Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Sclerotinia sclerotiorum Cultures and RNA Extraction
2.2. Analysis of S. sclerotiorum Transcriptome
2.3. Analysis of S. sclerotiorum Small RNA Populations
2.4. High-Throughput RNA Ligase-Mediated Rapid Amplification of cDNA Ends (HT-RACE) Analysis
3. Results
3.1. Changes in mRNA Accumulation Associated with Infection of S. sclerotiorum by SsHV2-L
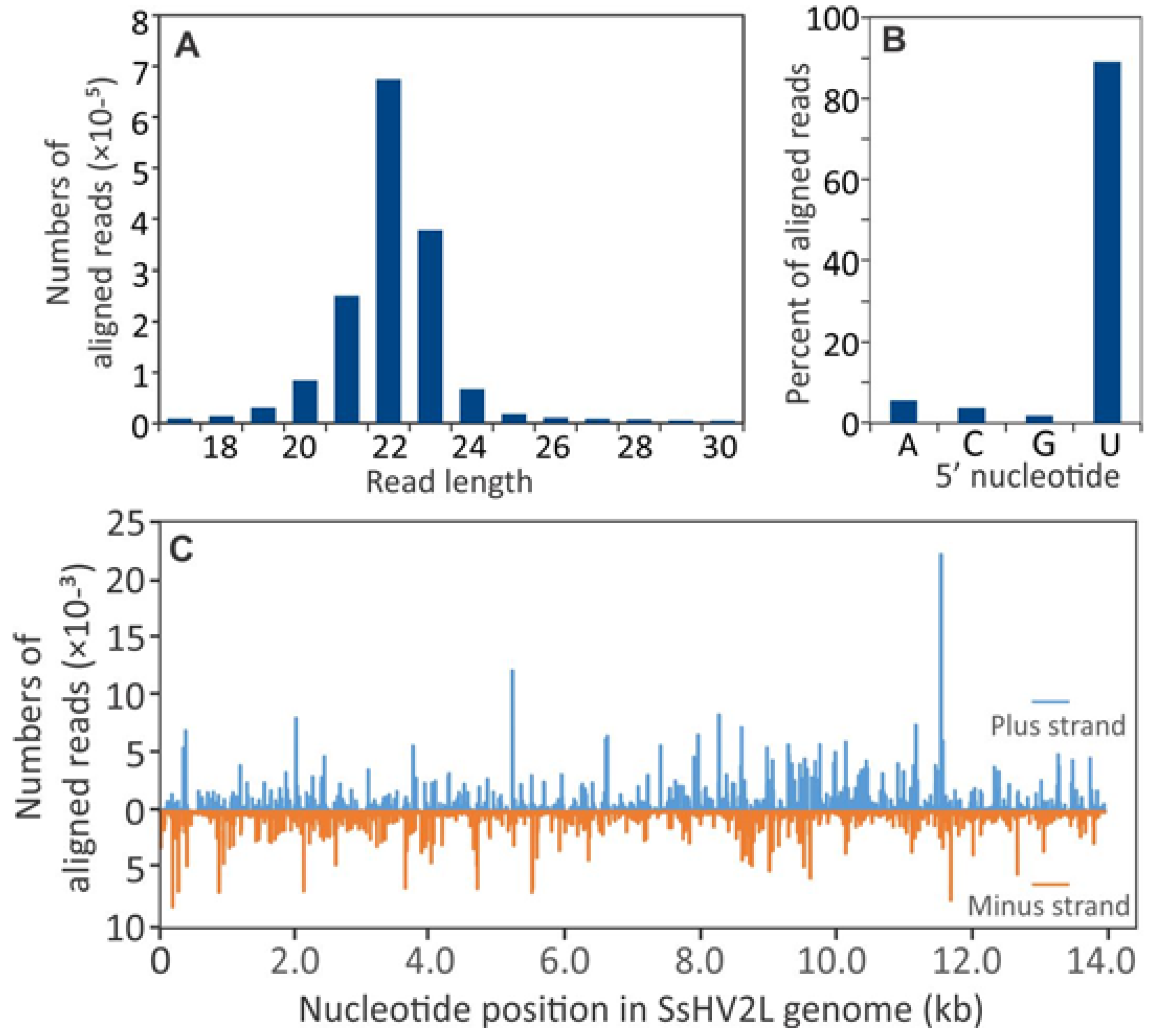
3.2. Small RNA Accumulation in Healthy and SsHV2-L-infected S. sclerotiorum Cultures
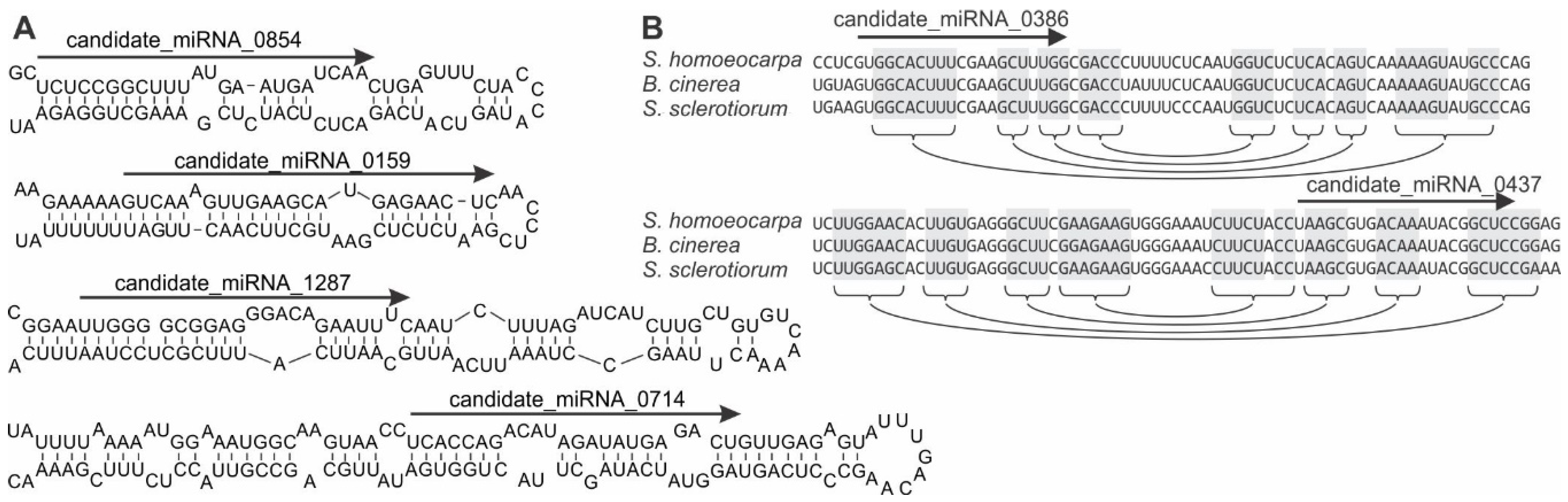
3.3. Identification of S. sclerotiorum Loci Producing microRNA-Like RNAs
3.4. Potential Targets for S. sclerotiorum sRNAs
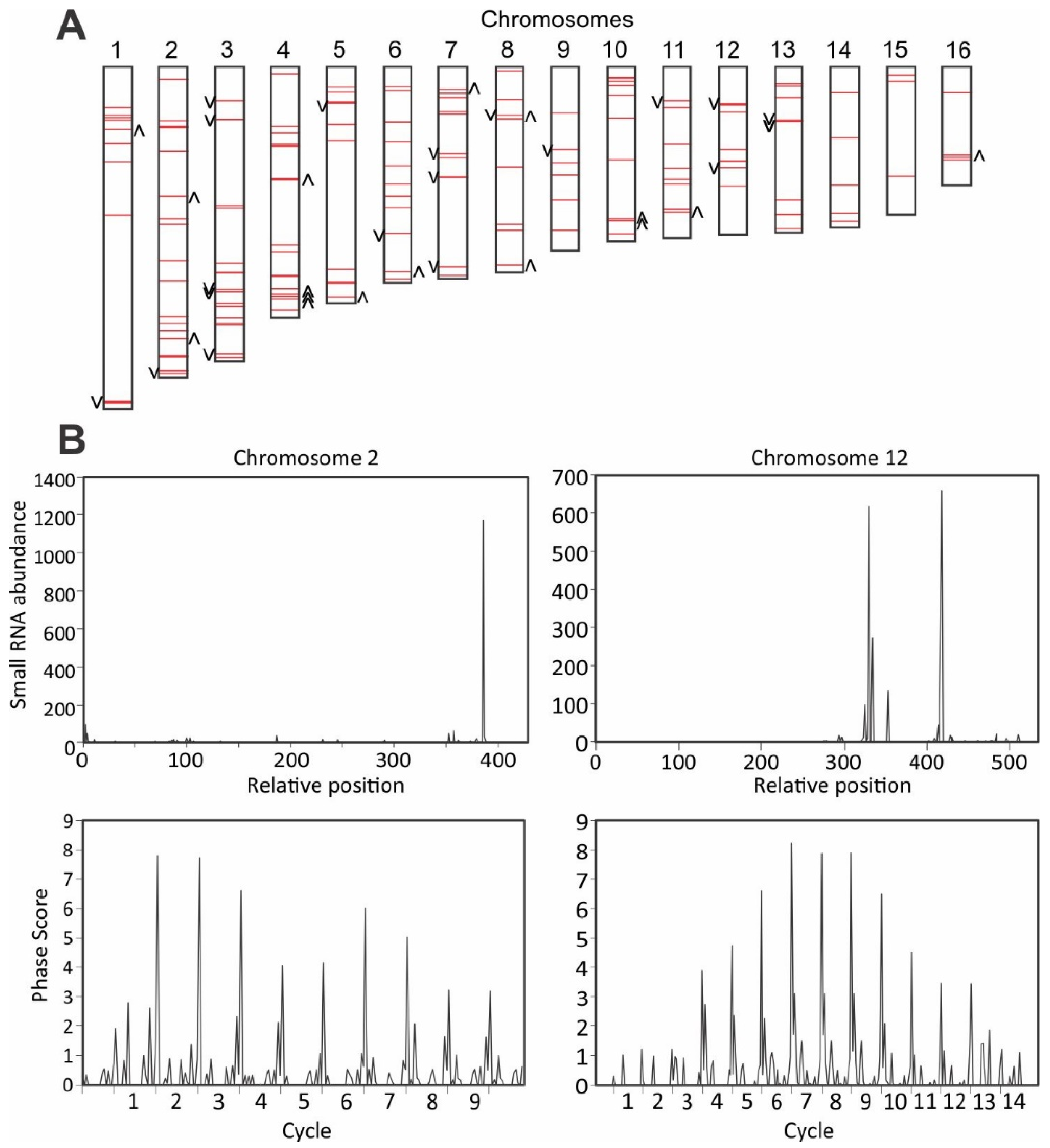
3.5. Detection of Phased siRNAs from S. sclerotiorum Noncoding RNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ghabrial, S.A.; Castón, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479–480, 356–368. [Google Scholar] [CrossRef]
- Nuss, D.L. Mycoviruses, RNA silencing, and viral RNA recombination. Adv. Virus Res. 2011, 80, 25–48. [Google Scholar]
- Baulcombe, D. RNA silencing. Trends Biochem. Sci. 2005, 30, 290–293. [Google Scholar] [CrossRef]
- Waterhouse, P.M.; Wang, M.B.; Lough, T. Gene silencing as an adaptive defence against viruses. Nature 2001, 411, 834–842. [Google Scholar] [CrossRef]
- Li, Y.; Lu, J.F.; Han, Y.H.; Fan, X.X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013, 342, 231–234. [Google Scholar] [CrossRef]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef]
- Nakayashiki, H.; Kadotani, N.; Mayama, S. Evolution and diversification of RNA silencing proteins in fungi. J. Mol. Evol. 2006, 63, 127–135. [Google Scholar] [CrossRef]
- Laurie, J.D.; Ali, S.; Linning, R.; Mannhaupt, G.; Wong, P.; Guldener, U.; Munsterkotter, M.; Moore, R.; Kahmann, R.; Bakkeren, G.; et al. Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species-specific presence of transposable elements. Plant Cell 2012, 24, 1733–1745. [Google Scholar] [CrossRef]
- Bernstein, D.A.; Vyas, V.K.; Weinberg, D.E.; Drinnenberg, I.A.; Bartel, D.P.; Fink, G.R. Candida albicans Dicer (CaDcr1) is required for efficient ribosomal and spliceosomal RNA maturation. Proc. Natl. Acad. Sci. USA 2012, 109, 523–528. [Google Scholar] [CrossRef]
- Axtell, M.J.; Westholm, J.O.; Lai, E.C. Vive la différence: Biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011, 12, 221. [Google Scholar] [CrossRef]
- Song, X.W.; Li, P.C.; Zhai, J.X.; Zhou, M.; Ma, L.J.; Liu, B.; Jeong, D.H.; Nakano, M.; Cao, S.Y.; Liu, C.Y.; et al. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012, 69, 462–474. [Google Scholar] [CrossRef]
- Arikit, S.; Zhai, J.X.; Meyers, B.C. Biogenesis and function of rice small RNAs from non-coding RNA precursors. Curr. Opin. Plant Biol. 2013, 16, 170–179. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.X.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Mueth, N.A.; Ramachandran, S.R.; Hulbert, S.H. Small RNAs from the wheat stripe rust fungus (Puccinia striiformis f.sp. tritici). BMC Genom. 2015, 16, 718. [Google Scholar] [CrossRef]
- Lin, R.M.; He, L.Y.; He, J.Y.; Qin, P.G.; Wang, Y.R.; Deng, Q.M.; Yang, X.T.; Li, S.C.; Wang, S.Q.; Wang, W.M.; et al. Comprehensive analysis of microRNA-Seq and target mRNAs of rice sheath blight pathogen provides new insights into pathogenic regulatory mechanisms. DNA Res. 2016, 23, 415–425. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, N.; Jiang, Q.Y.; Sun, X.J.; Wang, Y.; Zhang, H.; Hu, Z. Exploring microRNA-like small RNAs in the filamentous fungus Fusarium oxysporum. PLoS ONE 2014, 9, e104956. [Google Scholar] [CrossRef]
- Bai, Y.H.; Lan, F.X.; Yang, W.Q.; Zhang, F.; Yang, K.L.; Li, Z.G.; Gao, P.L.; Wang, S.H. SRNA profiling in Aspergillus flavus reveals differentially expressed miRNA-like RNAs response to water activity and temperature. Fungal Genet. Biol. 2015, 81, 113–119. [Google Scholar] [CrossRef]
- Thompson, D.M.; Parker, R. Stressing out over tRNA cleavage. Cell 2009, 138, 215–219. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, M.H.; Cao, Z.H.; Li, X.; Zhang, Y.F.; Shi, J.C.; Feng, G.H.; Peng, H.Y.; Zhang, X.D.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef]
- Martinez, G.; Choudury, S.G.; Slotkin, R.K. tRNA-derived small RNAs target transposable element transcripts. Nucleic Acids Res. 2017, 45, 5142–5152. [Google Scholar] [CrossRef]
- Nunes, C.C.; Gowda, M.; Sailsbery, J.; Xue, M.; Chen, F.; Brown, D.E.; Oh, Y.; Mitchell, T.K.; Dean, R.A. Diverse and tissue-enriched small RNAs in the plant pathogenic fungus, Magnaporthe oryzae. BMC Genom. 2011, 12, 288. [Google Scholar] [CrossRef]
- Wang, M.B.; Bian, X.Y.; Wu, L.M.; Liu, L.X.; Smith, N.A.; Isenegger, D.; Wu, R.M.; Masuta, C.; Vance, V.B.; Watson, J.M.; et al. On the role of RNA silencing in the pathogenicity and evolution of viroids and viral satellites. Proc. Natl. Acad. Sci. USA 2004, 101, 3275–3280. [Google Scholar] [CrossRef]
- Qi, X.; Bao, F.S.; Xie, Z. Small RNA deep sequencing reveals role for arabidopsis thaliana RNA-dependent RNA polymerases in viral siRNA biogenesis. PLoS ONE 2009, 4, e4971. [Google Scholar] [CrossRef]
- Shimura, H.; Pantaleo, V.; Ishihara, T.; Myojo, N.; Inaba, J.; Sueda, K.; Burgyan, J.; Masuta, C. A viral satellite RNA induces yellow symptoms on tobacco by targeting a gene involved in chlorophyll biosynthesis using the RNA silencing machinery. PLoS Pathog. 2011, 7, e1002021. [Google Scholar] [CrossRef]
- Smith, N.A.; Eamens, A.L.; Wang, M.B. Viral small interfering RNAs target host genes to mediate disease symptoms in plants. PLoS Pathog. 2011, 7, e1002022. [Google Scholar] [CrossRef]
- Cao, M.J.; Du, P.; Wang, X.B.; Yu, Y.Q.; Qiu, Y.H.; Li, W.X.; Gal-On, A.; Zhou, C.Y.; Li, Y.; Ding, S.W. Virus infection triggers widespread silencing of host genes by a distinct class of endogenous siRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 14613–14618. [Google Scholar] [CrossRef]
- McBride, R.C.; Boucher, N.; Park, D.S.; Turner, P.E.; Townsend, J.P. Yeast response to la virus indicates coadapted global gene expression during mycoviral infection. FEMS Yeast Res. 2013, 13, 162–179. [Google Scholar] [CrossRef]
- Allen, T.D.; Dawe, A.L.; Nuss, D.L. Use of cdna microarrays to monitor transcriptional responses of the chestnut blight fungus cryphonectria parasitica to infection by virulence-attenuating hypoviruses. Eukaryot. Cell 2003, 2, 1253–1265. [Google Scholar] [CrossRef]
- Wang, J.Z.; Shi, L.M.; He, X.P.; Lu, L.D.; Li, X.P.; Chen, B.S. Comparative secretome analysis reveals perturbation of host secretion pathways by a hypovirus. Sci. Rep. 2016, 6, 34308. [Google Scholar] [CrossRef]
- Kwon, S.J.; Cho, S.Y.; Lee, K.M.; Yu, J.; Son, M.; Kim, K.H. Proteomic analysis of fungal host factors differentially expressed by Fusarium graminearum infected with fusarium graminearum virus-DK21. Virus Res. 2009, 144, 96–106. [Google Scholar] [CrossRef]
- Marzano, S.L.; Hobbs, H.A.; Nelson, B.D.; Hartman, G.L.; Eastburn, D.E.; McCoppin, N.K.; Domier, L.L. Transfection of Sclerotinia sclerotiorum with in vitro transcripts of a naturally occurring interspecific recombinant of Sclerotinia sclerotiorum hypovirus 2 significantly reduces virulence of the fungus. J. Virol. 2015, 89, 5060–5071. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Braker1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mi, H.Y.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Mackowiak, S.D. Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr. Protoc. Bioinform. 2011, 36, 12.10.1–12.10.15. [Google Scholar]
- An, J.Y.; Lai, J.; Lehman, M.L.; Nelson, C.C. miRDeep*: An integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res. 2013, 41, 727–737. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W. Prediction of plant miRNA genes. In Plant MicroRNAs: Methods and Protocols; Humana Press: New York, NY, USA, 2010; pp. 19–30. [Google Scholar]
- An, J.Y.; Lai, J.; Sajjanhar, A.; Lehman, M.L.; Nelson, C.C. miRPlant: An integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinform. 2014, 15, 275. [Google Scholar] [CrossRef]
- Axtell, M.J. Shortstack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.R.; Yeoh, J.M.; Coruh, C.; Axtell, M.J. Improved placement of multi-mapping small RNAs. G3 2016, 6, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fahlgren, N.; Chapman, E.J.; Cumbie, J.S.; Sullivan, C.M.; Givan, S.A.; Kasschau, K.D.; Carrington, J.C. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell 2007, 19, 926–942. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Baker, B. Preparation of cDNA Library for dRNA-Seq. Bio-protocol 2012, 2, e302. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. Cleaveland: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef]
- Whitham, S.A.; Yang, C.L.; Goodin, M.M. Global impact: Elucidating plant responses to viral infection. Mol. Plant-Microbe Interact. 2006, 19, 1207–1215. [Google Scholar] [CrossRef]
- Spain, B.H.; Koo, D.; Ramakrishnan, M.; Dzudzor, B.; Colicelli, J. Truncated forms of a novel yeast protein suppress the lethality of a G-protein α subunit deficiency by interacting with the β subunit. J. Biol. Chem. 1995, 270, 25435–25444. [Google Scholar] [CrossRef]
- Wei, W. Transcriptomic Characterization of Soybean–Sclerotinia Sclerotiorum Interaction at Early Infection Stages. Ph.D. Thesis, University of Illinois at Urbana–Champaign, Champaign, IL, USA, 2017. [Google Scholar]
- Fei, Q.; Yu, Y.; Liu, L.; Zhang, Y.; Baldrich, P.; Dai, Q.; Chen, X.; Meyers, B.C. Biogenesis of a 22-nt microRNA in Phaseoleae species by precursor-programmed uridylation. Proc. Natl. Acad. Sci. USA 2018, 115, 8037–8042. [Google Scholar] [CrossRef]
- Hammond, T.M.; Spollen, W.G.; Decker, L.M.; Blake, S.M.; Springer, G.K.; Shiu, P.K.T. Identification of small RNAs associated with meiotic silencing by unpaired DNA. Genetics 2013, 194, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, G.; Song, N.; Wang, J.; Cao, L.; Jiang, H.; Ding, T. Complete mitochondrial genome sequence of the phytopathogenic fungi Sclerotinia sclerotiorum JX-21. Mitochondrial DNA Part B 2016, 1, 656–657. [Google Scholar] [CrossRef]
- Zhu, R.S.; Li, X.; Chen, Q.S. Discovering numerical laws of plant microRNA by evolution. Biochem. Biophys. Res. Commun. 2011, 415, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Bollmann, S.R.; Kasschau, K.D.; Cuperus, J.T.; Press, C.M.; Sullivan, C.M.; Chapman, E.J.; Hoyer, J.S.; Gilbert, K.B.; Grunwald, N.J.; et al. Phytophthora have distinct endogenous small RNA populations that include short interfering and microRNAs. PLoS ONE 2013, 8, e77181. [Google Scholar] [CrossRef] [PubMed]
- Pinzon, N.; Li, B.; Martinez, L.; Sergeeva, A.; Presumey, J.; Apparailly, F.; Seitz, H. microRNA target prediction programs predict many false positives. Genome Res. 2017, 27, 234–245. [Google Scholar] [CrossRef]
- Vaucheret, H.; Vazquez, F.; Crete, P.; Bartel, D.P. The action of argonaute1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Gene Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Dawe, A.L.; Van Voorhies, W.A.; Lau, T.A.; Ulanov, A.V.; Li, Z. Major impacts on the primary metabolism of the plant pathogen Cryphonectria parasitica by the virulence-attenuating virus CHV1-EP713. Microbiology 2009, 155, 3913–3921. [Google Scholar] [CrossRef]
- Wang, S.C.; Zhang, J.Z.; Li, P.F.; Qiu, D.W.; Guo, L.H. Transcriptome-based discovery of Fusarium graminearum stress responses to FgHV1 infection. Int. J. Mol. Sci. 2016, 17, 1922. [Google Scholar] [CrossRef]
- Meena, M.; Prasad, V.; Zehra, A.; Gupta, V.K.; Upadhyay, R.S. Mannitol metabolism during pathogenic fungal-host interactions under stressed conditions. Front. Microbiol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef]
- Laluk, K.; AbuQamar, S.; Mengiste, T. The Arabidopsis mitochondria-localized pentatricopeptide repeat protein PGN functions in defense against necrotrophic fungi and abiotic stress tolerance. Plant Physiol. 2011, 156, 2053–2068. [Google Scholar] [CrossRef]
- Zhu, M.K.; Chen, G.P.; Dong, T.T.; Wang, L.L.; Zhang, J.L.; Zhao, Z.P.; Hu, Z.L. SLDEAD31, a putative DEAD-box RNA helicase gene, regulates salt and drought tolerance and stress-related genes in tomato. PLoS ONE 2015, 10, e0133849. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ge, L.L.; Li, P.P.; Wang, Y.; Sun, M.X.; Huang, L.; Ishag, H.; Di, D.D.; Shen, Z.Q.; Fan, W.X.; et al. The DEAD-box RNA helicase DDX5 acts as a positive regulator of Japanese encephalitis virus replication by binding to viral 3′ UTR. Antivir. Res. 2013, 100, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, N.; Pogany, J.; Nagy, P.D. A co-opted dead-box RNA helicase enhances tombusvirus plus-strand synthesis. PLoS Pathog. 2012, 8, e1002537. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Suzuki, N. Highly activated RNA silencing via strong induction of dicer by one virus can interfere with the replication of an unrelated virus. Proc. Natl. Acad. Sci. USA 2015, 112, E4911–E4918. [Google Scholar] [CrossRef]
- Zhang, X.M.; Segers, G.C.; Sun, Q.H.; Deng, F.Y.; Nuss, D.L. Characterization of hypovirus-derived small RNAs generated in the chestnut blight fungus by an inducible DCL-2-dependent pathway. J. Virol. 2008, 82, 2613–2619. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Choi, G.H.; Nuss, D.L. A single argonaute gene is required for induction of RNA silencing antiviral defense and promotes viral RNA recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 17927–17932. [Google Scholar] [CrossRef] [PubMed]
- Yaegashi, H.; Shimizu, T.; Ito, T.; Kanematsu, S. Differential inductions of RNA silencing among encapsidated double-stranded RNA mycoviruses in the white root rot fungus Rosellinia necatrix. J. Virol. 2016, 90, 5677–5692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.H.; Fu, Y.P.; Xie, J.T.; Li, B.; Jiang, D.H.; Li, G.Q.; Cheng, J.S. Identification of microRNA-like RNAs in a plant pathogenic fungus Sclerotinia sclerotiorum by high-throughput sequencing. Mol. Genet. Genom. 2012, 287, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Chow, W.N.; Wong, A.Y.P.; Yeung, J.M.Y.; Bao, J.; Zhang, N.; Lok, S.; Woo, P.C.Y.; Yuen, K.Y. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei. PLoS Negl. Trop. Dis. 2013, 7, e2398. [Google Scholar] [CrossRef]
- Kang, K.; Zhong, J.S.; Jiang, L.; Liu, G.; Gou, C.Y.; Wu, Q.; Wang, Y.; Luo, J.; Gou, D.M. Identification of microRNA-like RNAs in the filamentous fungus Trichoderma reesei by Solexa sequencing. PLoS ONE 2013, 8, e76288. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, T.A.; Kuck, U. Dicer-dependent biogenesis of small RNAs and evidence for microRNA-like RNAs in the penicillin producing fungus Penicillium chrysogenum. PLoS ONE 2015, 10, e0125989. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Zhang, Z.Y.; Liu, Y. RNA interference pathways in fungi: Mechanisms and functions. Annu. Rev. Microbiol. 2012, 66, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Williamson, V.; Kim, A.; Xie, B.; McMichael, G.O.; Gao, Y.; Vladimirov, V. Detecting miRNAs in deep-sequencing data: A software performance comparison and evaluation. Brief. Bioinform. 2013, 14, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.M.; Hellens, R.P. Coding in non-coding RNAs. Nature 2015, 520, 41–42. [Google Scholar] [CrossRef]
- Wang, Q.H.; Li, T.T.; Xu, K.; Zhang, W.; Wang, X.L.; Quan, J.L.; Jin, W.B.; Zhang, M.X.; Fan, G.J.; Wang, M.B.; et al. The tRNA-derived small RNAs regulate gene expression through triggering sequence-specific degradation of target transcripts in the oomycete pathogen Phytophthora sojae. Front. Plant Sci. 2016, 7, 1938. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.P.; Hutvagner, G. tRNA-derived fragments (tRFs): Emerging new roles for an ancient RNA in the regulation of gene expression. Life 2015, 5, 1638–1651. [Google Scholar] [CrossRef] [PubMed]
- Komiya, R. Biogenesis of diverse plant phasiRNAs involves an miRNA-trigger and Dicer-processing. J. Plant Res. 2017, 130, 17–23. [Google Scholar] [CrossRef]
- Fei, Q.L.; Xia, R.; Meyers, B.C. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef]
- Da Silva, R.P.; Puccia, R.; Rodrigues, M.L.; Oliveira, D.L.; Joffe, L.S.; Cesar, G.V.; Nimrichter, L.; Goldenberg, S.; Alves, L.R. Extracellular vesicle-mediated export of fungal RNA. Sci. Rep. 2015, 5, 7763. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Qiao, L.; Wang, M.; He, B.; Lin, F.-M.; Palmquist, J.; Huang, H.-D.; Jin, H. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 2018, 360, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.W.; Zhang, Z.H.; Kaloshian, I.; Huang, H.D.; Jin, H.L. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Chen, L.T.; Patel, K.; Li, Y.H.; Baulcombe, D.C.; Wu, S.H. 22-nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. USA 2010, 107, 15269–15274. [Google Scholar] [CrossRef] [PubMed]
- Mochama, P.; Jadhav, P.; Neupane, A.; Marzano, S.-Y.L. Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum. Viruses 2018, 10, 214. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee Marzano, S.-Y.; Neupane, A.; Domier, L. Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus. Viruses 2018, 10, 713. https://doi.org/10.3390/v10120713
Lee Marzano S-Y, Neupane A, Domier L. Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus. Viruses. 2018; 10(12):713. https://doi.org/10.3390/v10120713
Chicago/Turabian StyleLee Marzano, Shin-Yi, Achal Neupane, and Leslie Domier. 2018. "Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus" Viruses 10, no. 12: 713. https://doi.org/10.3390/v10120713
APA StyleLee Marzano, S.-Y., Neupane, A., & Domier, L. (2018). Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus. Viruses, 10(12), 713. https://doi.org/10.3390/v10120713