef] [PubMed]
- Zernike, F. Phase contrast, a new method for the microscopic observation of transparent objects. Physica 1942, 9, 686–698. [Google Scholar] [CrossRef]
- Zernike, F. Phase contrast, a new method for the microscopic observation of transparent objects part II. Physica 1942, 9, 974–986. [Google Scholar] [CrossRef]
- Marquet, P.; Rappaz, B.; Magistretti, P.J.; Cuche, E.; Emery, Y.; Colomb, T.; Depeursinge, C. Digital holographic microscopy: A noninvasive contrast imaging technique allowing quantitative visualization of living cells with subwavelength axial accuracy. Opt. Lett. 2005, 30, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.; Yu, L.; Lo, C.-M.; Kim, M. High-resolution quantitative phase-contrast microscopy by digital holography. Opt. Express 2005, 13, 8693–8698. [Google Scholar] [CrossRef] [PubMed]
- Rappaz, B.; Marquet, P.; Cuche, E.; Emery, Y.; Depeursinge, C.; Magistretti, P. Measurement of the integral refractive index and dynamic cell morphometry of living cells with digital holographic microscopy. Opt. Express 2005, 13, 9361–9373. [Google Scholar] [CrossRef] [PubMed]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005, 307, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, S.; Gao, D.; Hu, D.; Gong, P.; Sheng, Z.; Deng, J.; Ma, Y.; Cai, L. Click-functionalized compact quantum dots protected by multidentate-imidazole ligands: Conjugation-ready nanotags for living-virus labeling and imaging. J. Am. Chem. Soc. 2012, 134, 8388–8391. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [PubMed]
- Pleiner, T.; Bates, M.; Trakhanov, S.; Lee, C.-T.; Schliep, J.E.; Chug, H.; Böhning, M.; Stark, H.; Urlaub, H.; Görlich, D. Nanobodies: Site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation. eLife 2015, 4, e11349. [Google Scholar] [CrossRef] [PubMed]
- Opazo, F.; Levy, M.; Byrom, M.; Schäfer, C.; Geisler, C.; Groemer, T.W.; Ellington, A.D.; Rizzoli, S.O. Aptamers as potential tools for super-resolution microscopy. Nat. Methods 2012, 9, 938–939. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.M.; Brinkley, B.R.; Boughter, J.M. Immunofluorescence of mitotic spindles by using monospecific antibody against bovine brain tubulin. Science 1975, 187, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.G.; Pardue, M.L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 63, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N.G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Leyton-Puig, D.; Kedziora, K.M.; Isogai, T.; van den Broek, B.; Jalink, K.; Innocenti, M. PFA fixation enables artifact-free super-resolution imaging of the actin cytoskeleton and associated proteins. Biol. Open 2016, 5, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.; Heaton, N.S.; Gao, Q.; Palese, P.; Singer, R.H.; Lionnet, T. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Mor, A.; White, A.; Zhang, K.; Thompson, M.; Esparza, M.; Muñoz-Moreno, R.; Koide, K.; Lynch, K.W.; García-Sastre, A.; Fontoura, B.M.A. Influenza virus mRNA trafficking through host nuclear speckles. Nat. Microbiol. 2016, 1, 16069. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Trehan, K.; Ong, M.-L.; Luna, J.M.; Hoffmann, H.-H.; Espiritu, C.; Sheahan, T.P.; Chandrasekar, H.; Schwartz, R.E.; Christine, K.S.; et al. Viral genome imaging of hepatitis C virus to probe heterogeneous viral infection and responses to antiviral therapies. Virology 2016, 494, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Player, A.N.; Shen, L.P.; Kenny, D.; Antao, V.P.; Kolberg, J.A. Single-copy gene detection using branched DNA (bDNA) in situ hybridization. J. Histochem. Cytochem. 2001, 49, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Kenny, D.; Shen, L.-P.; Kolberg, J.A. Detection of viral infection and gene expression in clinical tissue specimens using branched DNA (bDNA) in situ hybridization. J. Histochem. Cytochem. 2002, 50, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Nolte, F.S.; Boysza, J.; Thurmond, C.; Clark, W.S.; Lennox, J.L. Clinical comparison of an enhanced-sensitivity branched-DNA assay and reverse transcription-PCR for quantitation of human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 1998, 36, 716–720. [Google Scholar] [PubMed]
- Itzkovitz, S.; van Oudenaarden, A. Validating transcripts with probes and imaging technology. Nat. Methods 2011, 8, S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs inhibit Zika virus replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Makowska, Z.; Campana, B.; Calabrese, D.; Dill, M.T.; Chung, J.; Chisari, F.V.; Heim, M.H. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2014, 59, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, W.; Zheng, Y.; Wang, W.; Bai, L.; Chen, L.; Feng, Y.; Zhang, Z.; Yuan, Z. In situ analysis of intrahepatic virological events in chronic hepatitis B virus infection. J. Clin. Investig. 2016, 126, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Ukpo, O.C.; Ma, X.-J.; Flanagan, J.J.; Luo, Y.; Thorstad, W.L.; Chernock, R.D. Transcriptionally-active high-risk human papillomavirus is rare in oral cavity and laryngeal/hypopharyngeal squamous cell carcinomas—A tissue microarray study utilizing E6/E7 mRNA in situ hybridization. Histopathology 2012, 60, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, A.; Hochstrasser, R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef] [PubMed]
- Jungmann, R.; Steinhauer, C.; Scheible, M.; Kuzyk, A.; Tinnefeld, P.; Simmel, F.C. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010, 10, 4756–4761. [Google Scholar] [CrossRef] [PubMed]
- Jungmann, R.; Avendaño, M.S.; Woehrstein, J.B.; Dai, M.; Shih, W.M.; Yin, P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 2014, 11, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef] [PubMed]
- Auer, A.; Strauss, M.T.; Schlichthaerle, T.; Jungmann, R. Fast, Background-Free DNA-PAINT Imaging Using FRET-Based Probes. Nano Lett. 2017, 17, 6428–6434. [Google Scholar] [CrossRef] [PubMed]
- Jungmann, R.; Avendaño, M.S.; Dai, M.; Woehrstein, J.B.; Agasti, S.S.; Feiger, Z.; Rodal, A.; Yin, P. Quantitative super-resolution imaging with qPAINT. Nat. Methods 2016, 13, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Suomalainen, M.; Stidwill, R.P.; Boucke, K.; Ebersold, M.W.; Helenius, A. The role of the nuclear pore complex in adenovirus DNA entry. EMBO J. 1997, 16, 5998–6007. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Mosberger, N.; Fornerod, M.; Stidwill, R.P.; Greber, U.F. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 2001, 3, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, B.K. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Flatt, J.W.; Greber, U.F. Misdelivery at the Nuclear Pore Complex-Stopping a Virus Dead in Its Tracks. Cells 2015, 4, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Sekine, E.; Schmidt, N.; Gaboriau, D.; O’Hare, P. Spatiotemporal dynamics of HSV genome nuclear entry and compaction state transitions using bioorthogonal chemistry and super-resolution microscopy. PLoS Pathog. 2017, 13, e1006721. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Müller, B.; Kräusslich, H.-G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 2014, 3, e04114. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; DeLuca, N.A. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Cortez, D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 2012, 7, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Arend, K.C.; Li, Y.; Yamane, D.; McGivern, D.R.; Kato, T.; Wakita, T.; Moorman, N.J.; Lemon, S.M. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 2015, 17, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Kang, J.H.; Girona, G.E.; Aramburu, I.V.; Lemke, E.A. Labeling proteins on live mammalian cells using click chemistry. Nat. Protoc. 2015, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Prescher, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. USA 2010, 107, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Groskreutz, D.J.; Babor, E.C.; Monick, M.M.; Varga, S.M.; Hunninghake, G.W. Respiratory syncytial virus limits alpha subunit of eukaryotic translation initiation factor 2 (eIF2alpha) phosphorylation to maintain translation and viral replication. J. Biol. Chem. 2010, 285, 24023–24031. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, Q.; Satoh, T.; Arii, J.; Lanier, L.L.; Spear, P.G.; Kawaguchi, Y.; Arase, H. Binding of herpes simplex virus glycoprotein B (gB) to paired immunoglobulin-like type 2 receptor alpha depends on specific sialylated O-linked glycans on gB. J. Virol. 2009, 83, 13042–13045. [Google Scholar] [CrossRef] [PubMed]
- Sivan, G.; Weisberg, A.S.; Americo, J.L.; Moss, B. Retrograde Transport from Early Endosomes to the trans-Golgi Network Enables Membrane Wrapping and Egress of Vaccinia Virus Virions. J. Virol. 2016, 90, 8891–8905. [Google Scholar] [CrossRef] [PubMed]
- Click Chemistry Toolbox and Reagents | Click Chemistry Tools. Available online: https://clickchemistrytools.com/product-category/click-chemistry-toolbox/ (accessed on 7 March 2018).
- Bächi, T. Direct observation of the budding and fusion of an enveloped virus by video microscopy of viable cells. J. Cell Biol. 1988, 107, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Webster, P.; Weber, J.; Helenius, A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996, 15, 1766–1777. [Google Scholar] [PubMed]
- Suomalainen, M.; Nakano, M.Y.; Keller, S.; Boucke, K.; Stidwill, R.P.; Greber, U.F. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 1999, 144, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Zhang, L.; Yan, Z.; Engelhardt, J.F. Intracellular trafficking of adeno-associated viral vectors. Gene Ther. 2005, 12, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Ewers, H.; Rajamäki, M.-L.; Day, P.M.; Schiller, J.T.; Helenius, A. Human papillomavirus type 16 entry: Retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 2008, 4, e1000148. [Google Scholar] [CrossRef] [PubMed]
- Endress, T.; Lampe, M.; Briggs, J.A.G.; Kräusslich, H.-G.; Bräuchle, C.; Müller, B.; Lamb, D.C. HIV-1-cellular interactions analyzed by single virus tracing. Eur. Biophys. J. 2008, 37, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Greber, U.F. Principles of virus uncoating: Cues and the snooker ball. Traffic 2016, 17, 569–592. [Google Scholar] [CrossRef] [PubMed]
- Flatt, J.W.; Greber, U.F. Viral mechanisms for docking and delivering at nuclear pore complexes. Semin. Cell Dev. Biol. 2017, 68, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F. Virus and host mechanics support membrane penetration and cell entry. J. Virol. 2016, 90, 3802–3805. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate immunity to adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, N.; Greber, U.F. Adenovirus signalling in entry. Cell. Microbiol. 2013, 15, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Greber, U.F. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol. 2013, 21, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, C.J.; Greber, U.F. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 2009, 5, e1000621. [Google Scholar] [CrossRef] [PubMed]
- Fejer, G.; Freudenberg, M.; Greber, U.F.; Gyory, I. Adenovirus-triggered innate signalling pathways. Eur. J. Microbiol. Immunol. 2011, 1, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F. Viral trafficking violations in axons: The herpesvirus case. Proc. Natl. Acad. Sci. USA 2005, 102, 5639–5640. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F. Signalling in viral entry. Cell. Mol. Life Sci. 2002, 59, 608–626. [Google Scholar] [CrossRef] [PubMed]
- Meier, O.; Greber, U.F. Adenovirus endocytosis. J. Gene Med. 2003, 5, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Way, M. A superhighway to virus infection. Cell 2006, 124, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Hogue, I.B.; Bosse, J.B.; Engel, E.A.; Scherer, J.; Hu, J.-R.; Del Rio, T.; Enquist, L.W. Fluorescent protein approaches in alpha herpesvirus research. Viruses 2015, 7, 5933–5961. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Döhner, K.; Sodeik, B. Viral interactions with the cytoskeleton: A hitchhiker’s guide to the cell. Cell. Microbiol. 2006, 8, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.D.; Way, M. Arp2/3-mediated actin-based motility: A tail of pathogen abuse. Cell Host Microbe 2013, 14, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Way, M. Coupling viruses to dynein and kinesin-1. EMBO J. 2011, 30, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-H.; Burckhardt, C.J.; Yakimovich, A.; Morf, M.K.; Greber, U.F. The nuclear export factor CRM1 controls juxta-nuclear microtubule-dependent virus transport. J. Cell Sci. 2017, 130, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Shilagard, T.; Xiao, S.-Y.; Snyder, N.; Lau, D.; Cicalese, L.; Weiss, H.; Vargas, G.; Lemon, S.M. Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology 2009, 137, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Cella, L.N.; Biswas, P.; Yates, M.V.; Mulchandani, A.; Chen, W. Quantitative assessment of in vivo HIV protease activity using genetically engineered QD-based FRET probes. Biotechnol. Bioeng. 2014, 111, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Draz, M.S.; Fang, B.A.; Li, L.; Chen, Z.; Wang, Y.; Xu, Y.; Yang, J.; Killeen, K.; Chen, F.F. Hybrid nanocluster plasmonic resonator for immunological detection of hepatitis B virus. ACS Nano 2012, 6, 7634–7643. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-G.; Moon, S.; Kuritzkes, D.R.; Demirci, U. Quantum dot-based HIV capture and imaging in a microfluidic channel. Biosens. Bioelectron. 2009, 25, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Tripp, R.A.; Anderson, L.J.; Nie, S. Real-time detection of virus particles and viral protein expression with two-color nanoparticle probes. J. Virol. 2005, 79, 8625–8628. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, Y.; Kim, J.; Park, E.; Song, R. Positively charged compact quantum Dot-DNA complexes for detection of nucleic acids. Chemphyschem 2009, 10, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.-Y.; Zheng, L.-L.; Gao, P.-F.; Yang, X.-X.; Li, C.-M.; Li, Y.F.; Huang, C.Z. Real-time light scattering tracking of gold nanoparticles- bioconjugated respiratory syncytial virus infecting HEp-2 cells. Sci. Rep. 2014, 4, 4529. [Google Scholar] [CrossRef] [PubMed]
- Herod, M.R.; Pineda, R.G.; Mautner, V.; Onion, D. Quantum dot labelling of adenovirus allows highly sensitive single cell flow and imaging cytometry. Small 2015, 11, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, W.; Yin, W.; Guo, J.; Zhang, Z.-P.; Zeng, D.; Zhang, X.; Wu, Y.; Zhang, X.-E.; Cui, Z. Single-Particle Tracking of Human Immunodeficiency Virus Type 1 Productive Entry into Human Primary Macrophages. ACS Nano 2017, 11, 3890–3903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ke, X.; Zheng, Z.; Zhang, C.; Zhang, Z.; Zhang, F.; Hu, Q.; He, Z.; Wang, H. Encapsulating quantum dots into enveloped virus in living cells for tracking virus infection. ACS Nano 2013, 7, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Zhang, L.-J.; Wang, Z.-G.; Zhang, Z.-L.; Wu, Q.-M.; Sun, E.-Z.; Shi, Y.-B.; Pang, D.-W. Globally visualizing the microtubule-dependent transport behaviors of influenza virus in live cells. Anal. Chem. 2014, 86, 3902–3908. [Google Scholar] [CrossRef] [PubMed]
- Derfus, A.M.; Chan, W.C.W.; Bhatia, S.N. Intracellular delivery of quantum dots for live cell labeling and organelle tracking. Adv. Mater. Weinh. 2004, 16, 961–966. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, M.; Li, W.; Zhang, Z.; Zhang, X.; Tan, T.; Zhang, X.-E.; Cui, Z. Live cell imaging of single genomic loci with quantum dot-labeled TALEs. Nat. Commun. 2017, 8, 15318. [Google Scholar] [CrossRef] [PubMed]
- Arndt-Jovin, D.J.; Jovin, T.M. Analysis and sorting of living cells according to deoxyribonucleic acid content. J. Histochem. Cytochem. 1977, 25, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Roukos, V.; Pegoraro, G.; Voss, T.C.; Misteli, T. Cell cycle staging of individual cells by fluorescence microscopy. Nat. Protoc. 2015, 10, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Yakimovich, A.; Huttunen, M.; Zehnder, B.; Coulter, L.J.; Gould, V.; Schneider, C.; Kopf, M.; McInnes, C.J.; Greber, U.F.; Mercer, J. Inhibition of poxvirus gene expression and genome replication by bisbenzimide derivatives. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.J.; Blunt, N.; Wiltshire, M.; Hoy, T.; Teesdale-Spittle, P.; Craven, M.R.; Watson, J.V.; Amos, W.B.; Errington, R.J.; Patterson, L.H. Characteristics of a novel deep red/infrared fluorescent cell-permeant DNA probe, DRAQ5, in intact human cells analyzed by flow cytometry, confocal and multiphoton microscopy. Cytom. Part A 2000, 40, 280–291. [Google Scholar] [CrossRef]
- Mathur, A.; Hong, Y.; Kemp, B.K.; Barrientos, A.A.; Erusalimsky, J.D. Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc. Res. 2000, 46, 126–138. [Google Scholar] [CrossRef]
- Tran, M.N.; Rarig, R.-A.F.; Chenoweth, D.M. Synthesis and properties of lysosome-specific photoactivatable probes for live-cell imaging. Chem. Sci. 2015, 6, 4508–4512. [Google Scholar] [CrossRef] [PubMed]
- Lukinavičius, G.; Umezawa, K.; Olivier, N.; Honigmann, A.; Yang, G.; Plass, T.; Mueller, V.; Reymond, L.; Corrêa, I.R.; Luo, Z.-G.; et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lukinavičius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Göttfert, F.; Ta, H.; Güther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; English, B.P.; Chen, J.; Slaughter, J.P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J.J.; Normanno, D.; Singer, R.H.; et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 2015, 12, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Lukinavičius, G.; Reymond, L.; Umezawa, K.; Sallin, O.; D’Este, E.; Göttfert, F.; Ta, H.; Hell, S.W.; Urano, Y.; Johnsson, K. Fluorogenic probes for multicolor imaging in living cells. J. Am. Chem. Soc. 2016, 138, 9365–9368. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; Muthusamy, A.K.; Liang, Y.; Brown, T.A.; Lemon, W.C.; Patel, R.; Lu, R.; Macklin, J.J.; Keller, P.J.; Ji, N.; et al. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Cho, W.; Sung, J.; Kim, E.; Park, S.B. Monochromophoric Design Strategy for Tetrazine-Based Colorful Bioorthogonal Probes with a Single Fluorescent Core Skeleton. J. Am. Chem. Soc. 2018, 140, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.K.; Guchhait, N. Looking at the Green Fluorescent Protein (GFP) chromophore from a different perspective: A computational insight. Spectrochim. Acta A 2013, 103, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Baulcombe, D.C.; Chapman, S.; Santa Cruz, S. Jellyfish green fluorescent protein as a reporter for virus infections. Plant J. 1995, 7, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.G.; Cruz, S.S.; Roberts, I.M.; Prior, D.A.M.; Turgeon, R.; Oparka, K.J. Phloem Unloading in Sink Leaves of Nicotiana benthamiana: Comparison of a Fluorescent Solute with a Fluorescent Virus. Plant Cell 1997, 9, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Lampe, M.; Briggs, J.A.G.; Endress, T.; Glass, B.; Riegelsberger, S.; Kräusslich, H.-G.; Lamb, D.C.; Bräuchle, C.; Müller, B. Double-labelled HIV-1 particles for study of virus-cell interaction. Virology 2007, 360, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Zentgraf, H.; Kräusslich, H.-G. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 2004, 78, 10803–10813. [Google Scholar] [CrossRef] [PubMed]
- Puntener, D.; Engelke, M.F.; Ruzsics, Z.; Strunze, S.; Wilhelm, C.; Greber, U.F. Stepwise loss of fluorescent core protein V from human adenovirus during entry into cells. J. Virol. 2011, 85, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Person, S. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 1998, 72, 7563–7568. [Google Scholar] [PubMed]
- De Martin, R.; Raidl, M.; Hofer, E.; Binder, B.R. Adenovirus-mediated expression of green fluorescent protein. Gene Ther. 1997, 4, 493–495. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, B.; Manicassamy, S.; Belicha-Villanueva, A.; Pisanelli, G.; Pulendran, B.; García-Sastre, A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. USA 2010, 107, 11531–11536. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Hare, P. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J. Virol. 1999, 73, 4110–4119. [Google Scholar] [PubMed]
- Baumgärtel, V.; Ivanchenko, S.; Dupont, A.; Sergeev, M.; Wiseman, P.W.; Kräusslich, H.-G.; Bräuchle, C.; Müller, B.; Lamb, D.C. Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 2011, 13, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef] [PubMed]
- Benn, J.; Schneider, R.J. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc. Natl. Acad. Sci. USA 1995, 92, 11215–11219. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Kaida, A.; Sawai, N.; Sakaguchi, K.; Miura, M. Fluorescence kinetics in HeLa cells after treatment with cell cycle arrest inducers visualized with Fucci (fluorescent ubiquitination-based cell cycle indicator). Cell Biol. Int. 2011, 35, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, F.D.; Seyffert, M.; Vogel, R.; Yakimovich, A.; de Andrade Pereira, B.; Meier, A.F.; Sutter, S.O.; Tobler, K.; Vogt, B.; Greber, U.F.; et al. Cell Cycle-Dependent Expression of Adeno-Associated Virus 2 (AAV2) Rep in Coinfections with Herpes Simplex Virus 1 (HSV-1) Gives Rise to a Mosaic of Cells Replicating either AAV2 or HSV-1. J. Virol. 2017, 91, e00357-17. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Kotomura, N.; Ho, Y.-K.; Zhi, H.; Bixler, S.; Schell, M.J.; Giam, C.-Z. Complex cell cycle abnormalities caused by human T-lymphotropic virus type 1 Tax. J. Virol. 2011, 85, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.S.; Stangherlin, A.; Nagy, A.D.; Nicoll, M.P.; Efstathiou, S.; O’Neill, J.S.; Reddy, A.B. Cell autonomous regulation of herpes and influenza virus infection by the circadian clock. Proc. Natl. Acad. Sci. USA 2016, 113, 10085–10090. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Lam, A.J.; Badiee, R.K.; Oh, Y.-H.; Chu, J.; Zhou, X.X.; Kim, N.; Kim, B.B.; Chung, M.; Yablonovitch, A.L.; et al. Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 2016, 13, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Yo, M.; Komatsu, N.; Hiratsuka, T.; Kogure, T.; Hoshida, T.; Goshima, N.; Matsuda, M.; Miyoshi, H.; Miyawaki, A. Genetically encoded tools for optical dissection of the mammalian cell cycle. Mol. Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Lipman, Y.; Therkelsen, K.; Daubechies, I.; Enquist, L.W. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat. Commun. 2010, 1, 146. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Cohen, K.B.; Luo, T.; Lichtman, J.W.; Sanes, J.R. Improved tools for the Brainbow toolbox. Nat. Methods 2013, 10, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-R.; Zhao, H.; Abdul-Muneer, P.M.; Cao, H.; Li, X.; Geller, A.I. Neurons can be labeled with unique hues by helper virus-free HSV-1 vectors expressing Brainbow. J. Neurosci. Methods 2015, 240, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, J.B.; Michou, A.I.; Baker, A.; Saltik, M.; Cotten, M. Microtubule-independent motility and nuclear targeting of adenoviruses with fluorescently labeled genomes. J. Virol. 2001, 75, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Hedengren-Olcott, M.; Hruby, D.E. Conditional expression of vaccinia virus genes in mammalian cell lines expressing the tetracycline repressor. J. Virol. Methods 2004, 120, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.; Fussenegger, M. Approaches for trigger-inducible viral transgene regulation in gene-based tissue engineering. Curr. Opin. Biotechnol. 2004, 15, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Hashiguchi, N.; Janicki, S.M.; Tumbar, T.; Belmont, A.S.; Spector, D.L. Visualization of gene activity in living cells. Nat. Cell Biol. 2000, 2, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Mabit, H.; Nakano, M.Y.; Prank, U.; Saam, B.; Döhner, K.; Sodeik, B.; Greber, U.F. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 2002, 76, 9962–9971. [Google Scholar] [CrossRef] [PubMed]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Crivat, G.; Taraska, J.W. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 2012, 30, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Sakin, V.; Paci, G.; Lemke, E.A.; Müller, B. Labeling of virus components for advanced, quantitative imaging analyses. FEBS Lett. 2016, 590, 1896–1914. [Google Scholar] [CrossRef] [PubMed]
- Hinner, M.J.; Johnsson, K. How to obtain labeled proteins and what to do with them. Curr. Opin. Biotechnol. 2010, 21, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Idevall-Hagren, O.; Dickson, E.J.; Hille, B.; Toomre, D.K.; De Camilli, P. Optogenetic control of phosphoinositide metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, E2316–E2323. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Sadaghiani, A.M.; Hsueh, B.; Dolmetsch, R.E. Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 2009, 27, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Helma, J.; Schmidthals, K.; Lux, V.; Nüske, S.; Scholz, A.M.; Kräusslich, H.-G.; Rothbauer, U.; Leonhardt, H. Direct and dynamic detection of HIV-1 in living cells. PLoS ONE 2012, 7, e50026. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Ries, J.; Kaplan, C.; Platonova, E.; Eghlidi, H.; Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 2012, 9, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.I.; Ahmad, M.J.; Asif, A.R.; Adnan, M.; Iqbal, M.K.; Mehmood, K.; Muhammad, S.A.; Bhuiyan, A.A.; Elokil, A.; Du, X.; et al. A Review of CRISPR-Based Genome Editing: Survival, Evolution and Challenges. Curr. Issues Mol. Biol. 2018, 28, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Nelles, D.A.; Fang, M.Y.; O’Connell, M.R.; Xu, J.L.; Markmiller, S.J.; Doudna, J.A.; Yeo, G.W. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 2016, 165, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, M.; Li, W.; Zhang, Z.; Zhang, X.; Wu, G.; Tan, T.; Cui, Z.; Zhang, X.-E. Live Visualization of HIV-1 Proviral DNA Using a Dual-Color-Labeled CRISPR System. Anal. Chem. 2017, 89, 12896–12901. [Google Scholar] [CrossRef] [PubMed]
- Howell, D.N.; Miller, S.E. Identification of viral infection by confocal microscopy. Methods Enzymol. 1999, 307, 573–591. [Google Scholar] [PubMed]
- Goldmann, H. Spaltlampenphotographie und—Photometric. Ophthalmologica 1939, 98, 257–270. [Google Scholar] [CrossRef]
- Minsky, M. Memoir on inventing the confocal scanning microscope. Scanning 1988, 10, 128–138. [Google Scholar] [CrossRef]
- Davidovits, P.; Egger, M.D. Scanning laser microscope for biological investigations. Appl. Opt. 1971, 10, 1615–1619. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, J.; Brown, C.M. Any Way You Slice It-A Comparison of Confocal Microscopy Techniques. J. Biomol. Tech. 2015, 26, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, B.; Xu, J.; Wang, J.; Wu, J.; Shi, C.; Xu, Y.; Dong, J.; Wang, C.; Lai, W.; et al. Derivation of Pluripotent Stem Cells with In Vivo Embryonic and Extraembryonic Potency. Cell 2017, 169, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.H.; Jahnel, M.; Lauer, J.; Avellaneda, M.J.; Brouilly, N.; Cezanne, A.; Morales-Navarrete, H.; Perini, E.D.; Ferguson, C.; Lupas, A.N.; et al. An endosomal tether undergoes an entropic collapse to bring vesicles together. Nature 2016, 537, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.; Cao, L.; Lin, Z.; Wei, M.; Fang, M.; Li, S.; Zhang, J.; Xia, N.; Zhao, Q. Quantitative and epitope-specific antigenicity analysis of the human papillomavirus 6 capsid protein in aqueous solution or when adsorbed on particulate adjuvants. Vaccine 2016, 34, 4422–4428. [Google Scholar] [CrossRef] [PubMed]
- Rashed, K.; Sahuc, M.-E.; Deloison, G.; Calland, N.; Brodin, P.; Rouillé, Y.; Séron, K. Potent antiviral activity of Solanum rantonnetii and the isolated compounds against hepatitis C virus in vitro. J. Funct. Foods 2014, 11, 185–191. [Google Scholar] [CrossRef]
- Li, L.; Zhou, Q.; Voss, T.C.; Quick, K.L.; LaBarbera, D.V. High-throughput imaging: Focusing in on drug discovery in 3D. Methods 2016, 96, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Flottmann, B.; Gunkel, M.; Lisauskas, T.; Heilemann, M.; Starkuviene, V.; Reymann, J.; Erfle, H. Correlative light microscopy for high-content screening. BioTechniques 2013, 55, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 1931, 401, 273–294. [Google Scholar] [CrossRef]
- Hickman, H.D.; Bennink, J.R.; Yewdell, J.W. Caught in the act: Intravital multiphoton microscopy of host-pathogen interactions. Cell Host Microbe 2009, 5, 13–21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barlerin, D.; Bessière, G.; Domingues, J.; Schuette, M.; Feuillet, C.; Peixoto, A. Biosafety Level 3 setup for multiphoton microscopy in vivo. Sci. Rep. 2017, 7, 571. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.D.; Majewska, A.K.; Brown, E.B. Single- and two-photon fluorescence recovery after photobleaching. Cold Spring Harb. Protoc. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Ghukasyan, V.; Hsu, Y.-Y.; Kung, S.-H.; Kao, F.-J. Application of fluorescence resonance energy transfer resolved by fluorescence lifetime imaging microscopy for the detection of enterovirus 71 infection in cells. J. Biomed. Opt. 2007, 12, 024016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rochette, P.-A.; Laliberté, M.; Bertrand-Grenier, A.; Houle, M.-A.; Blache, M.-C.; Légaré, F.; Pearson, A. Visualization of mouse neuronal ganglia infected by Herpes Simplex Virus 1 (HSV-1) using multimodal non-linear optical microscopy. PLoS ONE 2014, 9, e105103. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Uckermann, O.; Andresen, E.F.; Geiger, K.D.; Koch, E.; Schackert, G.; Steiner, G.; Kirsch, M. Intrinsic indicator of photodamage during label-free multiphoton microscopy of cells and tissues. PLoS ONE 2014, 9, e110295. [Google Scholar] [CrossRef] [PubMed]
- Forster, T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 33, 166–175. [Google Scholar] [CrossRef]
- Zheng, J. Spectroscopy-based quantitative fluorescence resonance energy transfer analysis. Ion Channels 2006, 337, 65–77. [Google Scholar]
- Sood, C.; Francis, A.C.; Desai, T.M.; Melikyan, G.B. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J. Biol. Chem. 2017, 292, 20196–20207. [Google Scholar] [CrossRef] [PubMed]
- Marziali, F.; Bugnon Valdano, M.; Brunet Avalos, C.; Moriena, L.; Cavatorta, A.L.; Gardiol, D. Interference of HTLV-1 Tax Protein with Cell Polarity Regulators: Defining the Subcellular Localization of the Tax-DLG1 Interaction. Viruses 2017, 9, 355. [Google Scholar] [CrossRef]
- Jones, D.M.; Alvarez, L.A.; Nolan, R.; Ferriz, M.; Sainz Urruela, R.; Massana-Muñoz, X.; Novak-Kotzer, H.; Dustin, M.L.; Padilla-Parra, S. Dynamin-2 Stabilizes the HIV-1 Fusion Pore with a Low Oligomeric State. Cell Rep. 2017, 18, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Sweeney, T.R.; Goodfellow, I. A Cell-based Fluorescence Resonance Energy Transfer (FRET) Sensor Reveals Inter- and Intragenogroup Variations in Norovirus Protease Activity and Polyprotein Cleavage. J. Biol. Chem. 2015, 290, 27841–27853. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Momose, F.; Morikawa, Y. FRET analysis of HIV-1 Gag and GagPol interactions. FEBS Open Bio 2017, 7, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Monici, M. Cell and tissue autofluorescence research and diagnostic applications. Biotechnol. Annu. Rev. 2005, 11, 227–256. [Google Scholar] [PubMed]
- Ambrose, E.J. A Surface Contact Microscope for the study of Cell Movements. Nature 1956, 178, 1194. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Traffic 2001, 2, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Yakimovich, A.; Gumpert, H.; Burckhardt, C.J.; Lütschg, V.A.; Jurgeit, A.; Sbalzarini, I.F.; Greber, U.F. Cell-free transmission of human adenovirus by passive mass transfer in cell culture simulated in a computer model. J. Virol. 2012, 86, 10123–10137. [Google Scholar] [CrossRef] [PubMed]
- Hogue, I.B.; Bosse, J.B.; Hu, J.-R.; Thiberge, S.Y.; Enquist, L.W. Cellular mechanisms of alpha herpesvirus egress: Live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog. 2014, 10, e1004535. [Google Scholar] [CrossRef] [PubMed]
- Siedentopf, H.; Zsigmondy, R. Uber Sichtbarmachung und Größenbestimmung ultramikoskopischer Teilchen, mit besonderer Anwendung auf Goldrubingläser. Ann. Phys. 1902, 315, 1–39. [Google Scholar] [CrossRef]
- Voie, A.H.; Burns, D.H.; Spelman, F.A. Orthogonal-plane fluorescence optical sectioning: Three-dimensional imaging of macroscopic biological specimens. J. Microsc. 1993, 170, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H.K. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Preibisch, S.; Amat, F.; Stamataki, E.; Sarov, M.; Singer, R.H.; Myers, E.; Tomancak, P. Efficient Bayesian-based multiview deconvolution. Nat. Methods 2014, 11, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J.; Schmidt, A.D.; Wittbrodt, J.; Stelzer, E.H.K. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 2008, 322, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Ingold, E.; Vom Berg-Maurer, C.M.; Burckhardt, C.J.; Lehnherr, A.; Rieder, P.; Keller, P.J.; Stelzer, E.H.; Greber, U.F.; Neuhauss, S.C.F.; Gesemann, M. Proper migration and axon outgrowth of zebrafish cranial motoneuron subpopulations require the cell adhesion molecule MDGA2A. Biol. Open 2015, 4, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Tomer, R.; Ye, L.; Hsueh, B.; Deisseroth, K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat. Protoc. 2014, 9, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Treweek, J.B.; Kulkarni, R.P.; Deverman, B.E.; Chen, C.-K.; Lubeck, E.; Shah, S.; Cai, L.; Gradinaru, V. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell 2014, 158, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Renier, N.; Wu, Z.; Simon, D.J.; Yang, J.; Ariel, P.; Tessier-Lavigne, M. iDISCO: A simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 2014, 159, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.S.; Lichtman, J.W. Clarifying Tissue Clearing. Cell 2015, 162, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Treweek, J.B.; Gradinaru, V. Extracting structural and functional features of widely distributed biological circuits with single cell resolution via tissue clearing and delivery vectors. Curr. Opin. Biotechnol. 2016, 40, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, J.; Jo, Y.; Kim, J.Y.; Jang, Y.J.; Lee, H.M.; Kim, S.Y.; Lee, H.-J.; Cho, K.; Jung, N.; et al. ACT-PRESTO: Rapid and consistent tissue clearing and labeling method for 3-dimensional (3D) imaging. Sci. Rep. 2016, 6, 18631. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Cai, R.; Quacquarelli, F.P.; Ghasemigharagoz, A.; Lourbopoulos, A.; Matryba, P.; Plesnila, N.; Dichgans, M.; Hellal, F.; Ertürk, A. Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nat. Methods 2016, 13, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tillberg, P.W.; Boyden, E.S. Optical imaging. Expansion microscopy. Science 2015, 347, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Chozinski, T.J.; Halpern, A.R.; Okawa, H.; Kim, H.-J.; Tremel, G.J.; Wong, R.O.L.; Vaughan, J.C. Expansion microscopy with conventional antibodies and fluorescent proteins. Nat. Methods 2016, 13, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Asano, S.M.; Boyden, E.S. Q&A: Expansion microscopy. BMC Biol. 2017, 15, 50. [Google Scholar]
- Chang, J.-B.; Chen, F.; Yoon, Y.-G.; Jung, E.E.; Babcock, H.; Kang, J.S.; Asano, S.; Suk, H.-J.; Pak, N.; Tillberg, P.W.; et al. Iterative expansion microscopy. Nat. Methods 2017, 14, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bucur, O.; Irshad, H.; Chen, F.; Weins, A.; Stancu, A.L.; Oh, E.-Y.; DiStasio, M.; Torous, V.; Glass, B.; et al. Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nat. Biotechnol. 2017, 35, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kim, J.H.; Ranjan, P.; Metcalfe, M.G.; Cao, W.; Mishina, M.; Gangappa, S.; Guo, Z.; Boyden, E.S.; Zaki, S.; et al. Influenza virus exploits tunneling nanotubes for cell-to-cell spread. Sci. Rep. 2017, 7, 40360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Chang, J.-B.; Alvarez, M.M.; Trujillo-de Santiago, G.; Aleman, J.; Batzaya, B.; Krishnadoss, V.; Ramanujam, A.A.; Kazemzadeh-Narbat, M.; Chen, F.; et al. Hybrid Microscopy: Enabling Inexpensive High-Performance Imaging through Combined Physical and Optical Magnifications. Sci. Rep. 2016, 6, 22691. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ma, H.; Liu, Y. Stochastic optical reconstruction microscopy (STORM). Curr. Protoc. Cytom. 2017, 81. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Schumann, C.; Kraegeloh, A. STED microscopy and its applications: New insights into cellular processes on the nanoscale. Chemphyschem 2012, 13, 1986–2000. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, K.T.; Ding, J.B.; Sabatini, B.L. Live-cell superresolution imaging by pulsed STED two-photon excitation microscopy. Biophys. J. 2013, 104, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Stichling, N.; Suomalainen, M.; Flatt, J.W.; Schmid, M.; Pacesa, M.; Hemmi, S.; Jungraithmayr, W.; Maler, M.D.; Freudenberg, M.A.; Plückthun, A.; et al. Lung macrophage scavenger receptor SR-A6 (MARCO) is an adenovirus type-specific virus entry receptor. PLoS Pathog. 2018, 14, e1006914. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Zahn, J.; Maglione, M.; Sigrist, S.J.; Marquard, J.; Chojnacki, J.; Kräusslich, H.-G.; Sahl, S.J.; Engelhardt, J.; Hell, S.W. Ultrafast, temporally stochastic STED nanoscopy of millisecond dynamics. Nat. Methods 2015, 12, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Bailey, B.; Farkas, D.L.; Taylor, D.L.; Lanni, F. Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature 1993, 366, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.R. Microscopy: Ever-increasing resolution. Nature 2009, 462, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.M.; Beerli, C.; Pereira, P.M.; Scherer, K.M.; Samolej, J.; Bleck, C.K.E.; Mercer, J.; Henriques, R. VirusMapper: Open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci. Rep. 2016, 6, 29132. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Fiederling, F.; Bastmeyer, M. Practical limitations of superresolution imaging due to conventional sample preparation revealed by a direct comparison of CLSM, SIM and dSTORM. J. Microsc. 2016, 262, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Mönkemöller, V.; Hennig, S.; Hübner, W.; Huser, T. Open-source image reconstruction of super-resolution structured illumination microscopy data in ImageJ. Nat. Commun. 2016, 7, 10980. [Google Scholar] [CrossRef] [PubMed]
- Mandula, O.; Kielhorn, M.; Wicker, K.; Krampert, G.; Kleppe, I.; Heintzmann, R. Line scan—structured illumination microscopy super-resolution imaging in thick fluorescent samples. Opt. Express 2012, 20, 24167–24174. [Google Scholar] [CrossRef] [PubMed]
- York, A.G.; Chandris, P.; Nogare, D.D.; Head, J.; Wawrzusin, P.; Fischer, R.S.; Chitnis, A.; Shroff, H. Instant super-resolution imaging in live cells and embryos via analog image processing. Nat. Methods 2013, 10, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- York, A.G.; Parekh, S.H.; Dalle Nogare, D.; Fischer, R.S.; Temprine, K.; Mione, M.; Chitnis, A.B.; Combs, C.A.; Shroff, H. Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy. Nat. Methods 2012, 9, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Huff, J. The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, H. Structured illumination microscopy improves visualization of lytic granules in HIV-1 specific cytotoxic T-lymphocyte immunological synapses. AIDS Res. Hum. Retroviruses 2015, 31, 866–867. [Google Scholar] [CrossRef] [PubMed]
- Mühlbauer, D.; Dzieciolowski, J.; Hardt, M.; Hocke, A.; Schierhorn, K.L.; Mostafa, A.; Müller, C.; Wisskirchen, C.; Herold, S.; Wolff, T.; et al. Influenza virus-induced caspase-dependent enlargement of nuclear pores promotes nuclear export of viral ribonucleoprotein complexes. J. Virol. 2015, 89, 6009–6021. [Google Scholar] [CrossRef] [PubMed]
- Kner, P.; Chhun, B.B.; Griffis, E.R.; Winoto, L.; Gustafsson, M.G.L. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 2009, 6, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.B.; Enderlein, J. Image scanning microscopy. Phys. Rev. Lett. 2010, 104, 198101. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Pieper, C.; Clever, M.; Pfaff, J.; Ruhlandt, A.; Kehlenbach, R.H.; Wouters, F.S.; Großhans, J.; Bunt, G.; Enderlein, J. Resolution doubling in fluorescence microscopy with confocal spinning-disk image scanning microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 21000–21005. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, C.J.R.; Mehta, S.B.; Heintzmann, R. Superresolution by image scanning microscopy using pixel reassignment. Opt. Lett. 2013, 38, 2889–2892. [Google Scholar] [CrossRef] [PubMed]
- Remenyi, R.; Roberts, G.C.; Zothner, C.; Merits, A.; Harris, M. SNAP-tagged Chikungunya Virus Replicons Improve Visualisation of Non-Structural Protein 3 by Fluorescence Microscopy. Sci. Rep. 2017, 7, 5682. [Google Scholar] [CrossRef] [PubMed]
- De Abreu Manso, P.P.; Dias de Oliveira, B.C.E.P.; de Sequeira, P.C.; Maia de Souza, Y.R.; dos Santos Ferro, J.M.; da Silva, I.J.; Caputo, L.F.G.; Guedes, P.T.; dos Santos, A.A.C.; da Silva Freire, M.; et al. Yellow fever 17DD vaccine virus infection causes detectable changes in chicken embryos. PLoS Negl. Trop. Dis. 2015, 9, e0004064. [Google Scholar]
- Zheng, W.; Wu, Y.; Winter, P.; Fischer, R.; Nogare, D.D.; Hong, A.; McCormick, C.; Christensen, R.; Dempsey, W.P.; Arnold, D.B.; et al. Adaptive optics improves multiphoton super-resolution imaging. Nat. Methods 2017, 14, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Gregor, I.; Spiecker, M.; Petrovsky, R.; Großhans, J.; Ros, R.; Enderlein, J. Rapid nonlinear image scanning microscopy. Nat. Methods 2017, 14, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.N.; Pal, R. Image scanning microscopy: An overview. J. Microsc. 2017, 266, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Sage, D.; Donati, L.; Soulez, F.; Fortun, D.; Schmit, G.; Seitz, A.; Guiet, R.; Vonesch, C.; Unser, M. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods 2017, 115, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Itano, M.S.; Arnion, H.; Wolin, S.L.; Simon, S.M. Recruitment of 7SL RNA to assembling HIV-1 virus-like particles. Traffic 2018, 19, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Palankar, R.; Kohler, T.P.; Krauel, K.; Wesche, J.; Hammerschmidt, S.; Greinacher, A. Platelets kill bacteria by bridging innate and adaptive immunity via PF4 and FcγRIIA. J. Thromb. Haemost. 2018. [Google Scholar] [CrossRef] [PubMed]
- Alsteens, D.; Newton, R.; Schubert, R.; Martinez-Martin, D.; Delguste, M.; Roska, B.; Müller, D.J. Nanomechanical mapping of first binding steps of a virus to animal cells. Nat. Nanotechnol. 2017, 12, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, N.; Culley, S.; Ashdown, G.; Owen, D.M.; Pereira, P.M.; Henriques, R. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 2016, 7, 12471. [Google Scholar] [CrossRef] [PubMed]
- Culley, S.; Albrecht, D.; Jacobs, C.; Pereira, P.M.; Leterrier, C.; Mercer, J.; Henriques, R. NanoJ-SQUIRREL: Quantitative mapping and minimisation of super-resolution optical imaging artefacts. BioRxiv 2017, 158279. [Google Scholar] [CrossRef]
- Dertinger, T.; Colyer, R.; Iyer, G.; Weiss, S.; Enderlein, J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl. Acad. Sci. USA 2009, 106, 22287–22292. [Google Scholar] [CrossRef] [PubMed]
- Geissbuehler, S.; Dellagiacoma, C.; Lasser, T. Comparison between SOFI and STORM. Biomed. Opt. Express 2011, 2, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Rosten, E.; Monypenny, J.; Jovanovic-Talisman, T.; Burnette, D.T.; Lippincott-Schwartz, J.; Jones, G.E.; Heintzmann, R. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat. Methods 2011, 9, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Cyrklaff, M.; Risco, C.; Fernández, J.J.; Jiménez, M.V.; Estéban, M.; Baumeister, W.; Carrascosa, J.L. Cryo-electron tomography of vaccinia virus. Proc. Natl. Acad. Sci. USA 2005, 102, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Helmuth, J.A.; Burckhardt, C.J.; Koumoutsakos, P.; Greber, U.F.; Sbalzarini, I.F. A novel supervised trajectory segmentation algorithm identifies distinct types of human adenovirus motion in host cells. J. Struct. Biol. 2007, 159, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Helmuth, J.A.; Burckhardt, C.J.; Greber, U.F.; Sbalzarini, I.F. Shape reconstruction of subcellular structures from live cell fluorescence microscopy images. J. Struct. Biol. 2009, 167, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sbalzarini, I.F.; Greber, U.F. How computational models enable mechanistic insights into virus infection. In Influenza Virus: Methods & Protocols; Yamauchi, Y., Ed.; Springer: New York, NY, USA, 2018; in press. [Google Scholar]
- Krzywinski, M.; Altman, N. Points of significance: Power and sample size. Nat. Methods 2013, 10, 1139–1140. [Google Scholar] [CrossRef]
- Higginson, A.D.; Munafò, M.R. Current Incentives for Scientists Lead to Underpowered Studies with Erroneous Conclusions. PLoS Biol. 2016, 14, e2000995. [Google Scholar] [CrossRef] [PubMed]
- Challenges in Irreproducible Research: Nature News & Comment. Available online: https://www.nature.com/news/reproducibility-1.17552 (accessed on 7 November 2017).
- Baker, M.; Dolgin, E. Cancer reproducibility project releases first results. Nature 2017, 541, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.-Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Fatta, G.D.; Gabriel, T.R.; Georg, F.; Meinl, T.; Ohl, P.; Sieb, C.; Wiswedel, B. Technical Report Knime: The Konstanz Information Miner. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- De Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Dulbecco, R.; Vogt, M. Plaque formation and isolation of pure lines with poliomyelitis viruses. J. Exp. Med. 1954, 99, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Yakimovich, A.; Andriasyan, V.; Witte, R.; Wang, I.-H.; Prasad, V.; Suomalainen, M.; Greber, U.F. Plaque2.0-A High-Throughput Analysis Framework to Score Virus-Cell Transmission and Clonal Cell Expansion. PLoS ONE 2015, 10, e0138760. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Straehle, C.; Kothe, U.; Hamprecht, F.A. Ilastik: Interactive learning and segmentation toolkit. In Proceedings of the 2011 IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Chicago, IL, USA, 30 March–2 April 2011; pp. 230–233. [Google Scholar]
- Held, M.; Schmitz, M.H.A.; Fischer, B.; Walter, T.; Neumann, B.; Olma, M.H.; Peter, M.; Ellenberg, J.; Gerlich, D.W. CellCognition: Time-resolved phenotype annotation in high-throughput live cell imaging. Nat. Methods 2010, 7, 747–754. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Lecun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Xu, M.; Papageorgiou, D.P.; Abidi, S.Z.; Dao, M.; Zhao, H.; Karniadakis, G.E. A deep convolutional neural network for classification of red blood cells in sickle cell anemia. PLoS Comput. Biol. 2017, 13, e1005746. [Google Scholar] [CrossRef] [PubMed]
- Sadanandan, S.K.; Ranefall, P.; Le Guyader, S.; Wählby, C. Automated training of deep convolutional neural networks for cell segmentation. Sci. Rep. 2017, 7, 7860. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.; Jonas, E.; Babcock, H.P.; Recht, B. Deeploco: Fast 3D localization microscopy using neural networks. BioRxiv 2018, 267096. [Google Scholar] [CrossRef]
- De Boer, P.; Hoogenboom, J.P.; Giepmans, B.N.G. Correlated light and electron microscopy: Ultrastructure lights up! Nat. Methods 2015, 12, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Godman, G.C.; Morgan, C.; Breitenfeld, P.M.; Rose, H.M. A correlative study by electron and light microscopy of the development of type 5 adenovirus. II. Light microscopy. J. Exp. Med. 1960, 112, 383–402. [Google Scholar] [CrossRef] [PubMed]
- Tokuyasu, K.T. A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol. 1973, 57, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Agronskaia, A.V.; Valentijn, J.A.; van Driel, L.F.; Schneijdenberg, C.T.W.M.; Humbel, B.M.; van Bergen en Henegouwen, P.M.P.; Verkleij, A.J.; Koster, A.J.; Gerritsen, H.C. Integrated fluorescence and transmission electron microscopy. J. Struct. Biol. 2008, 164, 183–189. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.L. A review of high-pressure freezing preparation techniques for correlative light and electron microscopy of the same cells and tissues. J. Microsc. 2009, 235, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Briegel, A.; Chen, S.; Koster, A.J.; Plitzko, J.M.; Schwartz, C.L.; Jensen, G.J. Correlated Light and Electron Cryo-Microscopy. In Cryo-EM Part A Sample Preparation and Data Collection; Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 481, pp. 317–341. [Google Scholar]
- Giepmans, B.N.G.; Adams, S.R.; Ellisman, M.H.; Tsien, R.Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 312, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.R. Advances in electron microscopy: A qualitative view of instrumentation development for macromolecular imaging and tomography. Arch. Biochem. Biophys. 2015, 581, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Carroni, M.; Saibil, H.R. Cryo electron microscopy to determine the structure of macromolecular complexes. Methods 2016, 95, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Hampton, C.M.; Strauss, J.D.; Ke, Z.; Dillard, R.S.; Hammonds, J.E.; Alonas, E.; Desai, T.M.; Marin, M.; Storms, R.E.; Leon, F.; et al. Correlated fluorescence microscopy and cryo-electron tomography of virus-infected or transfected mammalian cells. Nat. Protoc. 2017, 12, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, P.; Kaufmann, R.; Siebert, C.A.; Hagen, C.; Wodrich, H.; Grünewald, K. High-precision correlative fluorescence and electron cryo microscopy using two independent alignment markers. Ultramicroscopy 2014, 143, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Van Rijnsoever, C.; Oorschot, V.; Klumperman, J. Correlative light-electron microscopy (CLEM) combining live-cell imaging and immunolabeling of ultrathin cryosections. Nat. Methods 2008, 5, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 2014, 343, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Ke, D.; Debiec, K.; Zhao, G.; Meng, X.; Ambrose, Z.; Gibson, G.A.; Watkins, S.C.; Zhang, P. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure 2011, 19, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Strunze, S.; Engelke, M.F.; Wang, I.-H.; Puntener, D.; Boucke, K.; Schleich, S.; Way, M.; Schoenenberger, P.; Burckhardt, C.J.; Greber, U.F. Kinesin-1-mediated capsid disassembly and disruption of the nuclear pore complex promote virus infection. Cell Host Microbe 2011, 10, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Berger, C.; Kallis, S.; Kolovou, A.; Paul, D.; Lohmann, V.; Bartenschlager, R. NS5A Domain 1 and Polyprotein Cleavage Kinetics Are Critical for Induction of Double-Membrane Vesicles Associated with Hepatitis C Virus Replication. MBio 2015, 6, e00759. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, P.; Cortese, M.; Chatel-Chaix, L.; Fischl, W.; Bartenschlager, R. Dengue Virus Non-structural Protein 1 Modulates Infectious Particle Production via Interaction with the Structural Proteins. PLoS Pathog. 2015, 11, e1005277. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Snapp, E.-L.; Perumal, G.S.; Macaluso, F.P.; Kielian, M. Imaging the alphavirus exit pathway. J. Virol. 2014, 88, 6922–6933. [Google Scholar] [CrossRef] [PubMed]
- Bykov, Y.S.; Cortese, M.; Briggs, J.A.G.; Bartenschlager, R. Correlative light and electron microscopy methods for the study of virus-cell interactions. FEBS Lett. 2016, 590, 1877–1895. [Google Scholar] [CrossRef] [PubMed]
- Cuche, E.; Marquet, P.; Depeursinge, C. Simultaneous amplitude-contrast and quantitative phase-contrast microscopy by numerical reconstruction of Fresnel off-axis holograms. Appl. Opt. 1999, 38, 6994–7001. [Google Scholar] [CrossRef] [PubMed]
- Cotte, Y.; Toy, F.; Jourdain, P.; Pavillon, N.; Boss, D.; Magistretti, P.; Marquet, P.; Depeursinge, C. Marker-free phase nanoscopy. Nat. Photonics 2013, 7, 113–117. [Google Scholar] [CrossRef]
- Pollaro, L.; Equis, S.; Dalla Piazza, B.; Cotte, Y. Stain-free 3D Nanoscopy of Living Cells. Opt. Photonik 2016, 11, 38–42. [Google Scholar] [CrossRef]
- Lai, Z.; Yang, X.; Li, A.; Qiu, Y.; Cai, J.; Yang, P. Facile preparation of full-color emissive carbon dots and their applications in imaging of the adhesion of erythrocytes to endothelial cells. J. Mater. Chem. B 2017, 5, 5259–5264. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
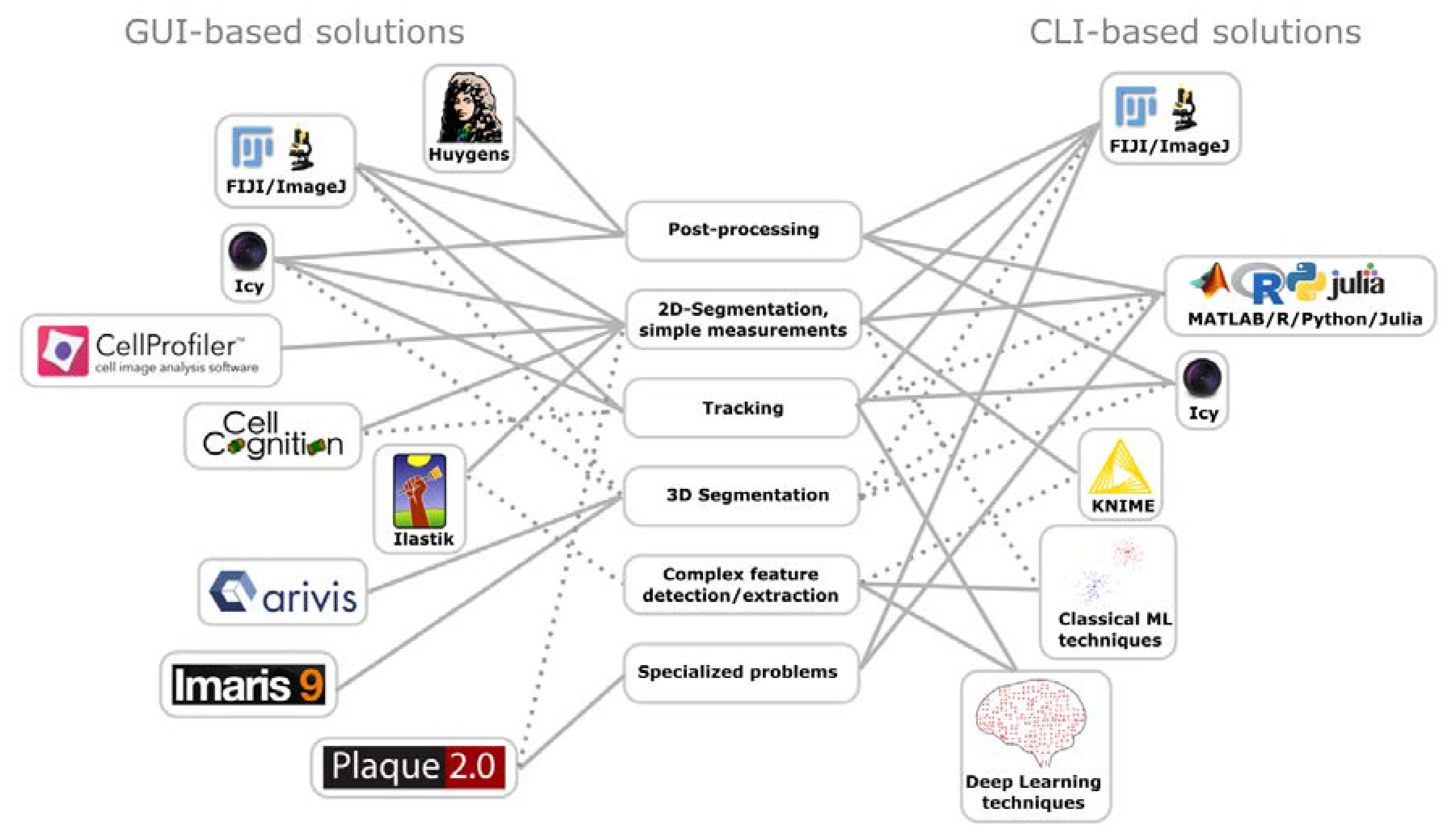{kind=link}
| Live Acquisition | Long Term Acquisition | 3D Acquisition | High-Throughput | Super-Resolution | Deep Tissue | FRET Compatible | |
|---|---|---|---|---|---|---|---|
| Widefield | + | + | + | ||||
| CLSM | + | (+) | + | ||||
| SDCM | + | + | + | + | + | ||
| 2-photon | + | + | + | + | |||
| Airyscan | + | + | + | + | + | + | |
| Lightsheet | + | + | + | + | |||
| STED | (+) | + | |||||
| PALM/STORM | + | ||||||
| SIM | (+) | + | + | ||||
| iSIM | + | + | + | (+) | + | (+) | |
| DHM | + | + | + | + |
| Ease of Use | Maintenance | File Sizes | Quantifiable | Postprocessing | |
|---|---|---|---|---|---|
| Widefield | Simple | User | Small | Yes | No |
| CLSM | Simple | Specialist | Moderate | Yes | No |
| SDCM | Simple | Specialist | Moderate | Yes | No |
| 2-photon | Expert | Engineer | Moderate | Yes | No |
| Airyscan | Simple | Engineer | Moderate | Yes | Yes |
| Lightsheet | Advanced | Specialist | Very large | Yes | Yes |
| STED | Advanced | Specialist | Small | Yes | No |
| PALM/STORM | Simple–Expert * | Specialist | Very large | No | Yes |
| SIM | Simple–Expert * | Engineer | Moderate | No | Yes |
| iSIM | Simple | Engineer | Moderate | Yes | No |
| DHM | Simple | User | Large | Yes | Yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witte, R.; Andriasyan, V.; Georgi, F.; Yakimovich, A.; Greber, U.F. Concepts in Light Microscopy of Viruses. Viruses 2018, 10, 202. https://doi.org/10.3390/v10040202
Witte R, Andriasyan V, Georgi F, Yakimovich A, Greber UF. Concepts in Light Microscopy of Viruses. Viruses. 2018; 10(4):202. https://doi.org/10.3390/v10040202
Chicago/Turabian StyleWitte, Robert, Vardan Andriasyan, Fanny Georgi, Artur Yakimovich, and Urs F. Greber. 2018. "Concepts in Light Microscopy of Viruses" Viruses 10, no. 4: 202. https://doi.org/10.3390/v10040202
APA StyleWitte, R., Andriasyan, V., Georgi, F., Yakimovich, A., & Greber, U. F. (2018). Concepts in Light Microscopy of Viruses. Viruses, 10(4), 202. https://doi.org/10.3390/v10040202