Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies



Abstract
1. Introduction
2. The Latent HIV-1 Reservoirs
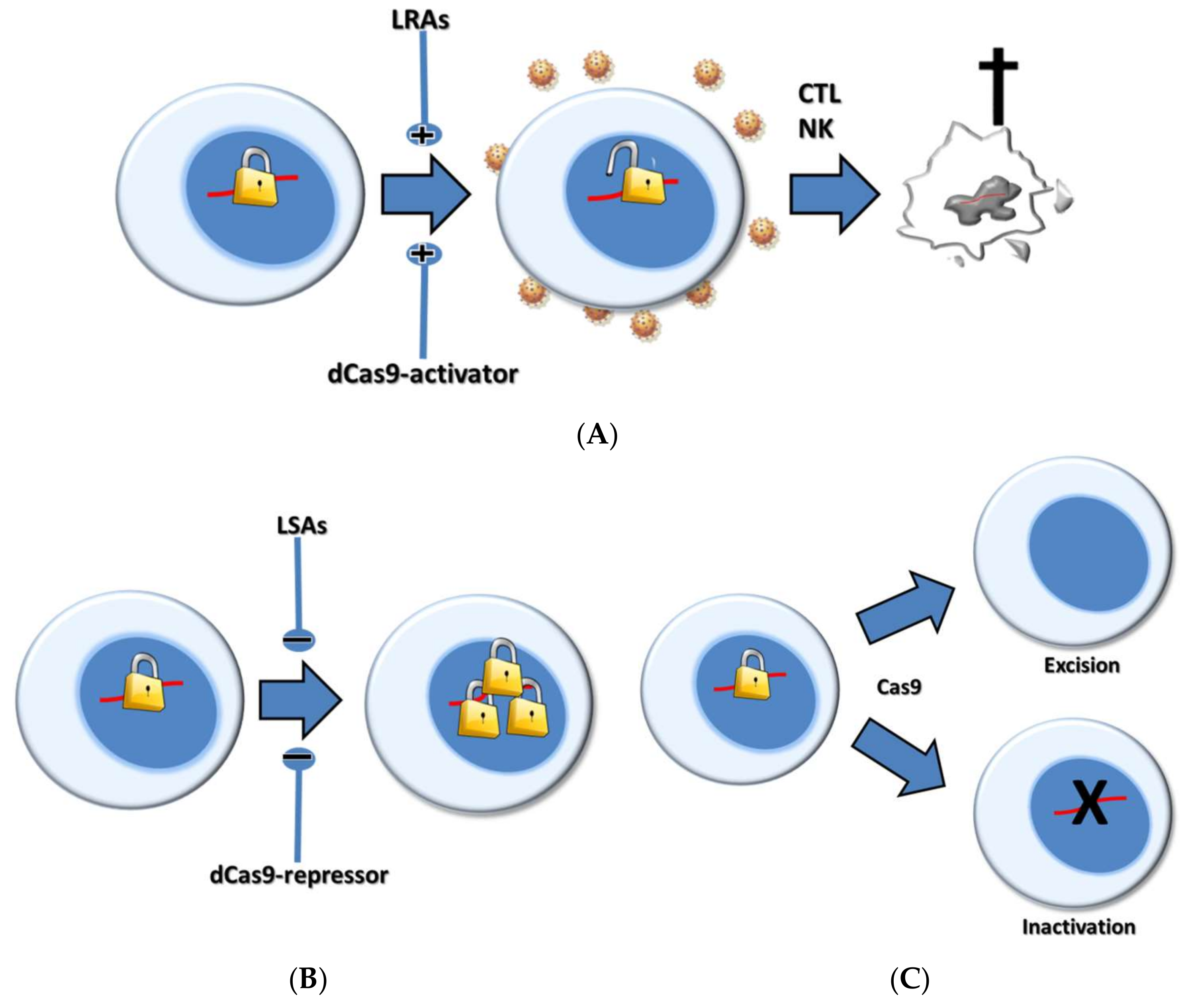
3. Pharmacologic Shock Strategies
3.1. Molecular Mechanisms of HIV Latency
3.2. Latency Reversing Agents
4. CRISPR-Based Shock Strategies
5. Pharmacological Versus CRISPR-Based Shock Strategies
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Davey, R.T., Jr.; Ostrowski, M.; Shawn Justement, J.; Engel, D.; Mullins, J.I.; Fauci, A.S. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 2000, 6, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Paiardini, M.; Muller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 2017, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Berkhout, B. HIV Reservoir: Finding the Right Needles in a Needlestack. Cell Host Microbe 2016, 20, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Hosmane, N.N.; Siliciano, R.F. Towards an HIV-1 cure: Measuring the latent reservoir. Trends Microbiol. 2015, 23, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Avettand-Fenoel, V.; Hocqueloux, L.; Ghosn, J.; Cheret, A.; Frange, P.; Melard, A.; Viard, J.P.; Rouzioux, C. Total HIV-1 DNA, a Marker of Viral Reservoir Dynamics with Clinical Implications. Clin. Microbiol. Rev. 2016, 29, 859–880. [Google Scholar] [CrossRef] [PubMed]
- Douek, D.C. Disrupting T-cell homeostasis: How HIV-1 infection causes disease. AIDS Rev. 2003, 5, 172–177. [Google Scholar] [PubMed]
- Baxter, A.E.; O’Doherty, U.; Kaufmann, D.E. Beyond the replication-competent HIV reservoir: Transcription and translation-competent reservoirs. Retrovirology 2018, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Q.; Lichterfeld, M. Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr. Opin. HIV AIDS 2016, 11, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.M.; de Leval, L.; Pantaleo, G.; Perreau, M. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016, 22, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Ruggiero, A.; Noto, A.; Ohmiti, K.; Cavassini, M.; Corpataux, J.M.; Paxton, W.A.; Pollakis, G.; Perreau, M. Blood CXCR3(+) CD4 T Cells Are Enriched in Inducible Replication Competent HIV in Aviremic Antiretroviral Therapy-Treated Individuals. Front. Immunol. 2018, 9, 144. [Google Scholar] [PubMed]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Gras, G.; Kaul, M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology 2010, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Canestri, A.; Lescure, F.X.; Jaureguiberry, S.; Moulignier, A.; Amiel, C.; Marcelin, A.G.; Peytavin, G.; Tubiana, R.; Pialoux, G.; Katlama, C. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin. Infect. Dis. 2010, 50, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Jenabian, M.A.; Costiniuk, C.T.; Mehraj, V.; Ghazawi, F.M.; Fromentin, R.; Brousseau, J.; Brassard, P.; Belanger, M.; Ancuta, P.; Bendayan, R.; et al. Immune tolerance properties of the testicular tissue as a viral sanctuary site in ART-treated HIV-infected adults. AIDS 2016, 30, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Coombs, R.W.; Van Lint, C. Exploring the anatomical HIV reservoirs: Role of the testicular tissue. AIDS 2016, 30, 2891–2893. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Shergill, A.K.; Ho, T.; Killian, M.; Girling, V.; Epling, L.; Li, P.; Wong, L.K.; Crouch, P.; Deeks, S.G.; et al. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: Implications for viral persistence. J. Infect. Dis. 2013, 208, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Costiniuk, C.T.; Jenabian, M.A. The lungs as anatomical reservoirs of HIV infection. Rev. Med. Virol. 2014, 24, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Edagwa, B.J.; Zhou, T.; McMillan, J.M.; Liu, X.M.; Gendelman, H.E. Development of HIV reservoir targeted long acting nanoformulated antiretroviral therapies. Curr. Med. Chem. 2014, 21, 4186–4198. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [PubMed]
- Le Douce, V.; Herbein, G.; Rohr, O.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, R.M.; van Capel, T.M.; Speijer, D.; Sanders, R.W.; Berkhout, B.; de Jong, E.C.; Jeeninga, R.E.; van Montfort, T. Dendritic cell type-specific HIV-1 activation in effector T cells: Implications for latent HIV-1 reservoir establishment. AIDS 2015, 29, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hatano, H.; Bacon, O.; Hogan, L.E.; Rutishauser, R.; Hill, A.; Kearney, M.F.; Anderson, E.M.; Buchbinder, S.P.; Cohen, S.E.; et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 2017, 14, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Saez-Cirion, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Laanani, M.; Ghosn, J.; Essat, A.; Melard, A.; Seng, R.; Gousset, M.; Panjo, H.; Mortier, E.; Girard, P.M.; Goujard, C.; et al. Impact of the Timing of Initiation of Antiretroviral Therapy During Primary HIV-1 Infection on the Decay of Cell-Associated HIV-DNA. Clin. Infect. Dis. 2015, 60, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 integration landscape during latent and active infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Simonetti, F.R.; Siliciano, R.F.; Laird, G.M. Measuring replication competent HIV-1: Advances and challenges in defining the latent reservoir. Retrovirology 2018, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Plantin, J.; Massanella, M.; Chomont, N. Inducible HIV RNA transcription assays to measure HIV persistence: Pros and cons of a compromise. Retrovirology 2018, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.J.; Fun, A.; Bandara, M.; Wills, M.R.; Mok, H.P.; Lever, A.M.L. Innovations in the quantitative virus outgrowth assay and its use in clinical trials. Retrovirology 2017, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Metcalf Pate, K.A.; Blankson, J.N. The mouse viral outgrowth assay: Avatars for the detection of HIV-1 reservoirs. Retrovirology 2017, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. Preclinical shock strategies to reactivate latent HIV-1: An update. Curr. Opin. HIV AIDS 2016, 11, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Bouchat, S.; Kirchhoff, F.; Van Lint, C. Molecular Control of HIV and SIV Latency. Curr. Top. Microbiol. Immunol. 2017. [Google Scholar] [CrossRef]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [PubMed]
- Jiang, G.; Nguyen, D.; Archin, N.M.; Yukl, S.A.; Mendez-Lagares, G.; Tang, Y.; Elsheikh, M.M.; Thompson, G.R., 3rd; Hartigan-O’Connor, D.J.; Margolis, D.M.; et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J. Clin. Investig. 2018, 128. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Thompson, M.A.; Brandt, S.J.; Hiebert, S.W. Histone deacetylase inhibitors induce the degradation of the t (8;21) fusion oncoprotein. Oncogene 2007, 26, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Sims, R.J., 3rd; Gottlieb, P.D.; Tucker, P.W. Identification and characterization of SMYD2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the SIN3 histone deacetylase complex. Mol. Cancer 2006, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Abu-Farha, M.; Lambert, J.P.; Al-Madhoun, A.S.; Elisma, F.; Skerjanc, I.S.; Figeys, D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell Proteom. 2008, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host Microbe 2017, 21, 569–579.e6. [Google Scholar] [CrossRef] [PubMed]
- Trejbalova, K.; Kovarova, D.; Blazkova, J.; Machala, L.; Jilich, D.; Weber, J.; Kucerova, D.; Vencalek, O.; Hirsch, I.; Hejnar, J. Development of 5’ LTR DNA methylation of latent HIV-1 provirus in cell line models and in long-term-infected individuals. Clin. Epigenet. 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology 2012, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.; Ackley, A.; Turner, A.W.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Campilongo, F.; Barclay, R.A.; DeMarino, C.; Iglesias-Ussel, M.D.; Kashanchi, F.; Romerio, F. The Human Immunodeficiency Virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology 2017, 506, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J. Virol. 2013, 87, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Rice, A.P. MicroRNA-mediated restriction of HIV-1 in resting CD4+ T cells and monocytes. Viruses 2012, 4, 1390–1409. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1 + JQ1 and Ingenol-B + JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Kollar, P.; Rajchard, J.; Balounova, Z.; Pazourek, J. Marine natural products: Bryostatins in preclinical and clinical studies. Pharm. Biol. 2014, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-AZA-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Palmer, B.E.; Nold, M.F.; Furlan, A.; Kassu, A.; Fossati, G.; Mascagni, P.; Dinarello, C.A. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J. Acquir. Immune Defic. Syndr. 2010, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Contreras, X.; Schweneker, M.; Chen, C.S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093. [Google Scholar] [CrossRef] [PubMed]
- Quivy, V.; Adam, E.; Collette, Y.; Demonte, D.; Chariot, A.; Vanhulle, C.; Berkhout, B.; Castellano, R.; de Launoit, Y.; Burny, A.; et al. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: Potential perspectives for the development of therapeutic strategies. J. Virol. 2002, 76, 11091–11103. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Andrade, A.; Eisele, E.; Hoh, R.; Bacchetti, P.; Bumpus, N.N.; Emad, F.; Buckheit, R., 3rd; McCance-Katz, E.F.; Lai, J.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin. Infect. Dis. 2014, 58, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.D.; Irrinki, A.; Tsai, A.; Kaur, J.; Lalezari, J.; Murry, J.; Cihlar, T. TLR7 Agonist GS-9620 Activates HIV-1 in PBMCs From HIV-Infected Patients on cART. In Proceedings of the 22nd Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 23–26 February 2015. [Google Scholar]
- Thibault, S.; Imbeault, M.; Tardif, M.R.; Tremblay, M.J. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology 2009, 389, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Novis, C.L.; Archin, N.M.; Buzon, M.J.; Verdin, E.; Round, J.L.; Lichterfeld, M.; Margolis, D.M.; Planelles, V.; Bosque, A. Reactivation of latent HIV-1 in central memory CD4(+) T cells through TLR-1/2 stimulation. Retrovirology 2013, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals With Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Berkhout, B. What do we measure when we measure cell-associated HIV RNA. Retrovirology 2018, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Crowther, D.; Prendiville, J.; McGown, A.T.; Scheid, C.; Stern, P.; Young, R.; Brenchley, P.; Chang, J.; Owens, S.; et al. A phase I trial of bryostatin 1 in patients with advanced malignancy using a 24 hour intravenous infusion. Br. J. Cancer 1995, 72, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Bouchat, S.; Kula, A.; Van Driessche, B.; Delacourt, N.; Vanhulle, C.; Avettand-Fenoel, V.; De Wit, S.; Rohr, O.; Rouzioux, C.; et al. Reactivation capacity by latency-reversing agents ex vivo correlates with the size of the HIV-1 reservoir. AIDS 2017, 31, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Yucha, R.W.; Hobbs, K.S.; Hanhauser, E.; Hogan, L.E.; Nieves, W.; Ozen, M.O.; Inci, F.; York, V.; Gibson, E.A.; Thanh, C.; et al. High-throughput Characterization of HIV-1 Reservoir Reactivation Using a Single-Cell-in-Droplet PCR Assay. EBioMedicine 2017, 20, 217–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, H.C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.; Thompson, G.R.; et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-κB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [PubMed]
- Rochat, M.A.; Schlaepfer, E.; Speck, R.F. Promising Role of Toll-Like Receptor 8 Agonist in Concert with Prostratin for Activation of Silent HIV. J. Virol. 2017, 91, e02084-16. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Ren, Y.; Thomas, A.S.; Chan, D.; Mueller, S.; Ward, A.R.; Patel, S.; Bollard, C.M.; Cruz, C.R.; Karandish, S.; et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J. Clin. Investig. 2018, 128, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Kwaa, A.K.; Goldsborough, K.; Walker-Sperling, V.E.; Pianowski, L.F.; Gama, L.; Blankson, J.N. The effect of Ingenol-B on the suppressive capacity of elite suppressor HIV-specific CD8+ T cells. PLoS ONE 2017, 12, e0174516. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Spivak, A.M.; Soriano-Sarabia, N.; Checkley, M.A.; Barker, E.; Karn, J.; Planelles, V.; Margolis, D.M. HIV Latency-Reversing Agents Have Diverse Effects on Natural Killer Cell Function. Front. Immunol. 2016, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Attacking HIV-1 RNA versus DNA by sequence-specific approaches: RNAi versus CRISPR-Cas. Biochem. Soc. Trans. 2016, 44, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Karg, M.; Herrera-Carrillo, E.; Berkhout, B. Towards Antiviral shRNAs Based on the AgoshRNA Design. PLoS ONE 2015, 10, e0128618. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Gene therapy strategies to block HIV-1 replication by RNA interference. Adv. Exp. Med. Biol. 2015, 848, 71–95. [Google Scholar] [PubMed]
- Herrera-Carrillo, E.; Liu, Y.P.; Berkhout, B. The impact of unprotected T cells in RNAi-based gene therapy for HIV-AIDS. Mol. Ther. 2014, 22, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Waheed, Y.; Ghazal, A.; Ullah, S.; Safi, S.Z.; Jamal, M.; Ali, M.; Atif, M.; Imran, M.; Ullah, F. Modern biotechnology-based therapeutic approaches against HIV infection. Biomed. Rep. 2017, 7, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. The impact of HIV-1 genetic diversity on the efficacy of a combinatorial RNAi-based gene therapy. Gene Ther. 2015, 22, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Novel AgoshRNA molecules for silencing of the CCR5 co-receptor for HIV-1 infection. PLoS ONE 2017, 12, e0177935. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S.; et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci. 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wainberg, M.A.; Das, A.T.; Berkhout, B. CRISPR/Cas9: A double-edged sword when used to combat HIV infection. Retrovirology 2016, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 696. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef] [PubMed]
- Bialek, J.K.; Dunay, G.A.; Voges, M.; Schafer, C.; Spohn, M.; Stucka, R.; Hauber, J.; Lange, U.C. Targeted HIV-1 Latency Reversal Using CRISPR/Cas9-Derived Transcriptional Activator Systems. PLoS ONE 2016, 11, e0158294. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; Ter-Ovanesyan, D.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Li, C.; Pan, H.; Fu, Z.; Qu, X.; Wang, P.; Deng, J.; et al. Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol. Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, F.; Dang, L.; Liang, C.; Wang, C.; He, B.; Liu, J.; Li, D.; Wu, X.; Xu, X.; et al. In Vivo Delivery Systems for Therapeutic Genome Editing. Int. J. Mol. Sci. 2016, 17, 626. [Google Scholar] [CrossRef] [PubMed]
- Tycko, J.; Myer, V.E.; Hsu, P.D. Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity. Mol. Cell 2016, 63, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Mays, L.E.; Wilson, J.M. The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol. Ther. 2011, 19, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [PubMed]
- De Solis, C.A.; Ho, A.; Holehonnur, R.; Ploski, J.E. The Development of a Viral Mediated CRISPR/Cas9 System with Doxycycline Dependent gRNA Expression for Inducible In vitro and In vivo Genome Editing. Front. Mol. Neurosci. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wu, L.; Zhang, S.M.; Lu, M.; Cheung, W.K.; Cai, W.; Gale, M.; Xu, Q.; Yan, Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016, 44, e149. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Zhu, Z.; Shi, Z.D.; Lelli, K.; Verma, N.; Li, Q.V.; Huangfu, D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 2014, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Shock Strategies to Cure HIV-1 | |
|---|---|
| Latency-Reversing Agents | CRISPR-dCas9 |
| Not specific | Sequence-specific |
| Toxicity at a high dose | Possible off-target effects not yet known |
| Weak as individual drugs | Potent (in vitro) |
| Already tested ex vivo and in vivo | Only tested in vitro thus far |
| Possible effects on other latent viruses (e.g., EBV, CMV) | Effect on other viruses unlikely |
| Diffusion via blood into the cells | Transduction of the multiple components required |
| Impact on immune system? | Host immune response due to bacterial origin |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darcis, G.; Das, A.T.; Berkhout, B. Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies. Viruses 2018, 10, 157. https://doi.org/10.3390/v10040157
Darcis G, Das AT, Berkhout B. Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies. Viruses. 2018; 10(4):157. https://doi.org/10.3390/v10040157
Chicago/Turabian StyleDarcis, Gilles, Atze T. Das, and Ben Berkhout. 2018. "Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies" Viruses 10, no. 4: 157. https://doi.org/10.3390/v10040157
APA StyleDarcis, G., Das, A. T., & Berkhout, B. (2018). Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies. Viruses, 10(4), 157. https://doi.org/10.3390/v10040157