1. Introduction
Melanoma is the leading cause (80%) of death from skin disease, although it accounts for only 4% of all dermatologic cancers [
1]. It is the fifth most frequently diagnosed malignancy in men and the seventh in women, with an estimation of 91,270 new cases in the United States (US) in 2018 [
2,
3].
The overall five-year survival rate for melanoma is 91.3%, but the prognosis is only good for patients with early stage disease, where surgical removal is possible, and tumors have not yet spread. In contrast, the five-year survival rate for patients with late stage melanoma is much lower, e.g., only 16% for metastatic stage IV melanoma [
4]. For this patient group, new immunotherapies have already considerably improved the prognosis. Monoclonal antibodies that block CTLA-4 or PD-1 act as checkpoint inhibitors and activate anti-tumoral T-cell responses [
5,
6]. However, only a fraction of tumor patients respond to checkpoint inhibitor therapy. A combination of two checkpoint inhibitors, e.g., ipilimumab and nivolumab, improves response rates. However, side effects are also enhanced, and still, a considerable proportion of patients does not respond [
7,
8].
To improve treatment efficacy, checkpoint inhibitors can be combined with other immunotherapies such as oncolytic viruses. At the end of 2015, the first oncolytic virus, the modified herpes simplex virus talimogene laherparepvec (T-VEC), was granted approval in the US and in the European Union (EU) for the treatment of advanced stage melanoma [
9]. T-VEC expresses the immunostimulatory cytokine granulocyte–macrophage colony-stimulating factor (GM-CSF), and has led to the enhanced survival of patients with unresected stage IIIB to IV melanoma [
10]. T-VEC is currently being tested in several clinical trials in combination with the checkpoint inhibitors ipilimumab or pembrolizumab (ClinicalTrials.gov identifier NCT03069378, NCT02626000, NCT02965716, NCT01740297, and NCT02263508). Initial small clinical studies indicate that T-VEC and ipilimumab [
11,
12], as well as T-VEC and pembrolizumab, act synergistically [
13].
However, pre-existing or vector-induced antiviral immunity are expected to limit the efficacy of the oncolytic viruses that are currently in clinical development [
14]. This is also true for the herpes simplex virus T-VEC, where pre-existing immunity even is a prerequisite for the safe application of the therapeutic dose. Virus-neutralizing antibodies are also expected to limit the systemic delivery of the virus.
We previously described a new oncolytic vesicular stomatitis virus, VSV-GP, in which the VSV glycoprotein G was substituted by the lymphocytic choriomeningitis virus (LCMV) glycoprotein GP. VSV-GP has several strengths, such as a fast replication cycle, no pre-existing immunity in the general population, and the capability to accommodate immunostimulatory cytokines or tumor antigens. We showed that systemic, intracranial, and intratumoral virus application is safe, even in immunodefective mice [
15]. Further, VSV-GP lacks VSV’s inherent neurotoxicity, and does not readily induce neutralizing antibodies [
16]. These features, and its favorable safety profile, make VSV-GP an ideal candidate for the treatment of advanced cancers [
16,
17,
18].
We have previously shown in mouse models that intravenous treatment with VSV-GP is effective against subcutaneous as well as intracranial malignant glioblastoma [
16]. As previously shown for the parental recombinant VSV, the oncolytic activity of VSV-GP depends on a defect type I IFN signaling in many tumors. Using interferon-competent ovarian cancer models, we could improve treatment efficacy by inhibiting the antiviral innate immune response with ruxolitinib, a Jak1/2 inhibitor [
15].
Here, we analyze the efficacy of VSV-GP in cell lines and primary cultures as well as mouse models for malignant melanoma. We show that VSV-GP was not only effective in subcutaneous tumor models, it was also effective against developing lung metastases upon systemic treatment.
2. Materials and Methods
2.1. Ethics Statement
Animal experiments were performed in compliance with the national animal experimentation law (“Tierversuchsgesetz”), and animal trial permission was granted by national authorities (Bundesministerium für Wissenschaft und Forschung, #BMWF-66.011/0119-II/3b/2012 (31 August 2012) and BMWFW-66.011/0041-WF/V/3b/2016 (4 March 2016)).
2.2. Primary Cultures
Short-term cultures of human melanoma cells were a kind gift from Drs. R. Halaban and A. Bacchiocchi from the Specimen Resource Core of Yale University (SPORE) in Skin Cancer, and were designated with a YU prefix and an arbitrary letter code that was non-related to the patient's identity, in accordance with the Health Insurance Portability and Accountability Act (HIPAA) and the institutional Human Investigative Committee protocol. Melanoma tissue was derived from primary and metastatic sites with patients’ informed consent [
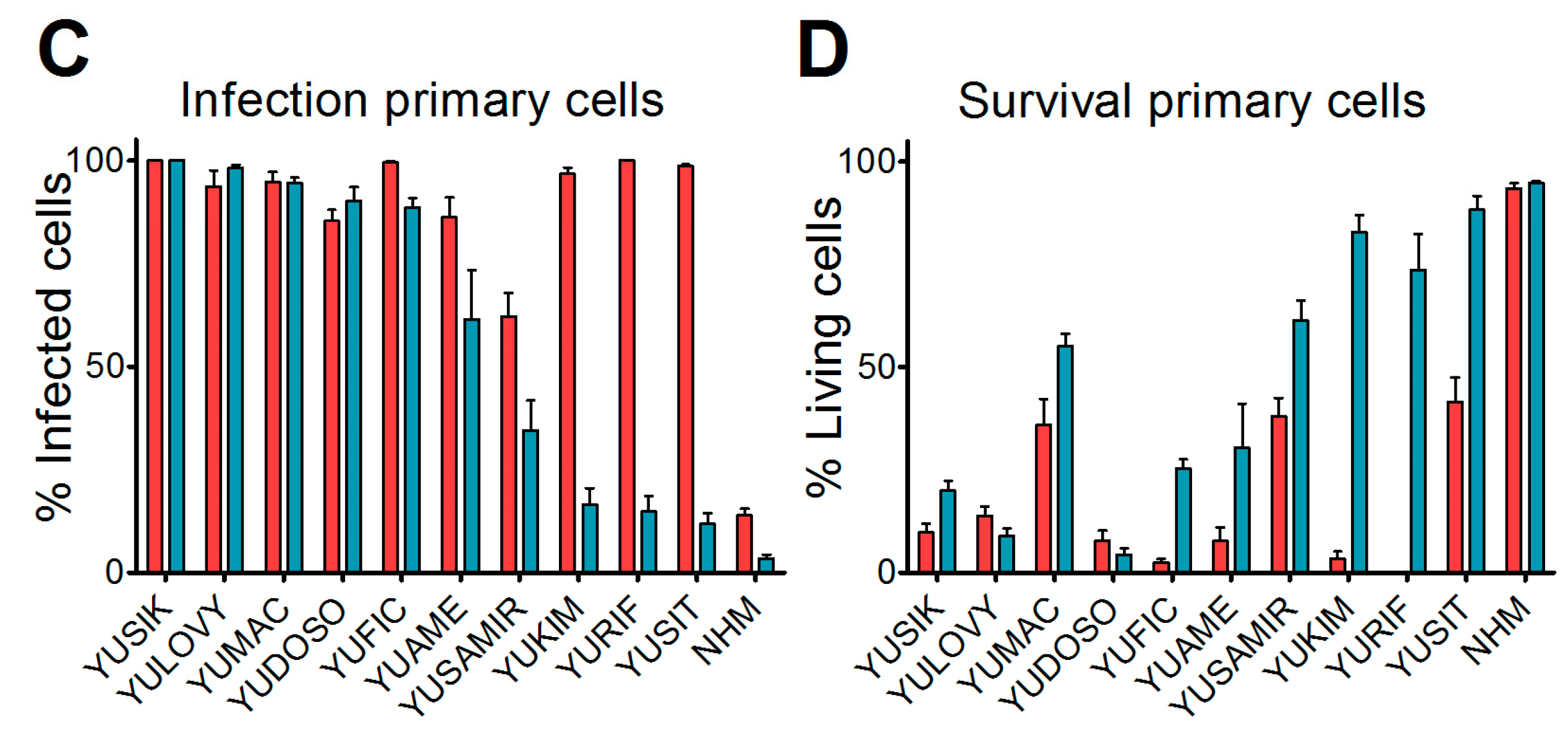
19]. Cultures were maintained in Opti-MEM supplemented with 5% fetal calf serum (FCS). At 36 hours before virus inoculation, cells were seeded in 24-well plates. Medium was replaced at low volume with medium containing either VSV-GFP or VSV-GP-GFP for a final multiplicity of infection (MOI) of 0.1. At 24 h post-infection, cultures were analyzed for GFP expression and the presence of cytopathic effects using a fluorescence microscope. Ten microscopic fields were assessed per condition.
2.3. Cell Lines
A375 (from European Collection of Cell cultures) were maintained in Dulbecco’s modified medium (PAA) supplemented with 15% FCS (PAA), 4 mM l-Glutamine (Gibco, Waltham, MA, USA), 100 U/mL penicillin, and 100 mg/mL Streptomycin (Gibco). NW-1539 (kind gift from Prof. Dr. E. Jäger, II.Medizinische Klinik/Onkologie, Nordwestkrankenhaus Frankfurt/Main, Germany), B16-OVA (kind gift from Dr. Edith Lord and Dr. John Frelinger, University of Rochester Medical Center, USA) and MDA-MB-435 were maintained in Dulbeco’s modified medium supplemented with 10% FCS, 4 mM l-Glutamine, 100 U/mL penicillin, and 100 mg/mL Streptomycin. Mel-Juso and SK-MEL5 were maintained in RPMI-1640 medium (Gibco) supplemented with 10% FCS, 4 mM l-Glutamine, 100 U/mL penicillin, and 100 mg/mL Streptomycin. SK-MEL3 (from DSMZ) were maintained in McCoy’s A5 medium (Gibco) supplemented with 10% FCS, 100 U/mL penicillin, and 100 mg/mL Streptomycin. BHK-21 (ACCT) were maintained in GMEM (Gibco) supplemented with 10% FCS, 5% Tryptose (Gibco), 100 U/mL penicillin, and 100 mg/mL Streptomycin.
2.4. Virus Variants
VSV, VSV-GP, VSV-GFP, VSV-GP-GFP, VSV-GP-Luciferase, VSV*ΔG, and VSV*M
QΔG (recombinant attenuated VSV with four mutations in the M protein and a deletion of the G protein), were described previously [
15,
16,
18,
20,
21]. L929 or BHK-21 cells were used for the amplification of replication-competent VSV variants and BHK-21 or 293T cells expressing LCMV-GP or VSV-G for ΔG variants. Titers of replication-competent viruses were determined on BHK-21 cells, as described previously.
2.5. Tropism Assay
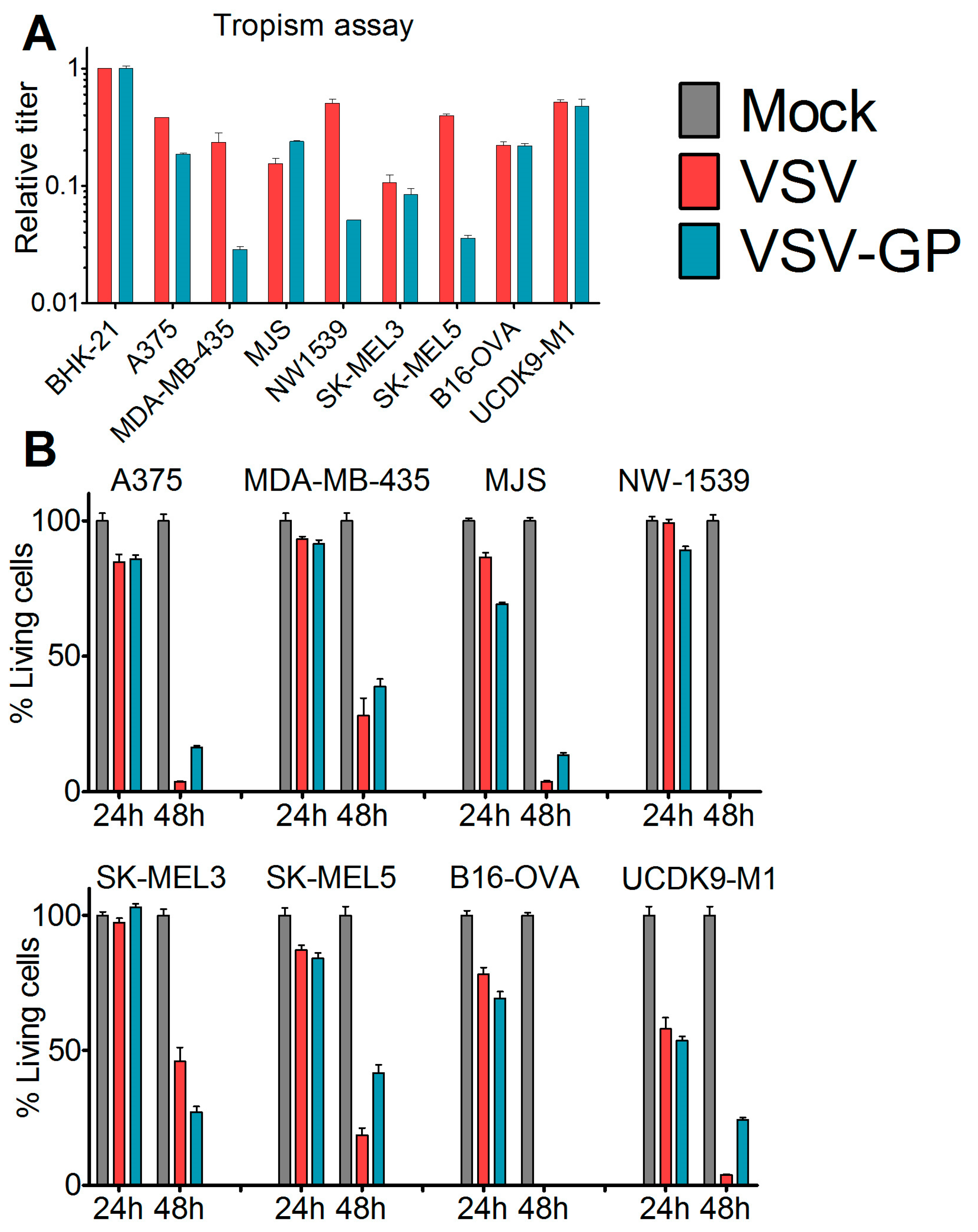
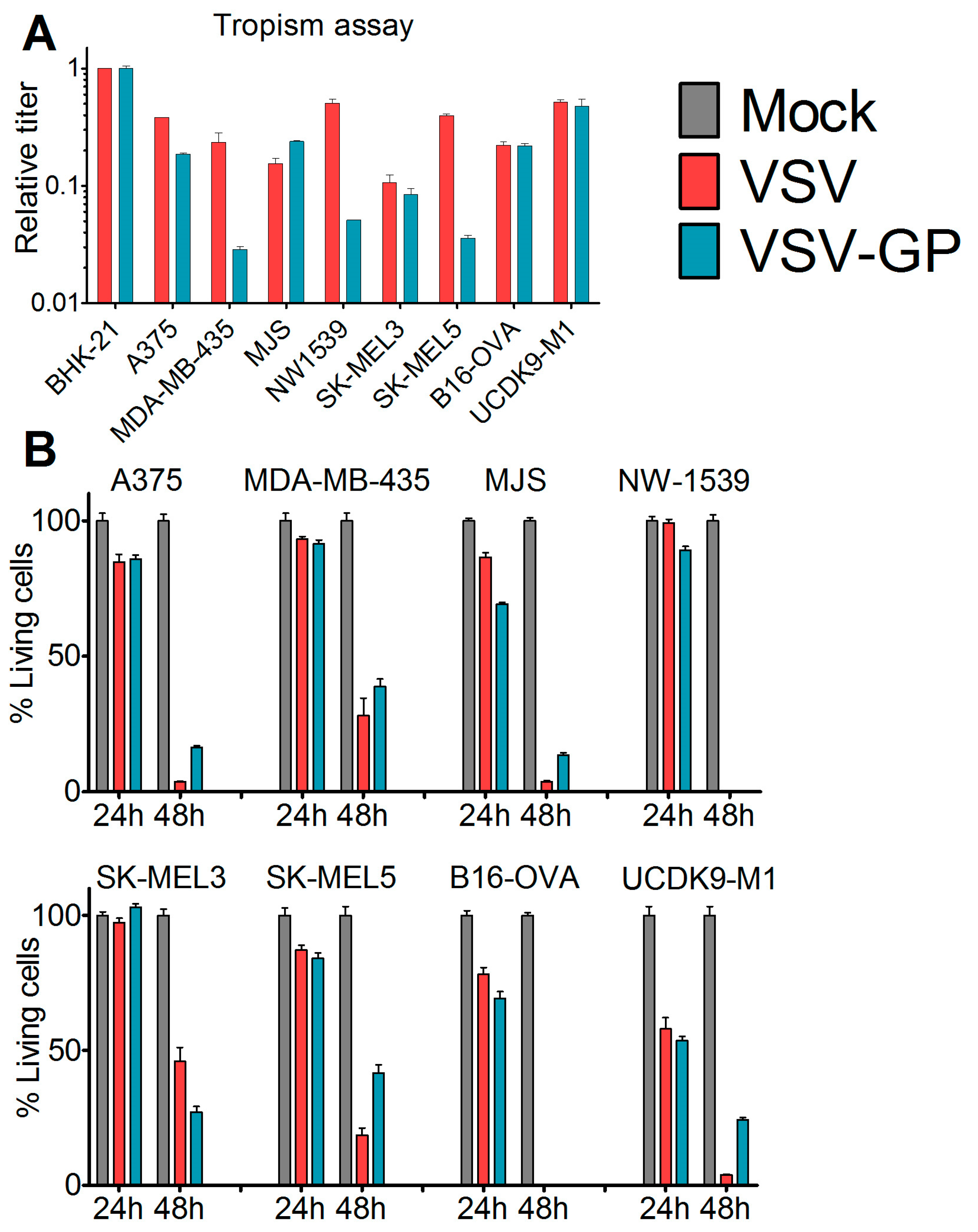
Cells were seeded in 24-well plates and were infected eight hours post-seeding with 10-fold serial dilutions of VSV*MQΔG-GP and VSV*MQΔG-G or mock-treated with phosphate buffered saline (PBS). After 16 hours, cells were analyzed via flow cytometry for GFP expression. The viral titer in each cell line was calculated and normalized to the obtained titer in the reference cell line BHK-21.
2.6. Killing Assay (WST-1 Assay)
Cells were seeded in 96-well plates. At 80% confluence, cells (12 technical replicates) were either infected with VSV wild-type or VSV-GP at an MOI of 0.1 or mock-treated with PBS. At indicated time points, 10 µL cell proliferation reagent WST-1 (Roche) were added per well. After four hours, color change was measured in a plate reader (Microplate reader Model 680, Biorad, Hercules, CA, USA) at 450 nm. For each well, a reference value, the wavelength at 650 nm, was subtracted. Obtained values were normalized to the mock-infected sample, and represented as a percentage of viable cells.
2.7. IFN Response
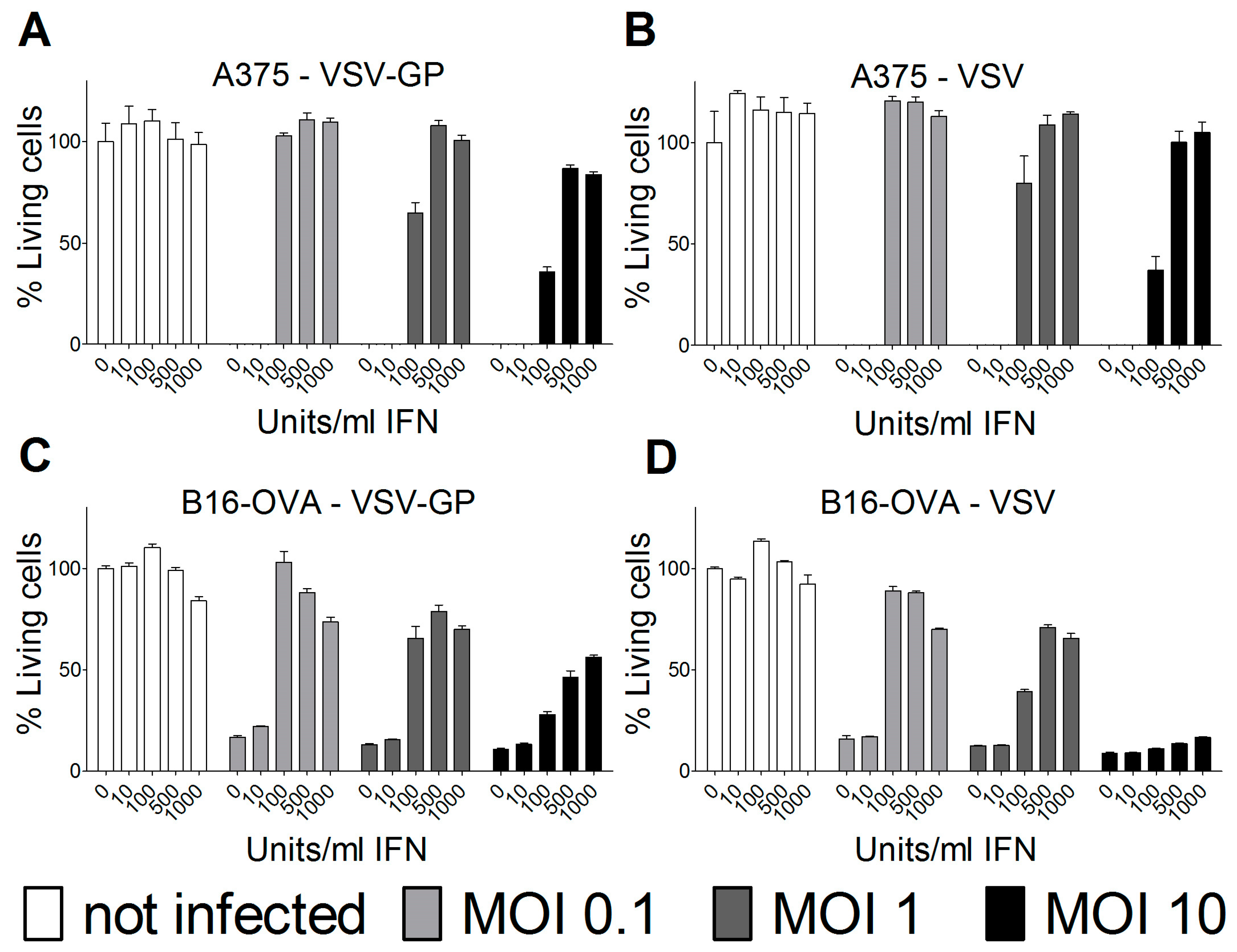
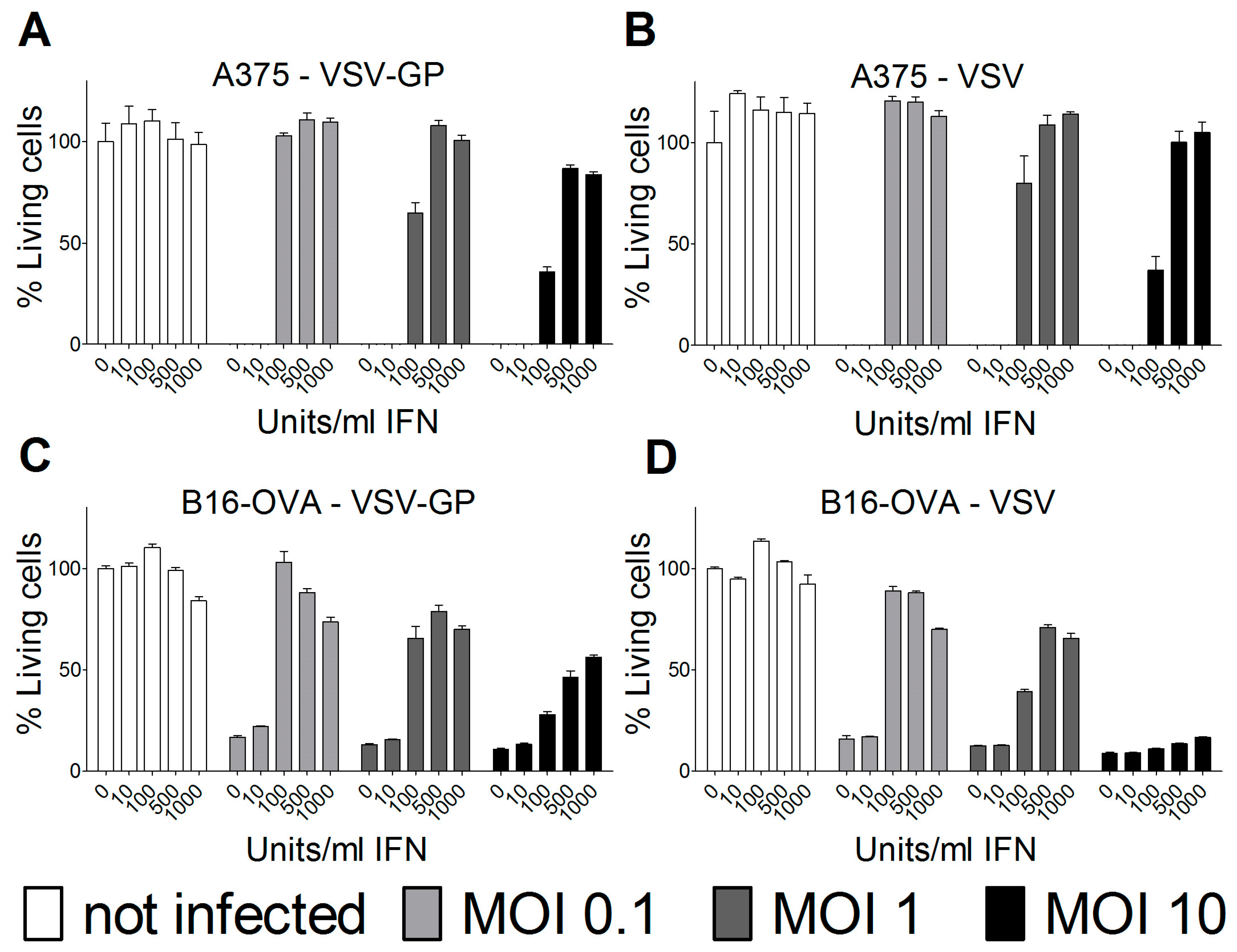
Cells were seeded in 96-well plates and pre-incubated overnight with the indicated concentrations of universal type-1 IFN (PBL, Piscataway, NJ, USA). The following morning, cells were infected with VSV wild-type or VSV-GP at an MOI of 0.1, 1, or 10. For each condition, quadruplicate samples were performed. As a positive, killing control cells were incubated, with a final concentration of 6.67 mM of H2O2. Three days after infection, cells were analyzed for viability using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-based in vitro cytotoxicity assay (Sigma-Aldrich, Saint Louis, MI, USA), according to the manufacturer’s recommendations. Plates were measured at 550 nm, and blank values of wells without cells were subtracted. Values were normalized to mock-infected cells that were not pre-treated with interferon (IFN), and represented as a percentage of viable cells.
2.8. Neutralization Assay
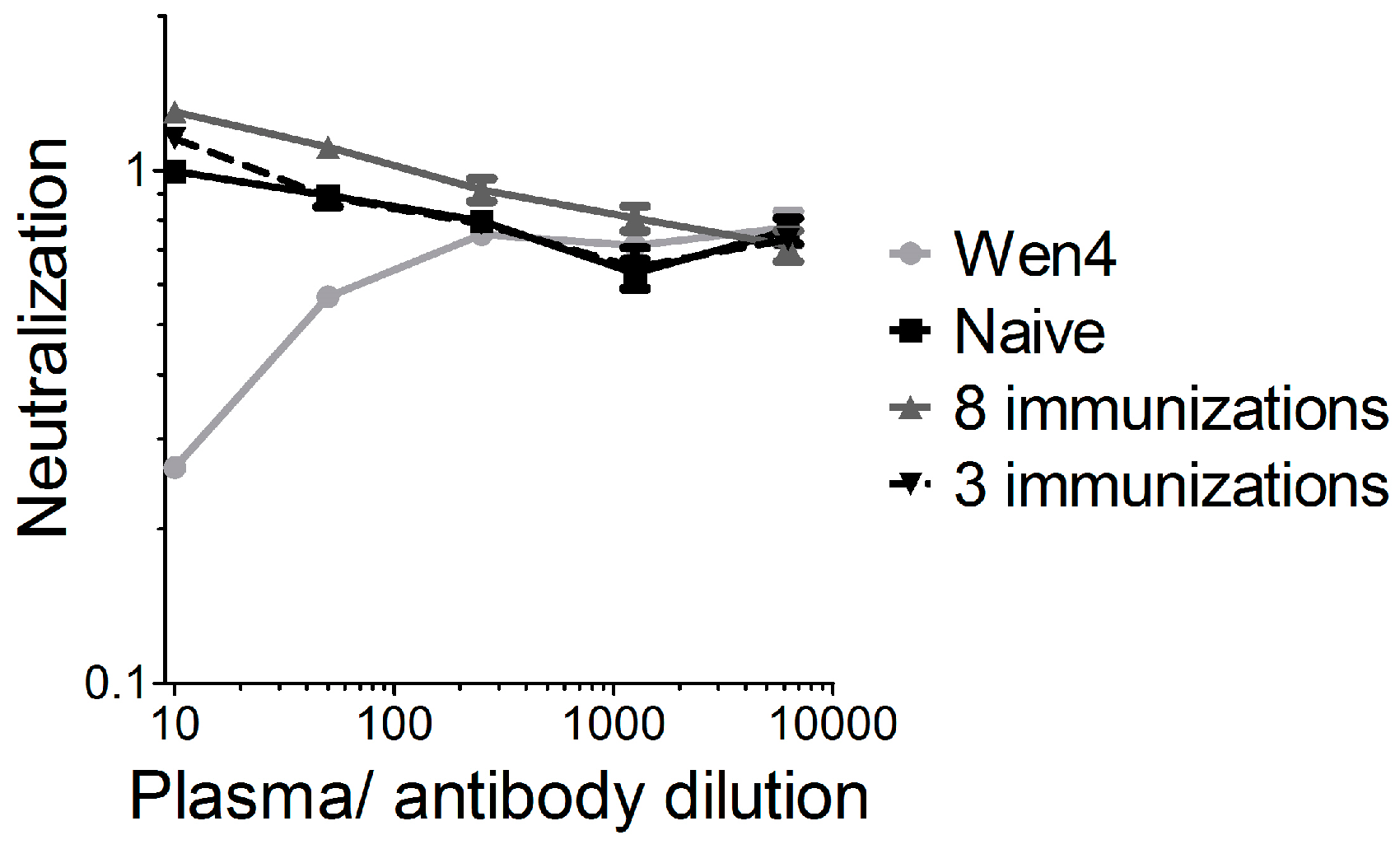
Heat-inactivated mouse plasma or the LCMV-neutralizing antibody Wen4, was serially diluted 1:5 fold starting with a 1:10 dilution. Dilutions were pre-incubated with the single-cycle infectious VSV*ΔG-GP virus (VSV*ΔG produced on cells expressing the LCMV GP) for one hour on ice. Subsequently, BHK-21 cells were infected in triplicate samples with the mixtures. Cells were incubated overnight at 37 °C. Cells were analyzed via flow cytometry for GFP expression. The mean value of the 1:10 dilution of the naïve plasma was set to 1, and all of the other values were given relative to this.
2.9. Fluorescence-Activated Cell Scanning Analysis
Blood from treated animals was collected two days after the third treatment using EDTA-coated tubes. 20 µL of blood per tube was stained with an APC (allophycocyanin)-conjugated SIINFEKL-specific tetramer (MBL) and fluorescence-conjugated antibodies against CD3, CD8, CD43, and CD44. Erythrocytes were lysed using ACK (ammonium-chloride-potassium) buffer, and samples were measured using a fluorescence-activated cell scanning (FACS) Canto II. Samples were analyzed using FlowJo software (FlowJo, Franklin Lakes, NJ, USA).
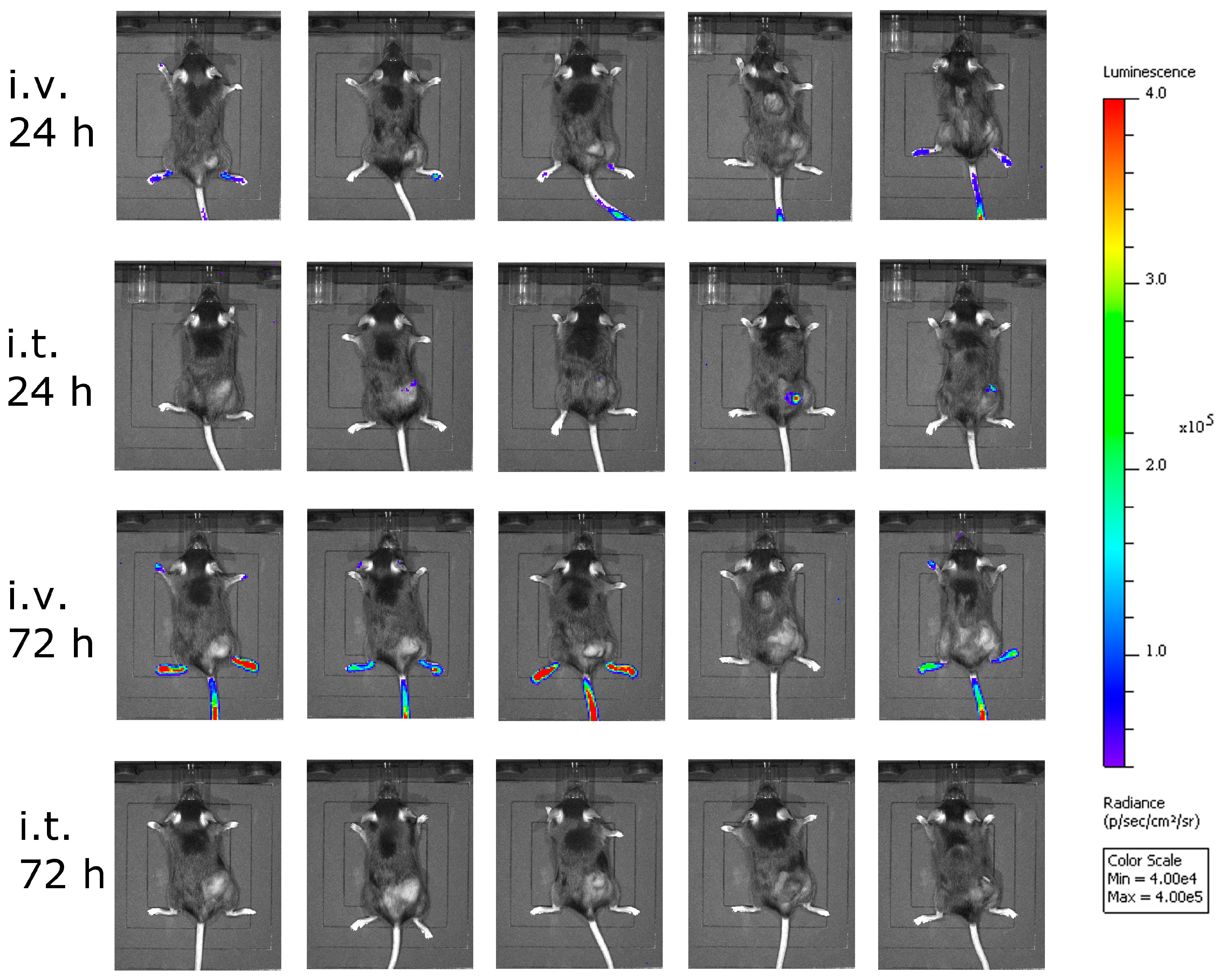
2.10. Bioluminescence Imaging (BLI)
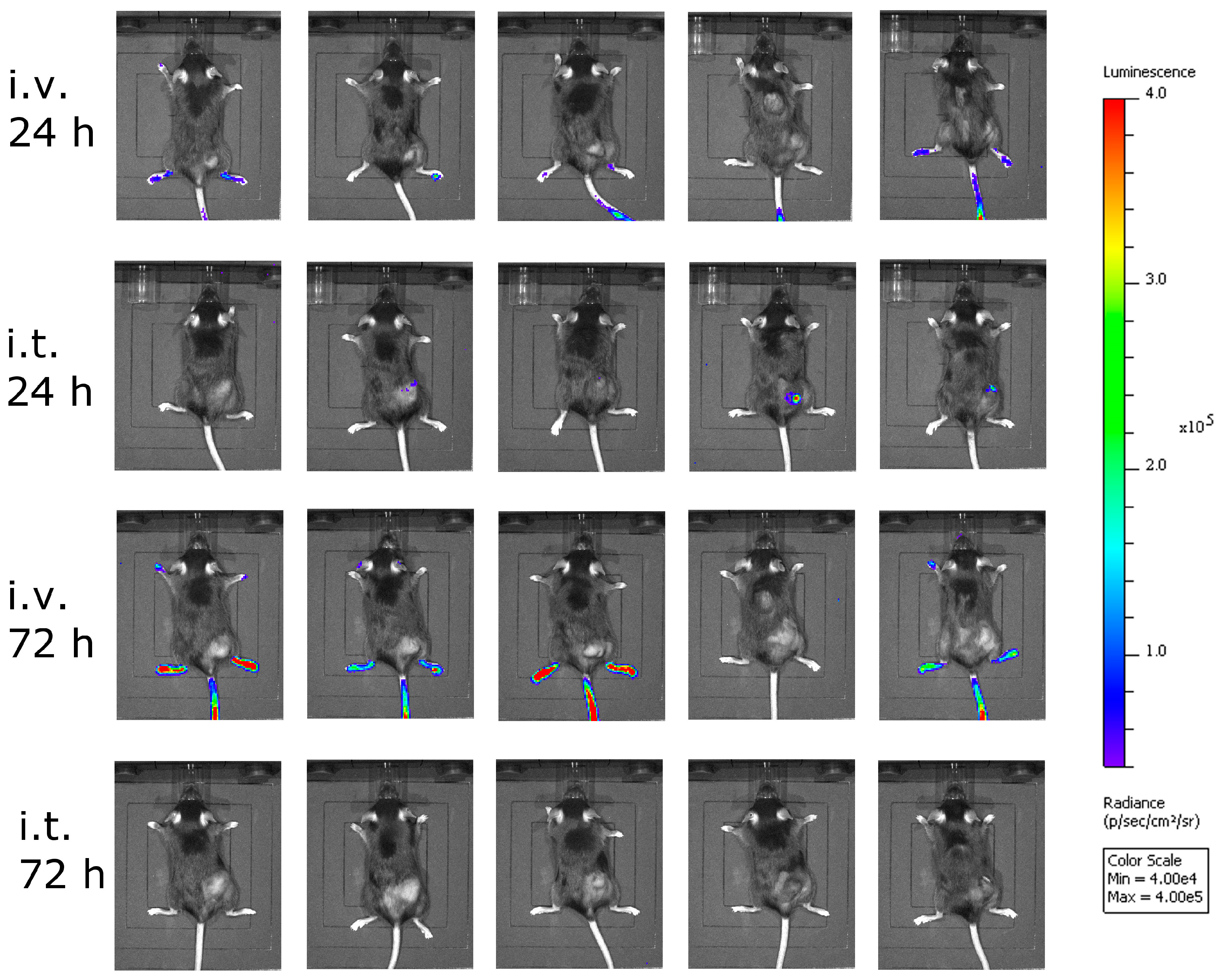
Bioluminescence imaging (BLI) was performed using the Lumina In Vivo Imaging System (IVIS Lumina, Perkin Elmer). Mice were injected intraperitoneally with 1.5 mg d-luciferin (Promega, Madison, WI, USA).
2.11. Xenograft Mouse Model
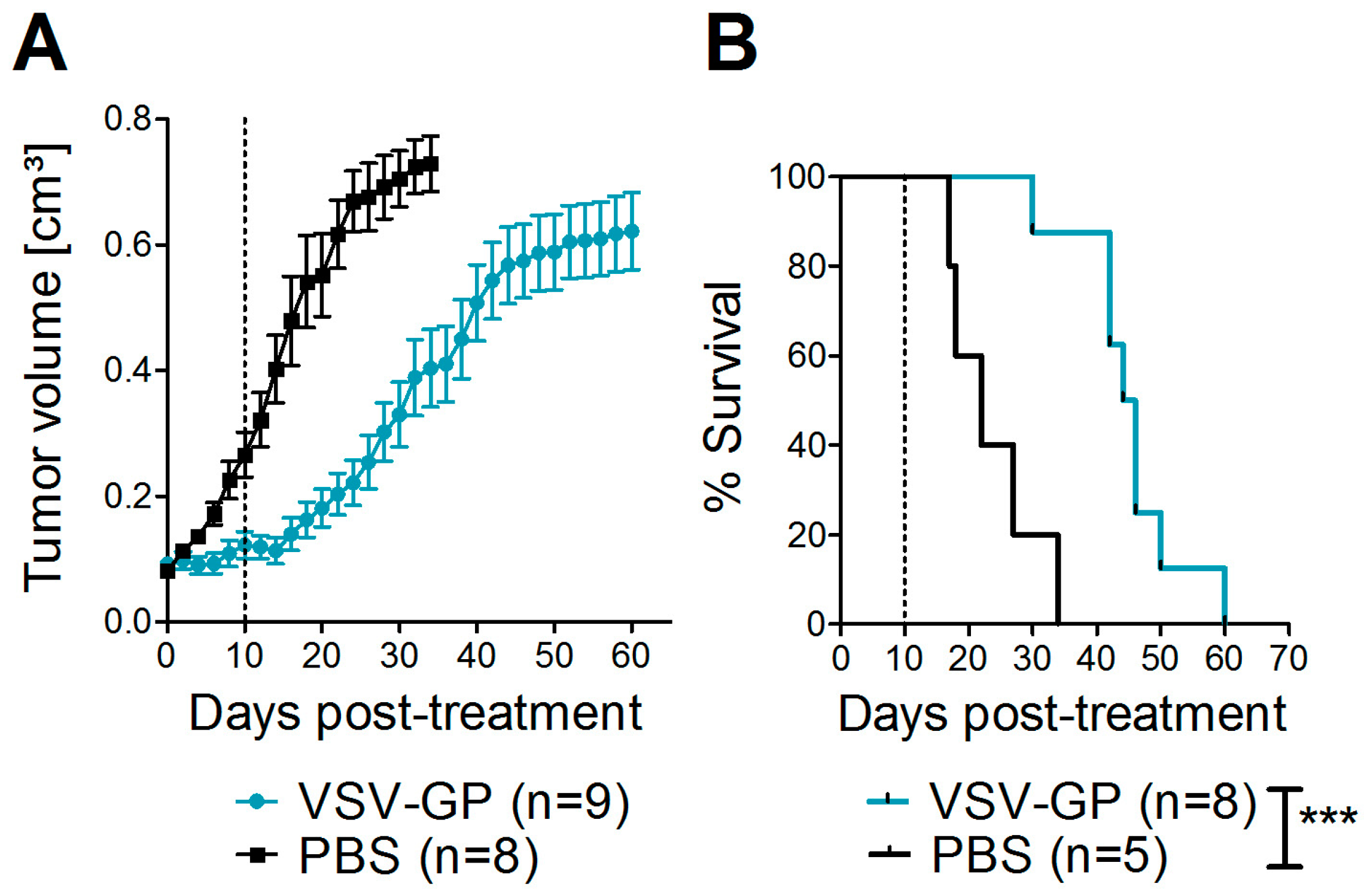
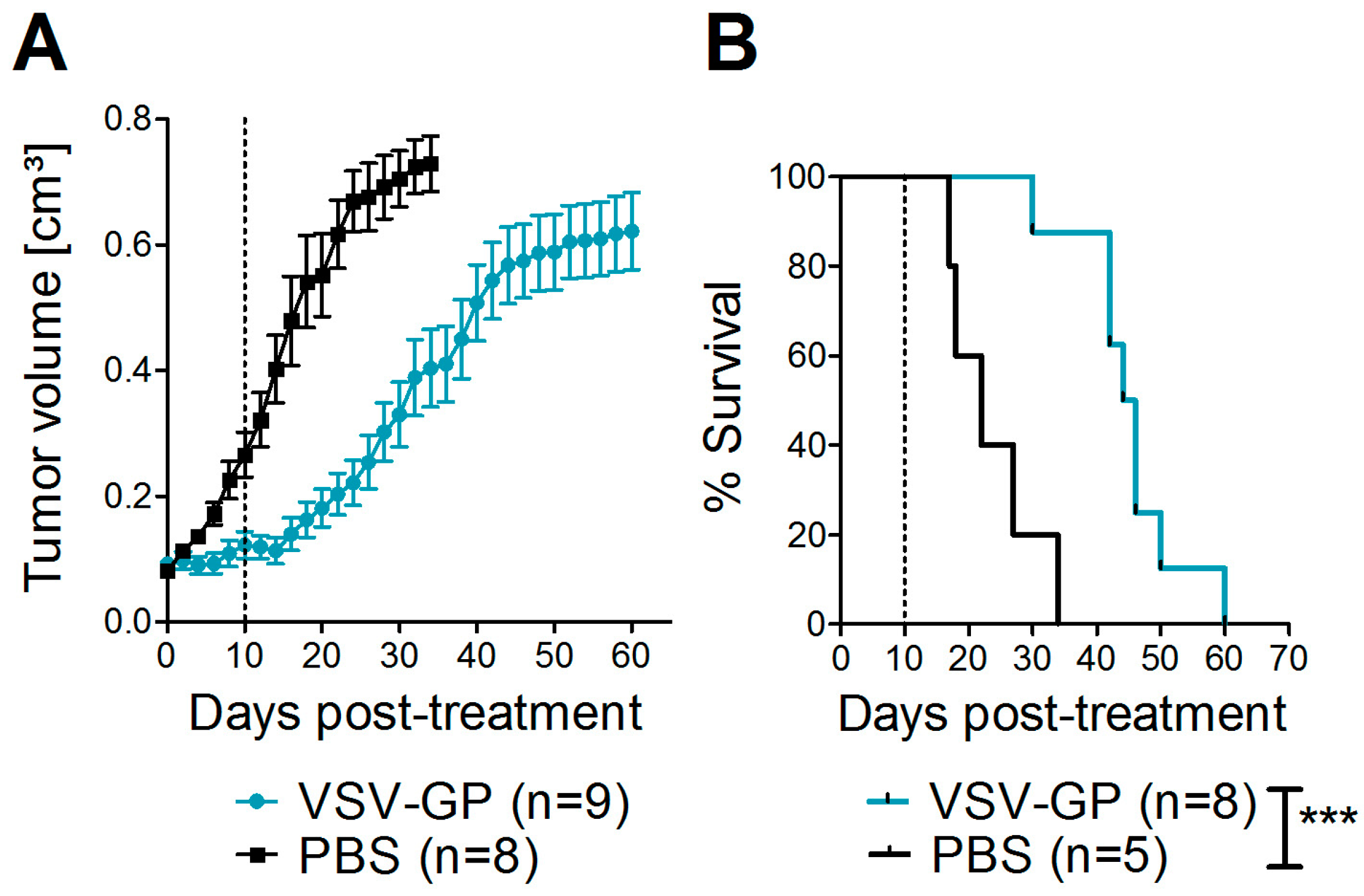
Bilateral tumors (5 × 106 A375 cells/tumor in 50 µL PBS) were injected subcutaneously into the flanks of NOD.CB17-Prkdcscid/J mice (NOD/SCID mice). Tumor growth was measured every other day with a caliper. Tumor volume was calculated as length × width2 × 0.4. When one tumor reached 0.07 cm3, both tumors were treated with intratumoral injections of either 107 PFU of VSV-GP in 30 µL PBS, or mock-treated with 30 µL of PBS. A second, identical round of treatment was given 10 days after the first treatment. Mice were sacrificed by cervical dislocation when moribund or when tumor size exceeded 0.8 cm³.
2.12. Syngeneic Subcutaneous Mouse Model
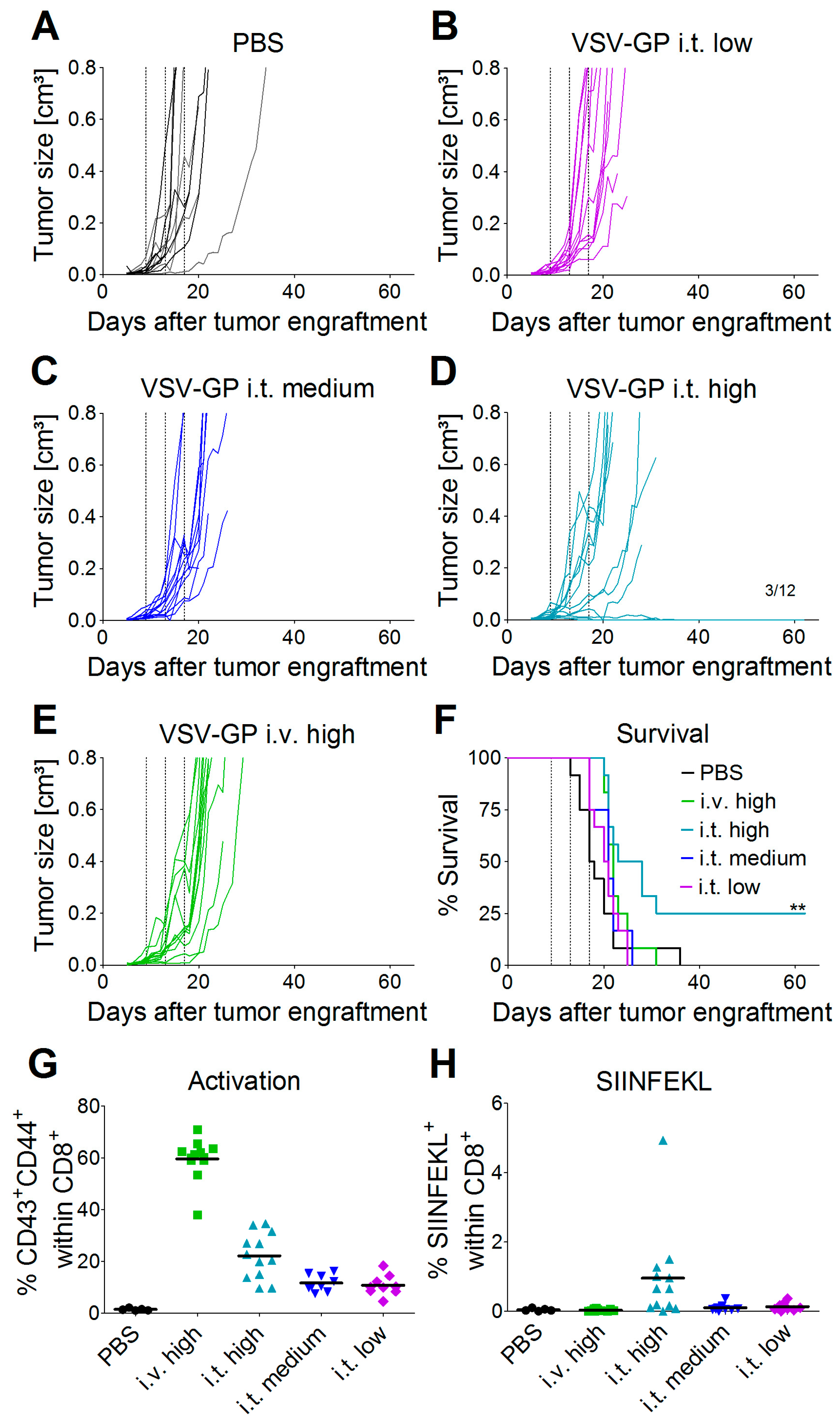
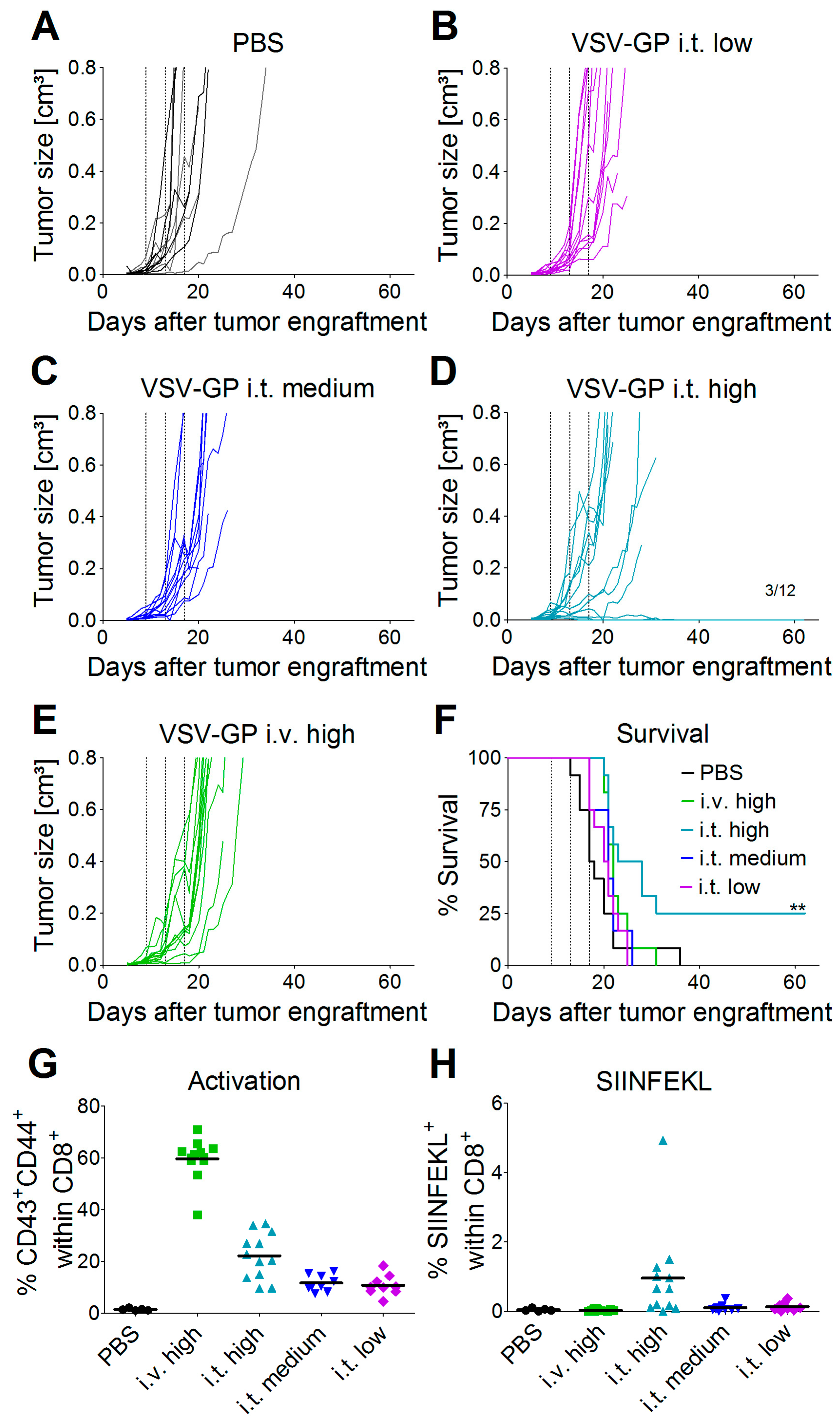
C57BL/6J mice (Janvier) were transplanted subcutaneously (s.c.) in the right flank with 5 × 105 murine B16-OVA melanoma cells in 100 µL PBS under isoflurane anesthesia. Tumor growth was monitored with a caliper starting three to five days post-injection, when the first tumors were palpable and visible. Tumor volumes were calculated as length × width2 × 0.4. Mice were sacrificed by cervical dislocation when either moribund, when tumor size exceeded 0.8 cm³, or when tumors ulcerated. Mice were divided into groups with similar average tumor volumes (n = 12 mice per group) on day 9 post-tumor cell transplantation. Mice were treated on days 9, 13, and 17 after tumor cell transplantation with either a low (2.36 × 104 PFU), medium (4.72 × 105 PFU), or high (2.36 × 107 PFU) dose of VSV-GP-GFP intratumorally, or a high (2.36 × 107 PFU) dose of VSV-GP-GFP intravenously. Virus was administered intratumorally in a volume of 30 µL PBS and intravenously in 100 µL PBS. PBS control mice were divided into two groups of six mice each, and one group was treated on days 9, 13, and 17 with 30 µL of PBS intratumorally, and the other with 100 µL of PBS intravenously.
2.13. Syngeneic Lung Metastasis Model
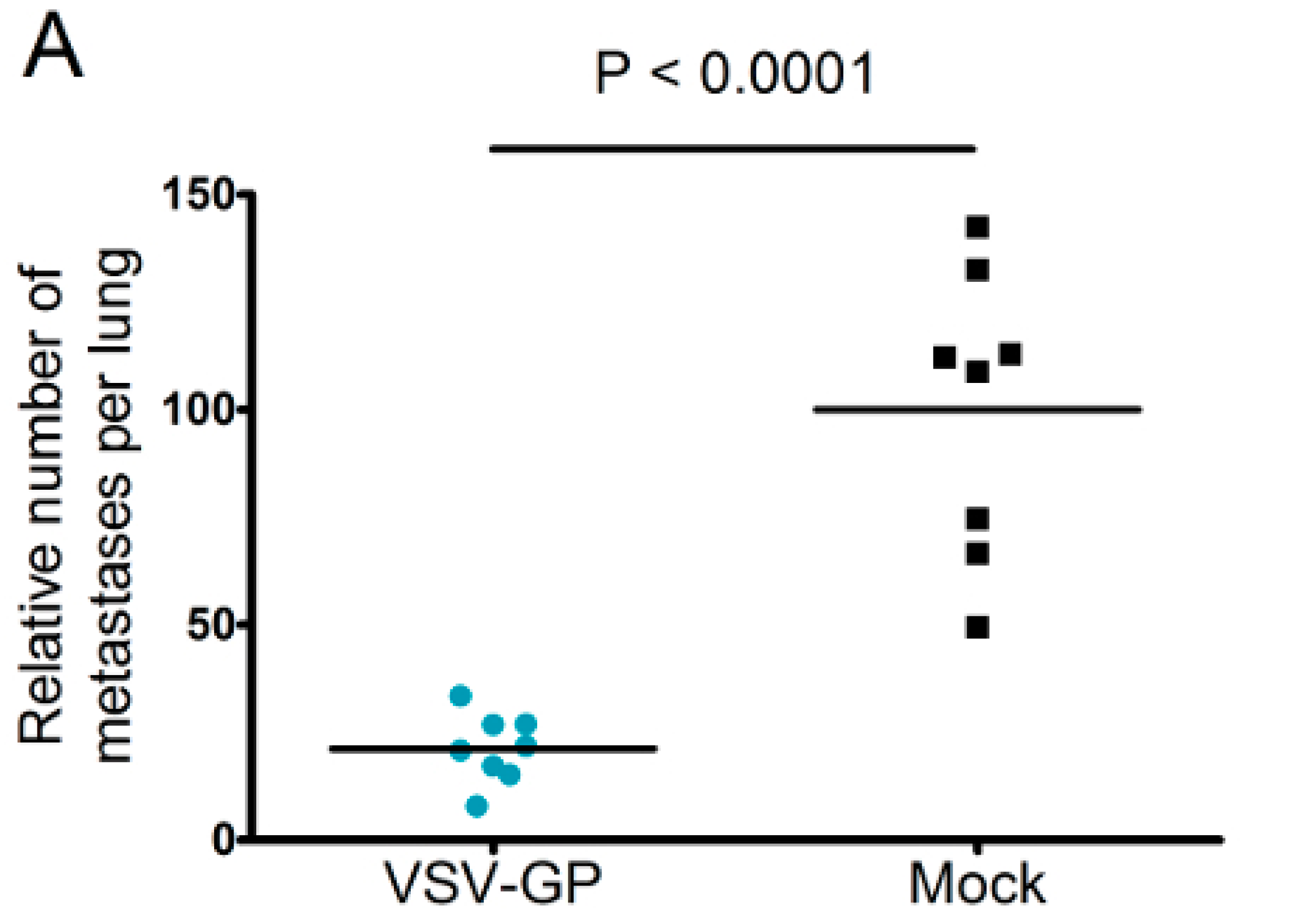
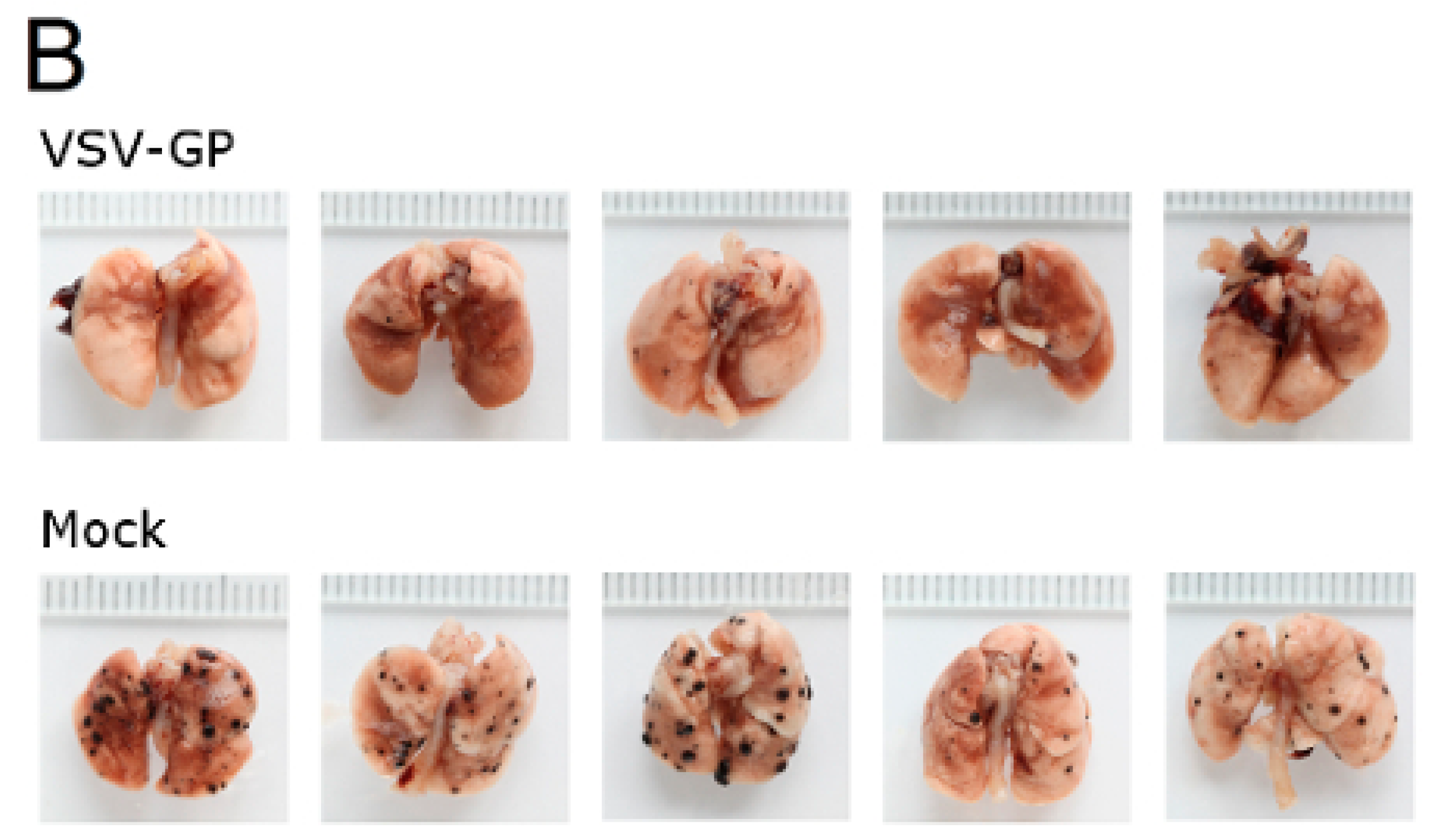
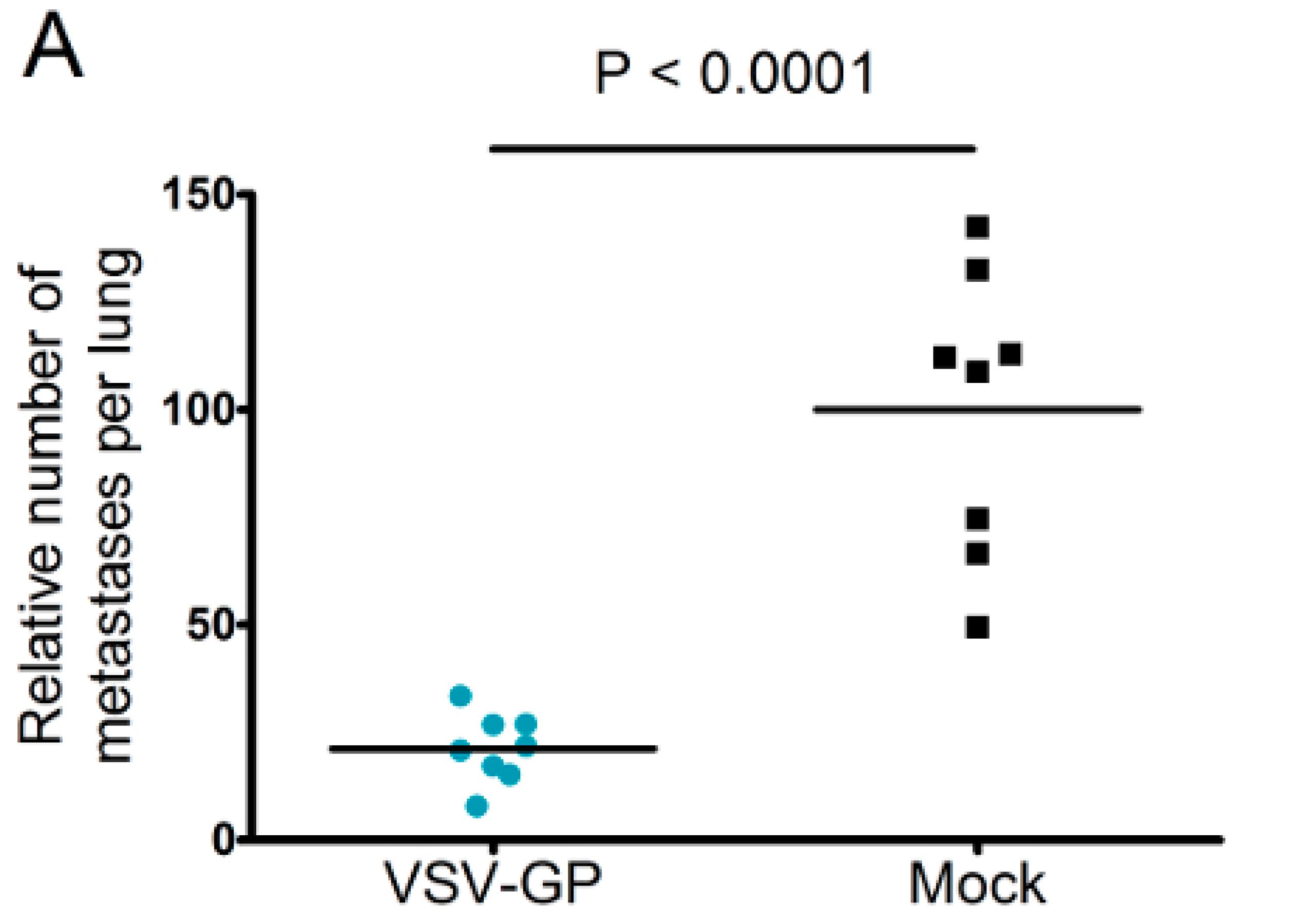
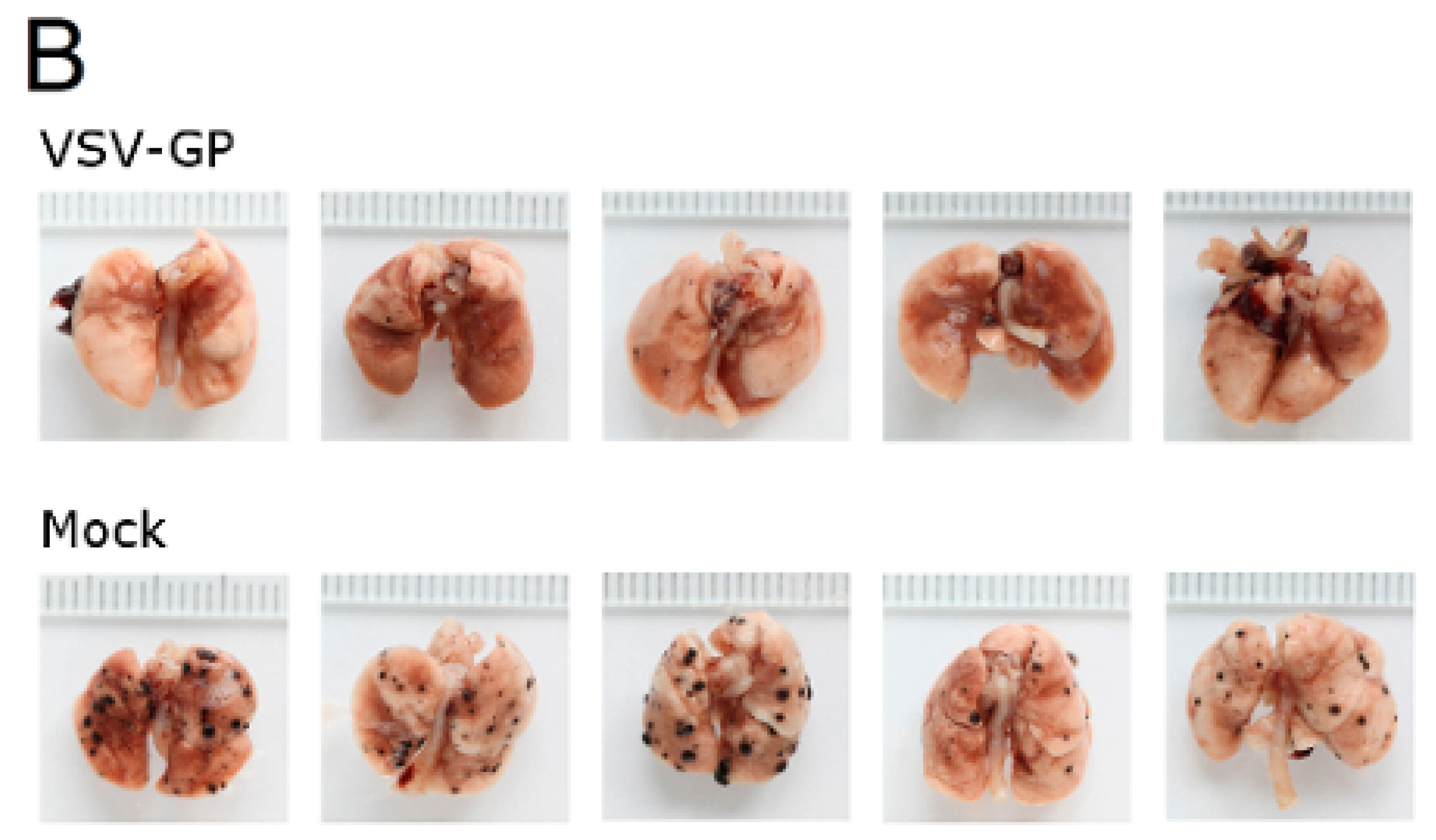
Murine melanoma lung metastases were established in C57BL/6 mice by intravenous injection of 1 × 106 B16-OVA cells on day 0. Mice were treated intravenously with 5 × 108 PFU VSV-GP on days 2, 4, 6, 8, and 10, or left untreated. On day 14, mice were sacrificed, and lungs were collected and stored in 1.5% formaldehyde. Lungs were dissected into individual lobes, and the number of visible metastases per lung was counted using a stereomicroscope.
2.14. Statistical Analysis
Statistical analysis was performed using GraphPad prism software (version 5, GraphPad Software, Inc., La Jolla, CA, USA), as indicated in the figure legends.
4. Discussion
We previously described VSV-GP as a potent oncolytic virus for the treatment of glioblastoma and ovarian cancer [
15,
16]. Here, we explored malignant melanoma as a new target for VSV-GP oncolytic virotherapy. Advanced malignant melanomas often metastasize into the brain. In this setting, the lack of neurotropism/neurotoxicity, which is one of the strengths of VSV-GP compared to the parental VSV wild-type virus, is highly relevant. However, even in patients without brain metastases, the safety profile of a non-modified VSV will not be acceptable, and the virus will need to have further attenuations, e.g., the expression of IFNβ.
We found that in vitro, all of the melanoma cell lines from our panel, including a murine and a canine cell line, were susceptible to VSV-GP infection and VSV-GP-mediated cell killing. As established cell lines differ from primary tumors in many aspects, we also evaluated VSV-GP-mediated oncolysis in primary cultures of malignant melanoma. For the primary cultures, the situation was more diverse, as some samples were highly susceptible to VSV-GP infection and killing, whereas others seemed to be more resistant. Infection correlated well with the survival of cells. Cultures with a high infection rate were efficiently lysed by VSV-GP, whereas primary cultures with a low infection rate, such as YUKIM, YURIF, or YUSIT, showed a higher level of surviving cells one day after infection. It is of special interest that normal human melanocytes were not infected and lysed, neither by VSV-GP, nor by the parental VSV wild-type virus. This is in accordance with our previous data for ovarian cells, where benign immortalized human ovarian surface epithelial cells were not lysed by VSV-GP [
15].
Despite the high susceptibility of melanoma cell lines and primary tumor cultures in vitro, we observed limitations of VSV-GP therapy in vivo in melanoma mouse models, especially when compared to the treatment efficacy in brain cancer [
16]. Although in both melanoma models the tumor growth was delayed upon VSV-GP treatment, only a few animals with long-term tumor remission were observed. We and others have shown that type I interferon sensitivity of a tumor can limit efficacy of oncolytic virus treatment in mouse cancer models [
15,
22,
23,
24].
Therefore, we analyzed the IFN sensitivity of the two lines used for the mouse models and indeed found that both human A375 and mouse B16-OVA cells mounted a protective response against VSV and VSV-GP infection upon stimulation with universal IFN. This is in accordance with previous studies for these two cell lines [
25,
26]. Others have shown that A375 cells also produce high amounts of IFN in vitro [
27]. This is especially important for the xenograft model, as the murine IFN produced by the tumor microenvironment will not sufficiently activate the signaling pathway in the human tumor cells to induce an antiviral state. In contrast, in the syngeneic B16-OVA mouse model, IFN produced by the tumor stroma cells can also induce an antiviral state in the sensitive B16-OVA cells, and thus limit the virus replication independent of IFN production by the tumor cells themselves.
In study by Stojdl et al., more than 80% of human tumor cell lines from the NCI60 panel had defects in their IFN response, and were therefore highly susceptible to VSV mediated killing in vitro [
28]. Therefore, the IFN responsiveness in human A375 melanoma cells is a less common feature, which we also previously described for ovarian cancer cell lines [
15]. Despite an efficient lysis of the human A2780 ovarian cancer cells by VSV-GP, the in vitro efficacy in an in vivo mouse model is limited. Similar to the A375 melanoma cells, the ovarian cancer line A2780 has an intact IFN responsiveness and production. In the latter, we showed that efficacy in the mouse model can be improved by inhibiting the downstream signaling of the IFNα receptor by the JAK1/2-inhibitor ruxolitinib [
15]. We propose that a similar approach might also enhance the efficacy of VSV-GP in melanoma mouse models.
Thus, especially for the A375 melanoma, the IFN response might have limited virus spread and long-term therapeutic efficacy. The IFN response most likely also limited VSV-GP spread in B16 tumors, but as this is an immunocompetent, syngeneic model, the situation is more complex. B16 cells have been described to be rather resistant to VSV-mediated oncolysis in in vivo mouse models. In the B16 tumor model, replication of the virus and direct oncolysis seem to be of minor importance, as replication-defective VSV is as effective as a replication-competent virus [
29]. Stimulation of innate immune mechanisms and the induction of anti-tumoral CTLs are more crucial factors for tumor control [
30]. Janelle et al. showed that VSV replicates in subcutaneous B16 tumors for only a few days after intratumoral injection [
31]. We made similar observations for VSV-GP using a virus variant expressing luciferase. One day after intratumoral virus injection, we detected only a low level of luciferase expression in the tumor, which was gone three days after treatment. Despite this low virus replication, and presumably also a low level of direct oncolysis, we could detect tumor-specific, i.e., OVA-specific, CTLs in the blood of mice treated intratumorally with the highest dose of VSV-GP. Only in this group, long-term tumor free survival was observed. In addition, these animals were protected from a rechallenge with the same B16-OVA tumor cells, indicating a long-lasting protection that is most likely mediated primarily by OVA-specific CTLs. To enhance this immunologic effect, arming the virus with immunostimulatory cytokines or tumor antigens is an interesting approach [
26].
Although intravenous treatment led to the highest general activation of CD8+ T-cells in the blood of the animals, tumor-specific CTLs were not detected. This might be explained by the low level of virus replication in tumor tissue, and thus the low oncolysis and tumor antigen release after intravenous virus application.
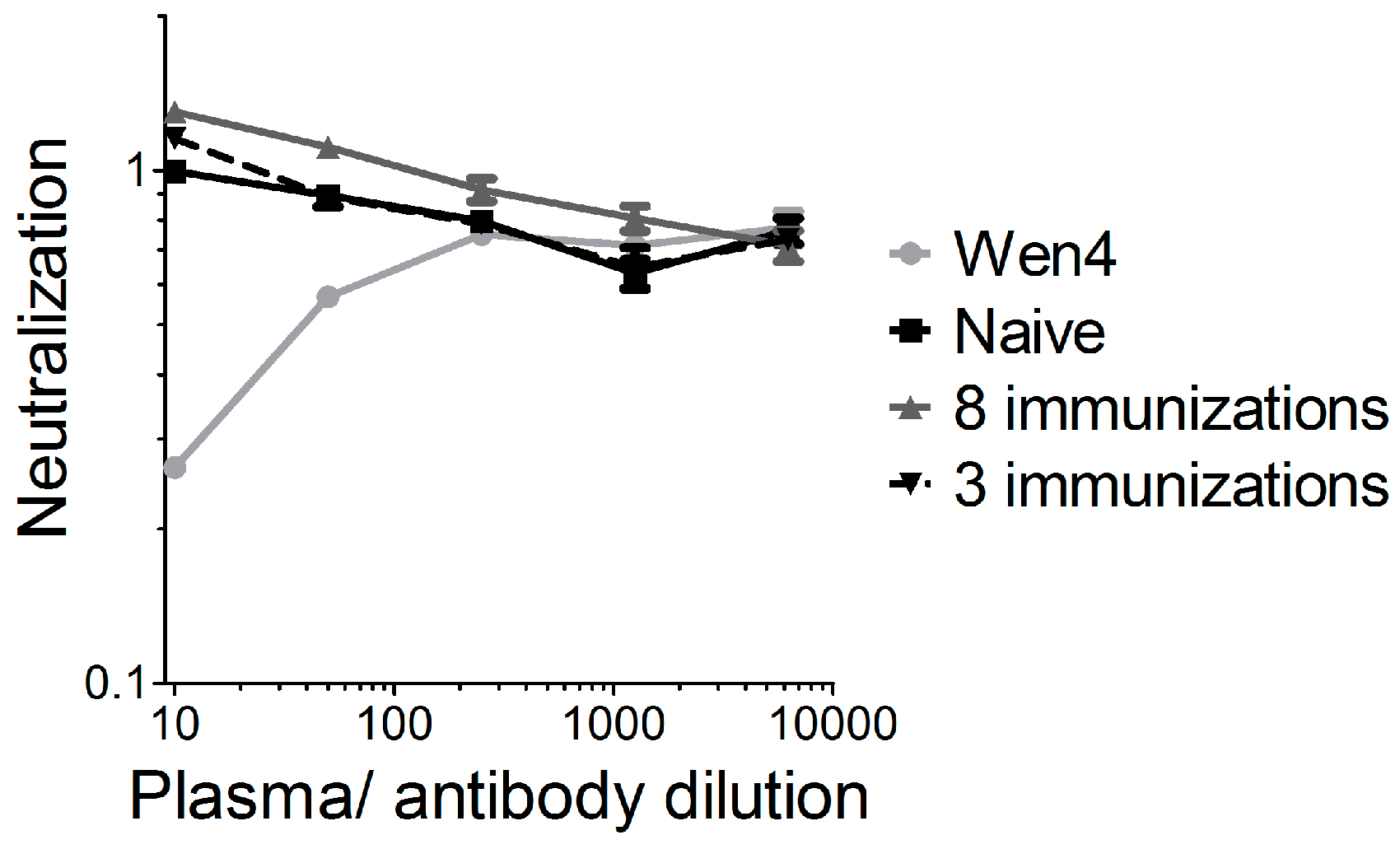
For oncolytic viruses, such as herpes virus, polio, adenovirus, or a non-neurotoxic attenuated VSV variant expressing IFNβ, which are currently in clinical or pre-clinical testing, neutralizing antibodies against the vector backbone are either present in the general population, or are induced readily after the first administration, which limits the efficacy of repeated administration. We previously showed for VSV-GP that vector-neutralizing antibodies are not induced after two immunizations in mice, and that therefore, the immune response against a foreign antigen can be boosted by homologous immunizations [
17]. For LCMV, the donor of the glycoprotein in our chimeric VSV-GP, neutralizing antibodies are induced only after long-term chronic infection [
32]. It is speculated that it is difficult to induce neutralizing antibodies against LCMV due to the high glycosylation of the glycoprotein. We therefore increased the observation time after the immunization, and also the number of immunizations. In addition, we used a highly sensitive assay for the detection of neutralizing antibodies. However, even after eight subsequent immunizations, we could not detect any VSV-GP-neutralizing antibodies. Therefore, neutralizing antibodies did not limit virus homing to the tumor or local virus replication. Here, in addition to the IFN response as discussed above, poor tumor vascularization, hypoxia, and high intratumoral pressure might have limited intratumoral virus spread [
33]. Further studies will be required in order to define potential additional relevant viral restriction factors in the B16 model for VSV-GP.
Despite the limited virus replication in B16 tumor tissue, the intravenous treatment of developing B16 lung micro-metastases with VSV-GP was effective. Thus, VSV-GP could have an impact on the growth of the metastases, or the development of new ones. In the metastases model, we could not compare VSV-GP’s efficacy to wild-type VSV, as in a preceding experiment, two-thirds of the tumor-free animals that were treated intravenously with a single high dose (5 × 10
8 PFU) of wild-type VSV succumbed from neurotoxicity [
16]. However, it will be interesting to compare the efficacy of VSV-GP in future studies with the attenuated VSV variant VSV-IFNβ, which is currently being tested in clinical trials for different tumor entities (ClinicalTrials.gov Identifier: NCT02923466, NCT01628640).
Taken together, VSV-GP is a promising new treatment option for malignant melanoma. Factors restricting VSV-GP’s efficacy will need to be further explored prior to clinical development. Understanding these restrictions is the basis for defining predictive biomarkers that will be essential for the selection of a target patient population that will benefit most from oncolytic virotherapy.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}