Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine


 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Phages and Bacterial Strains
2.2. Phage Techniques
2.3. Transmission Electron Microscopy
2.4. DNA Isolation and Restriction Analysis
2.5. Genome Sequencing and Analysis
2.6. Analysis of Structural Proteins
2.7. Nucleotide Sequence Accession Numbers
3. Results
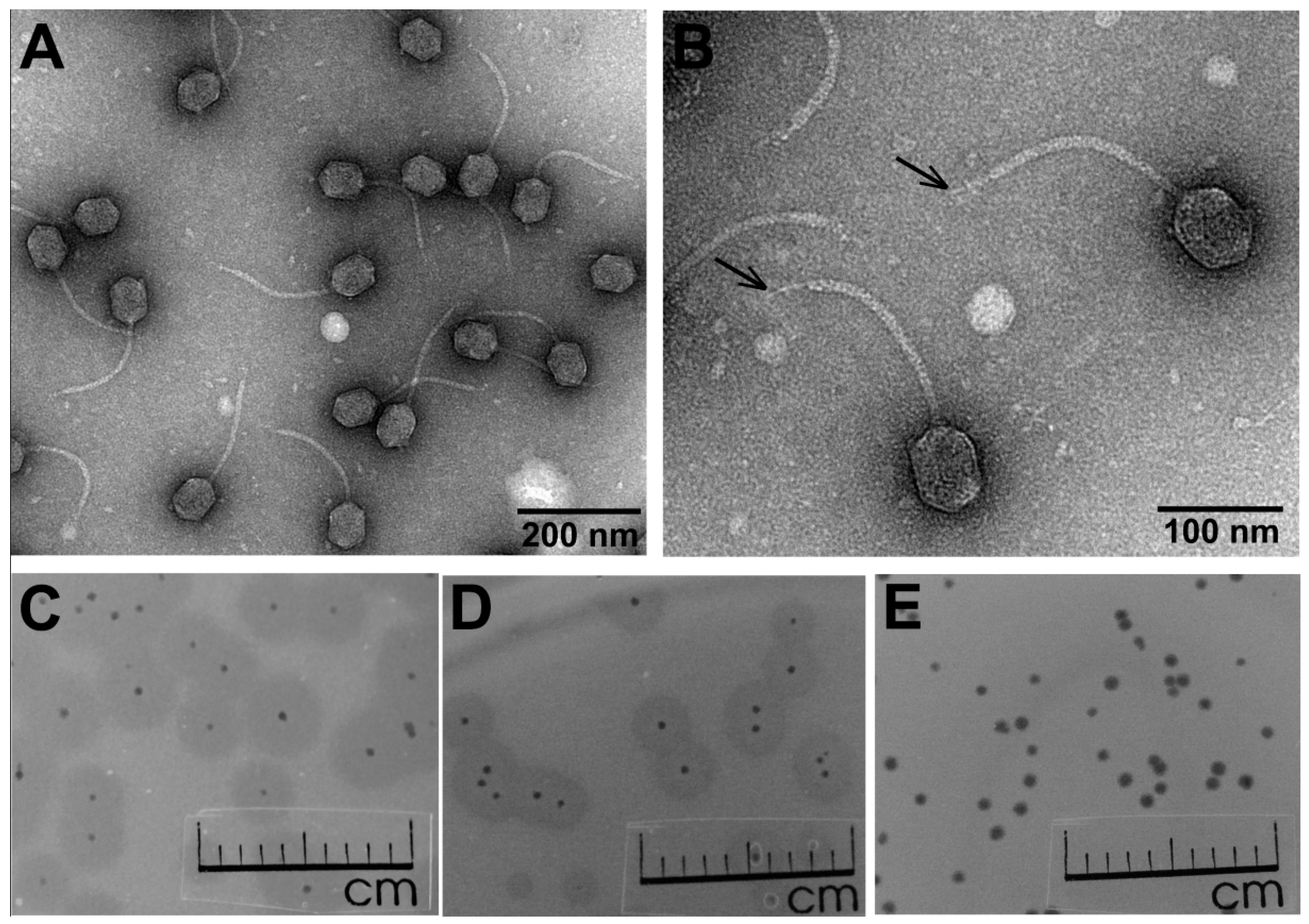
3.1. Phage Morphology, Host Range, and Physiological Characteristics
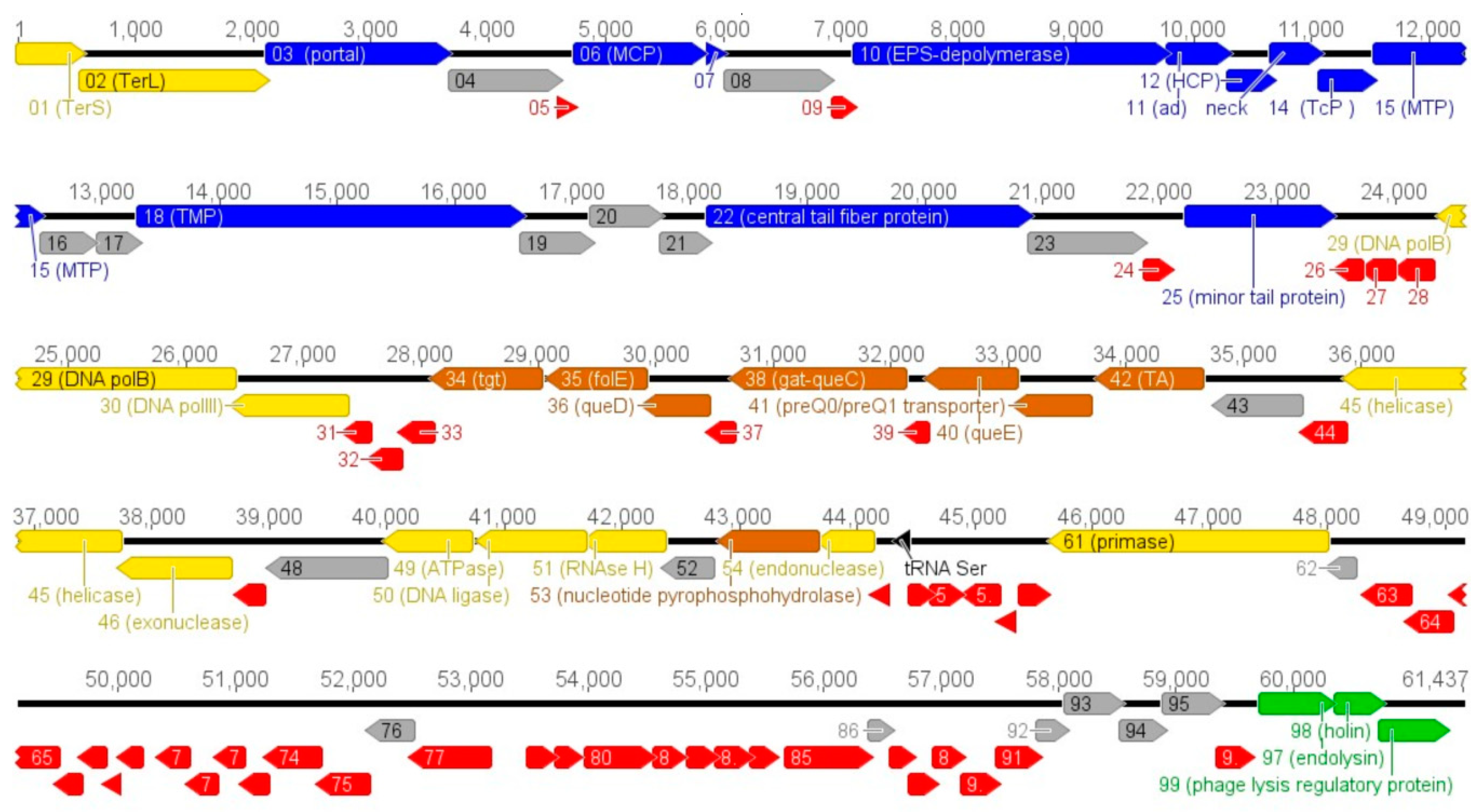
3.2. Vid5 Genomic and Proteomic Analysis
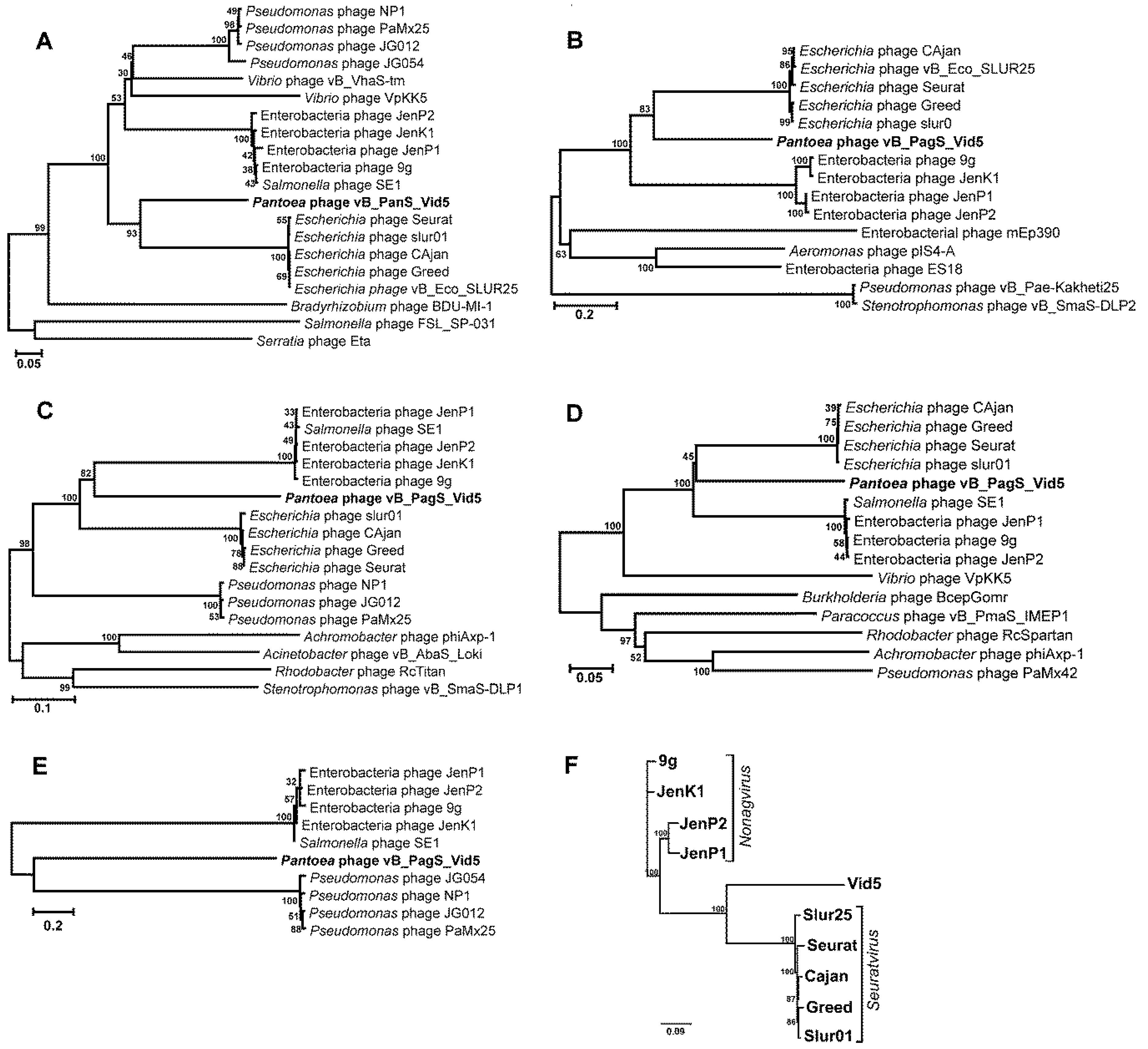
3.3. Phylogenetic Analysis
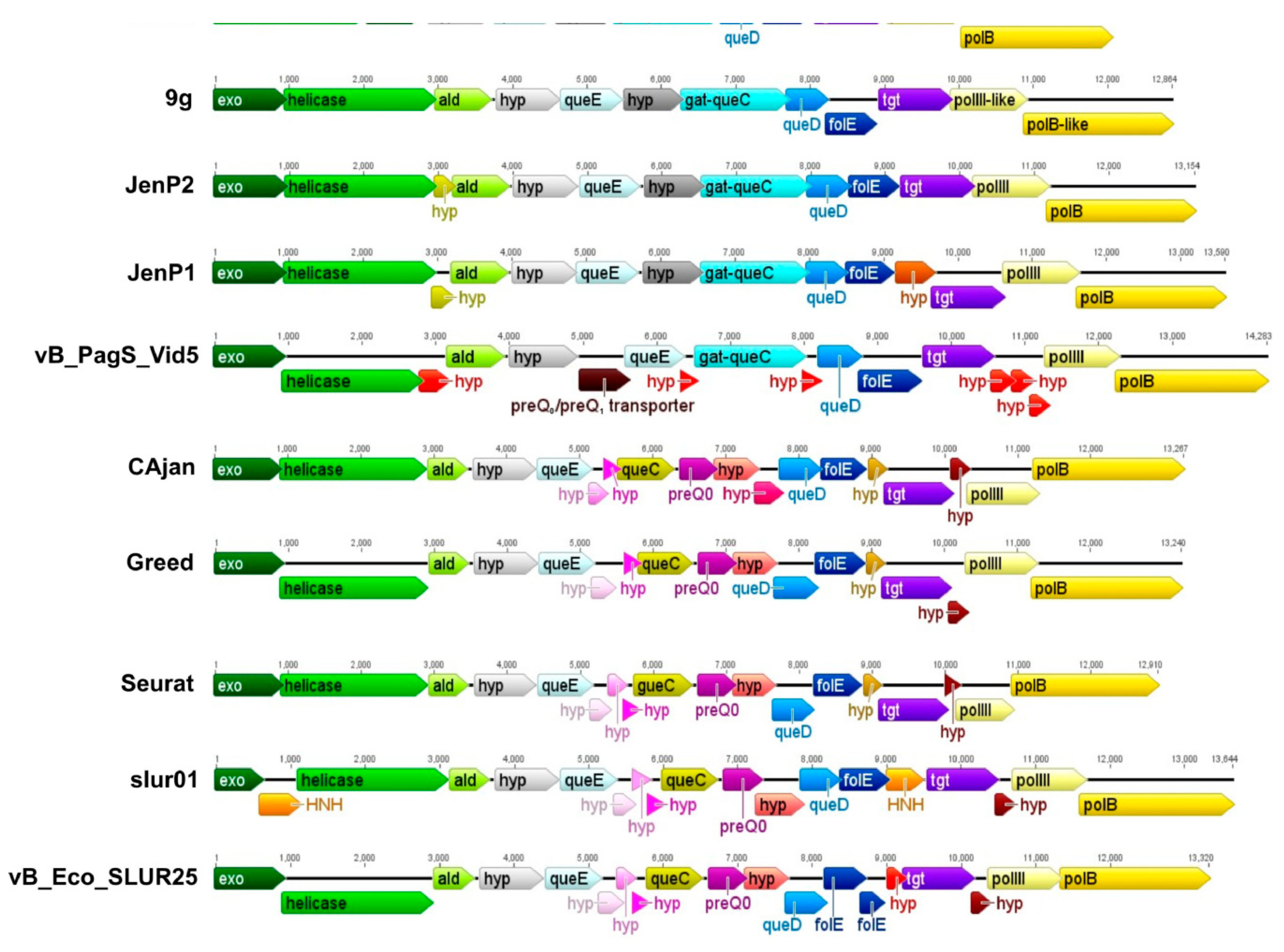
3.4. The Gene Cluster Potentially Involved in the Biosynthesis of 7-Deazaguanine Derivatives
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Walterson, A.M.; Stavrinides, J. Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 2015, 39, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, J.; Mackiewicz, B.; Lemieszek, M.; Golec, M.; Milanowski, J. Pantoea agglomerans—A mysterious bacterium of evil and good. Part I. Deleterious effects: Dust-borne endotoxins and allergens—Focus on cotton dust. Ann. Agric. Environ. Med. 2015, 22, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; Van Vaerenbergh, J.; Maes, M.; De Proft, M.; Lavigne, R. Bacteriophages LIME-light and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses”. Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Lurz, R.; Kube, M.; Quedenau, C.; Jelkmann, W.; Geider, K. Molecular and physiological properties of bacteriophages from North America and Germany affecting the fire blight pathogen Erwinia amylovora. Microb. Biotechnol. 2011, 4, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Born, Y.; Fiesler, L.; Marazzi, J.; Lurz, R.; Duffy, B.; Loessner, M.J. Novel virulent and broad host range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and relationship to Enterobacteriacea phages. Appl. Environ. Microbiol. 2011, 77, 5945–5954. [Google Scholar] [CrossRef] [PubMed]
- Boulé, J.; Sholberg, P.L.; Lehman, S.M.; O’gorman, D.T.; Svircev, A.M. Isolation and characterization of eight bacteriophages infecting Erwinia amylovora and their potential as biological control agents in British Columbia, Canada. Can. J. Plant Pathol. 2011, 33, 308–317. [Google Scholar] [CrossRef]
- Lagonenko, A.L.; Sadovskaya, O.; Valentovich, L.N.; Evtushenkov, A.N. Characterization of a new Vil-like Erwinia amylovora bacteriophage phiEa2809. FEMS Microbiol. Lett. 2015, 362, fnv031. [Google Scholar] [CrossRef] [PubMed]
- Schwarczinger, I.; Kolozsváriné Nagy, J.; Künstler, A.; Szabó, L.; Geider, K.; Király, L.; Pogány, M. Characterization of Myoviridae and Podoviridae family bacteriophages of Erwinia amylovora from Hungary—Potential of application in biological control of fire blight. Eur. J. Plant Pathol. 2017, 149, 639–652. [Google Scholar] [CrossRef]
- Buttimer, C.; Born, Y.; Lucid, A.; Loessner, M.J.; Fieseler, L.; Coffey, A. Erwinia amylovora phage vB_EamM_Y3 represents another lineage of hairy Myoviridae. Res. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.L.; Venter, S.N.; Cleenwerck, I.; Vandemeulebroecke, K.; De Vos, P.; Coutinho, T.A. Transfer of Pantoea citrea, Pantoea punctata and Pantoea terrea to the genus Tatumella emend. as Tatumella citrea comb. nov., Tatumella punctate comb. nov and Tatumella terrea comb. nov and description of Tatumella morbirosei sp nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 484–494. [Google Scholar] [CrossRef]
- Palmer, M.; Steenkamp, E.T.; Coetzee, M.P.A.; Chan, W.Y.; van Zyl, E.; De Maayer, P.; Coutinho, T.A.; Blom, J.; Smits, T.H.M.; Duffy, B.; et al. Phylogenomic resolution of the bacterial genus Pantoea and its relationship with Erwinia and Tatumella. Antonie van Leeuwenhoek 2017, 110, 1287–1309. [Google Scholar] [CrossRef] [PubMed]
- Meczker, K.; Domotor, D.; Vass, J.; Rakhely, G.; Schneider, G.; Kovacs, T. The genome of the Erwinia amylovora phage PhiEaH1 reveals greater diversity and broadens the applicability of phages for the treatment of fire blight. FEMS Microbiol. Lett. 2014, 350, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, D.; Becságh, P.; Rákhely, G.; Schneider, G.; Kovács, T. Complete genomic sequence of Erwinia amylovora phage PhiEaH2. J. Virol. 2012, 86, 10899. [Google Scholar] [CrossRef] [PubMed]
- Faidiuk, Y.V.; Boyko, А.А.; Muchnyk, F.V.; Tovkach, F.I. Virion morphology and structural organization of polyvalent bacteriophages TT10-27 and KEY. Mikrobiol. Z. 2015, 77, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.J.; Crow, M.A.; Williamson, N.R.; Orme, W.; Thomson, N.R.; Komitopoulou, E.; Salmond, G.P.C. Characterization of a broad-host-range flagellum-dependent phage that mediates high-efficiency generalized transduction in, and between, Serratia and Pantoea. Microbiology 2010, 156, 240–247. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Godon, J.J.; Zumstein, E.; Dabert, P.; Habouzit, F.; Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997, 63, 2802–2813. [Google Scholar] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Šimoliūnas, E.; Kaliniene, L.; Stasilo, M.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Nainys, J.; Kaupinis, A.; Valius, M.; Meškys, R. Isolation and characterization of vB_ArS-ArV2—First Arthrobacter sp. infecting bacteriophage with completely sequenced genome. PLoS ONE 2014, 9, e111230. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russel, D. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Šimoliūnas, E.; Kaliniene, L.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Kaupinis, J.; Ger, M.; Valius, M.; Meškys, R. Klebsiella phage vB_KleM-RaK2—A giant singleton virus of the family Myoviridae. PLoS ONE 2013, 8, e60717. [Google Scholar] [CrossRef] [PubMed]
- Carlson, K.; Miller, E. Experiments in T4 genetics. In Bacteriophage T4; Karam, J.D., Ed.; ASM Press: Washington, DC, USA, 1994; pp. 419–483. [Google Scholar]
- Alva, V.; Nam, S.Z.; Söding, J.; Lupas, A.N. The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated identification of tailed bacteriophages and classification according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.E. Ultrastructure of bacteriophages and bacteriocins. Bacteriol. Rev. 1967, 31, 230–314. [Google Scholar] [PubMed]
- Ackermann, H.W.; Eisenstark, A. The present state of phage taxonomy. Intervirology 1974, 3, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Oliveira, H.; Melo, L.D.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, A.; Ceyssens, P.J.; T’Syen, J.; Van Praet, H.; Noben, J.P.; Shaburova, O.V.; Krylov, V.N.; Volckaert, G.; Lavigne, R. The T7-related Pseudomonas putida phage phi 15 displays virion-associated biofilm degradation properties. PLoS ONE 2011, 6, e18597. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and phage-derived proteins‒application approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Cowan, D.A. Using signature genes as tools to assess environmental viral ecology and diversity. Appl. Environ. Microbiol. 2014, 80, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, E.E.; Golomidova, A.K.; Letarova, M.A.; Kostryukova, E.S.; Zelenin, A.S.; Prokhorov, N.S.; Letarov, A.V. Genomic sequencing and biological characteristics of a novel Escherichia coli bcteriophage 9g, a putative representative of a new Siphoviridae genus. Viruses 2014, 6, 5077–5092. [Google Scholar] [CrossRef] [PubMed]
- Carstens, A.B.; Kot, W.; Hansen, L.H. Complete genome sequences of four novel Escherichia coli bacteriophages belonging to new phage groups. Genome Announc. 2015, 3, e00741-15. [Google Scholar] [CrossRef] [PubMed]
- Doan, D.P.; Lessor, L.E.; Hernandez, A.C.; Kuty Everett, G.F. Complete genome sequence of enterotoxigenic Escherichia coli siphophage Seurat. Genome Announc. 2015, 3, e00044-15. [Google Scholar] [CrossRef] [PubMed]
- Sazinas, P.; Redgwell, T.; Rihtman, B.; Grigonyte, A.; Michniewski, S.; Scanlan, D.J.; Hobman, J.; Millard, A. Comparative genomics of bacteriophage of the genus Seuratvirus. Genome Biol. Evol. 2018, 10, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; O’Hara, M.; Hobman, J.L.; Millard, A.D. Draft genome sequences of 14 Escherichia coli phages isolated from cattle slurry. Genome Announc. 2015, 3, e01364-15. [Google Scholar] [CrossRef] [PubMed]
- Malki, K.; Sible, E.; Cooper, A.; Garretto, A.; Bruder, K.; Watkins, S.C.; Putonti, C. Seven bacteriophages isolated from the female urinary microbiota. Genome Announc. 2016, 4, e01003-16. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, J.J.; Kellner, S.M.; Yuan, Y.; Hutinet, G.; Thiaville, P.C.; Jumpathong, W.; Mohapatra, S.; Brochier-Armanet, C.; Letarov, A.V.; Hillebrand, R.; et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, e1452–e1459. [Google Scholar] [CrossRef] [PubMed]
- Hutinet, G.; Swarjo, M.A.; de Crécy-Lagard, V. Deazaguanine derivatives, examples of crosstalk between RNA and DNA modification pathways. RNA Biol. 2017, 14, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.; Corrêa, I.R.; Xu, M.Y.; Xu, S.Y. Restriction and modification of deoxy-archaeosine (dG+)-containing phage 9 g DNA. Sci. Rep. 2017, 7, 8348. [Google Scholar] [CrossRef] [PubMed]
- Zallot, R.; Ross, R.; Chen, W.H.; Bruner, S.D.; Limbach, P.A.; de Crécy-Lagard, V. Identification of a novel epoxyqueuosine reductase family by comparative genomics. ACS Chem. Biol. 2017, 12, 844–851. [Google Scholar] [CrossRef] [PubMed]
- McCarty, R.M.; Bandarian, V. Biosynthesis of pyrrolopyrimidines. Bioorg. Chem. 2012, 43, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kaliniene, L.; Truncaitė, L.; Šimoliūnas, E.; Zajančkauskaitė, A.; Vilkaitytė, M.; Kaupinis, A.; Skapas, M.; Meškys, R. Molecular analysis of the low-temperature Escherichia coli phage vB_EcoS_NBD2. Arch. Virol. 2017, 163, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Kaliniene, L.; Klausa, V.; Truncaite, L. Low-temperature T4-like coliphages vB_EcoM-VR5, vB_EcoM-VR7 and vB_EcoM-VR20. Arch. Virol. 2010, 155, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Jurczak-Kurek, A.; Gasior, T.; Nejman-Falenczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimoliūnas, E.; Šimoliūnienė, M.; Kaliniene, L.; Zajančkauskaitė, A.; Skapas, M.; Meškys, R.; Kaupinis, A.; Valius, M.; Truncaitė, L. Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses 2018, 10, 583. https://doi.org/10.3390/v10110583
Šimoliūnas E, Šimoliūnienė M, Kaliniene L, Zajančkauskaitė A, Skapas M, Meškys R, Kaupinis A, Valius M, Truncaitė L. Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses. 2018; 10(11):583. https://doi.org/10.3390/v10110583
Chicago/Turabian StyleŠimoliūnas, Eugenijus, Monika Šimoliūnienė, Laura Kaliniene, Aurelija Zajančkauskaitė, Martynas Skapas, Rolandas Meškys, Algirdas Kaupinis, Mindaugas Valius, and Lidija Truncaitė. 2018. "Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine" Viruses 10, no. 11: 583. https://doi.org/10.3390/v10110583
APA StyleŠimoliūnas, E., Šimoliūnienė, M., Kaliniene, L., Zajančkauskaitė, A., Skapas, M., Meškys, R., Kaupinis, A., Valius, M., & Truncaitė, L. (2018). Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses, 10(11), 583. https://doi.org/10.3390/v10110583