βTrCP is Required for HIV-1 Vpu Modulation of CD4, GaLV Env, and BST-2/Tetherin

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Lines
2.3. Western Blotting
2.4. Infectivity Assays
2.5. Surface Labeling
2.6. Imaging
3. Results
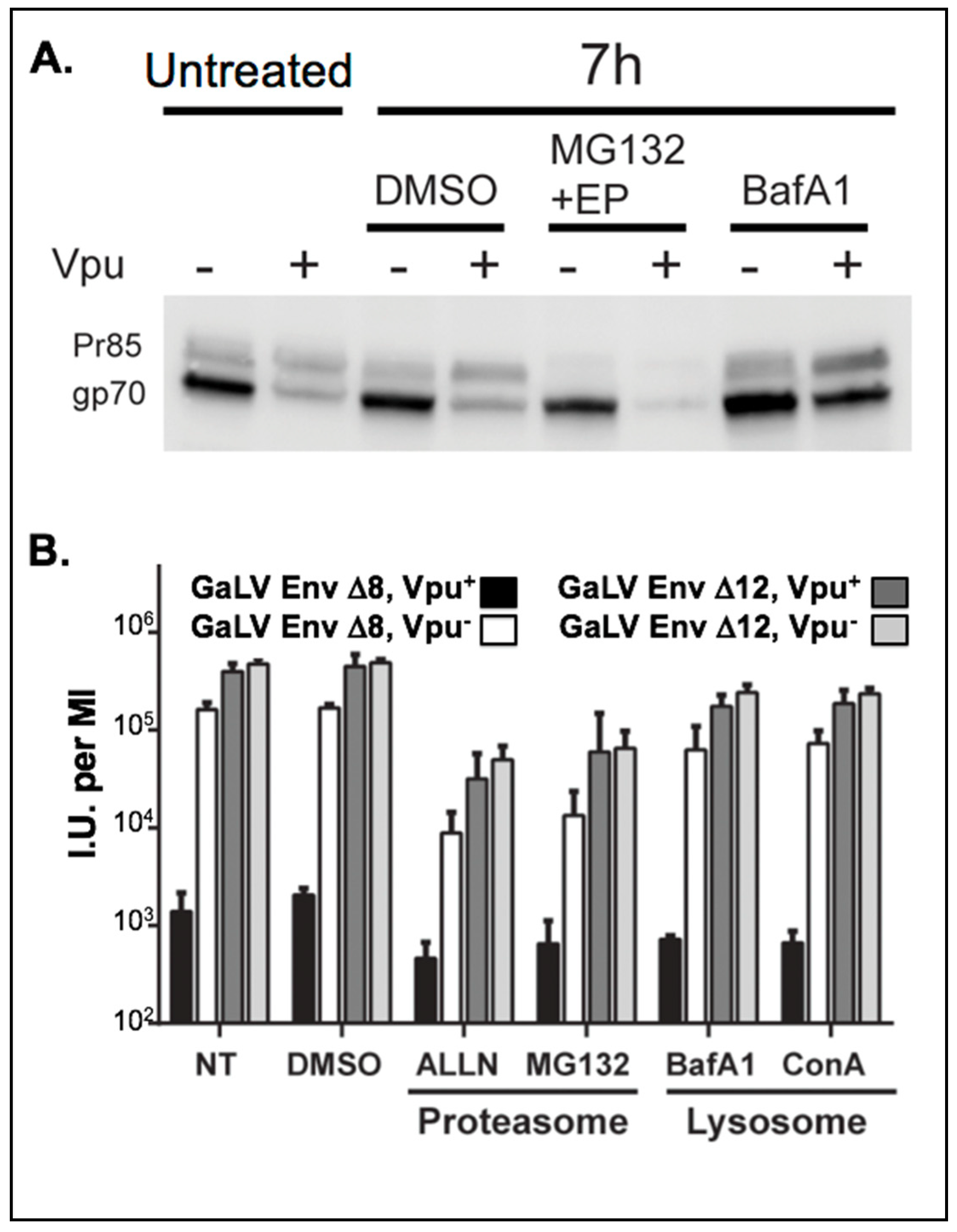
3.1. GaLV Env ∆8 Is Targeted for Lysosomal Degradation
3.2. Inhibition of Lysosomal Degradation Does Not Restore Infectious Particle Production with GaLV Env ∆8
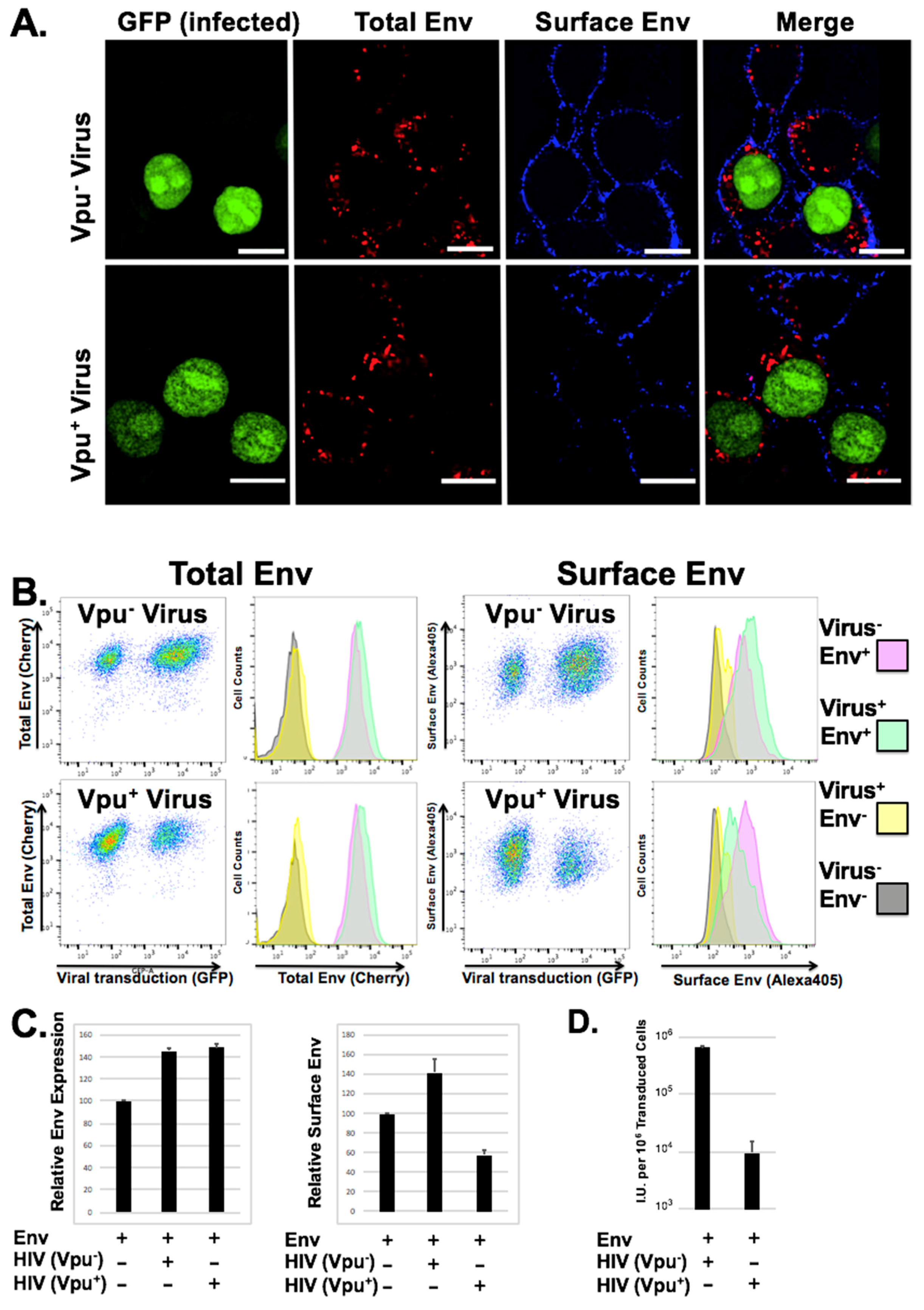
3.3. Loss of Surface Expression Weakly Correlates with Loss of Infectious Particle Production
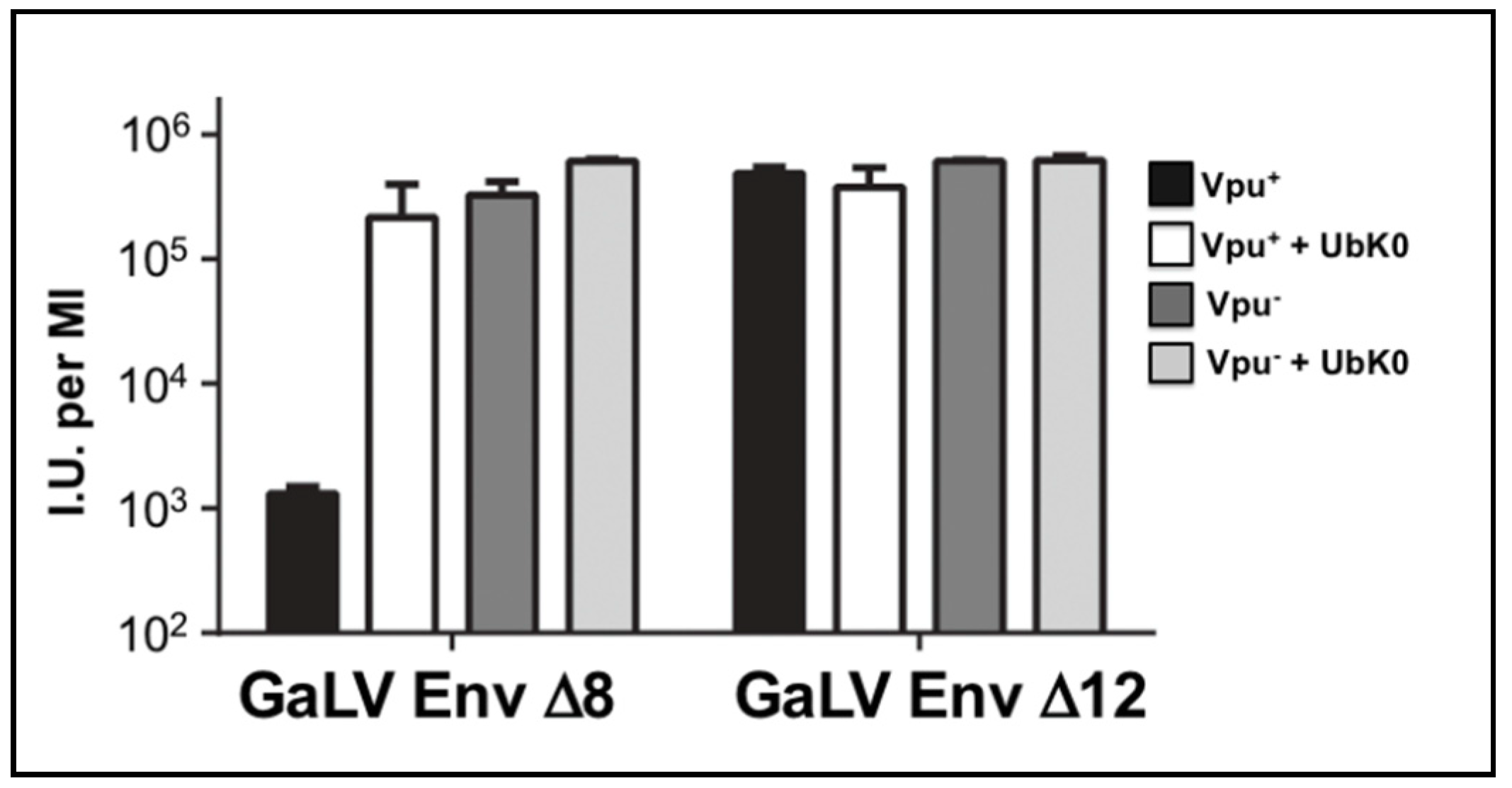
3.4. Vpu Inhibition of GaLV Env ∆8 Infectious Particle Production Is Polyubiquitin Dependent
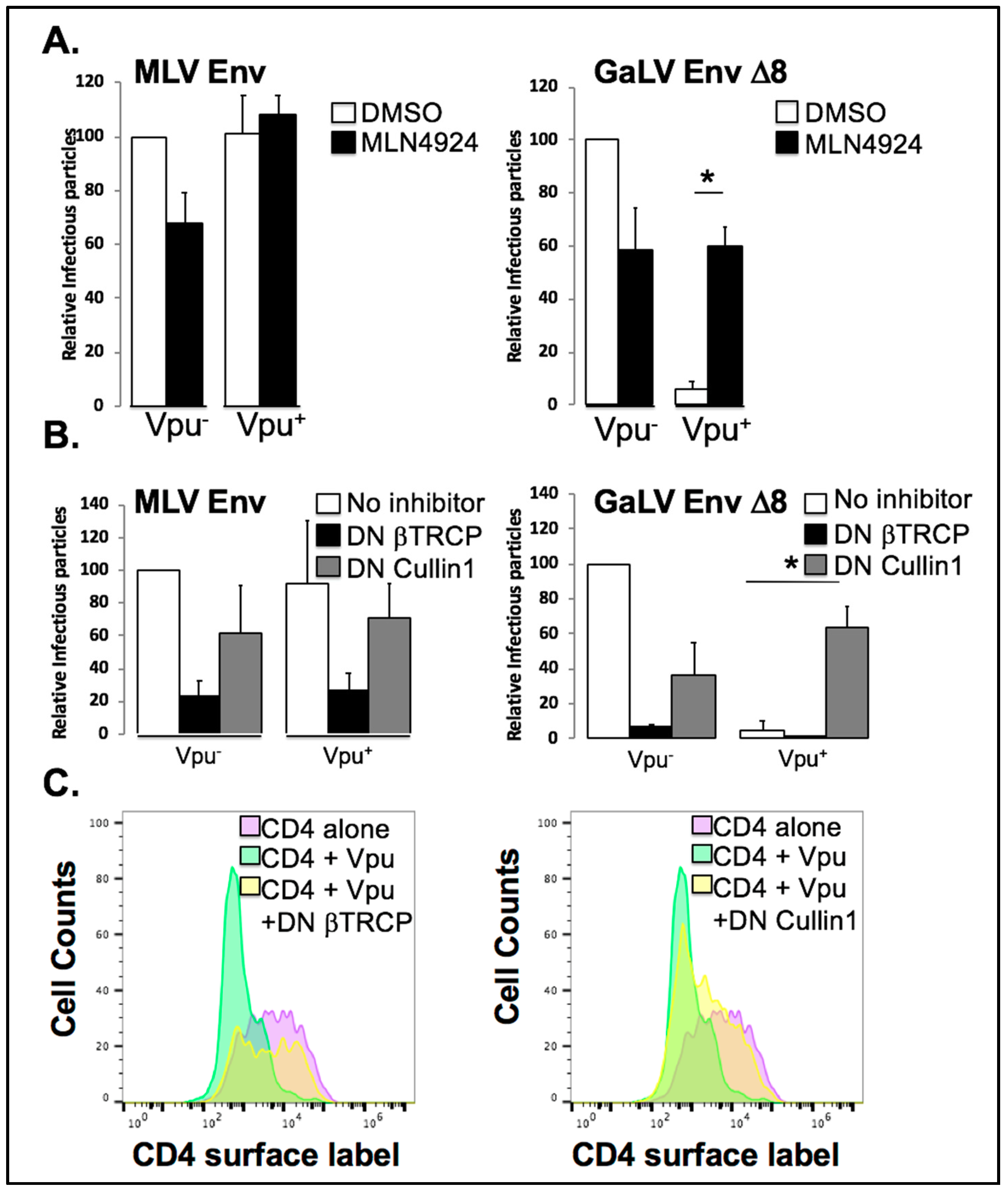
3.5. Vpu Inhibition of GaLV Env ∆8 Infectious Particle Production Is Restored by Neddylation Inhibitor MLN4924
3.6. Vpu Inhibition of GaLV Env ∆8 Infectious Particle Production Is Restored by Dominant Negative (DN) Cullin 1, But Not a DN βTrCP
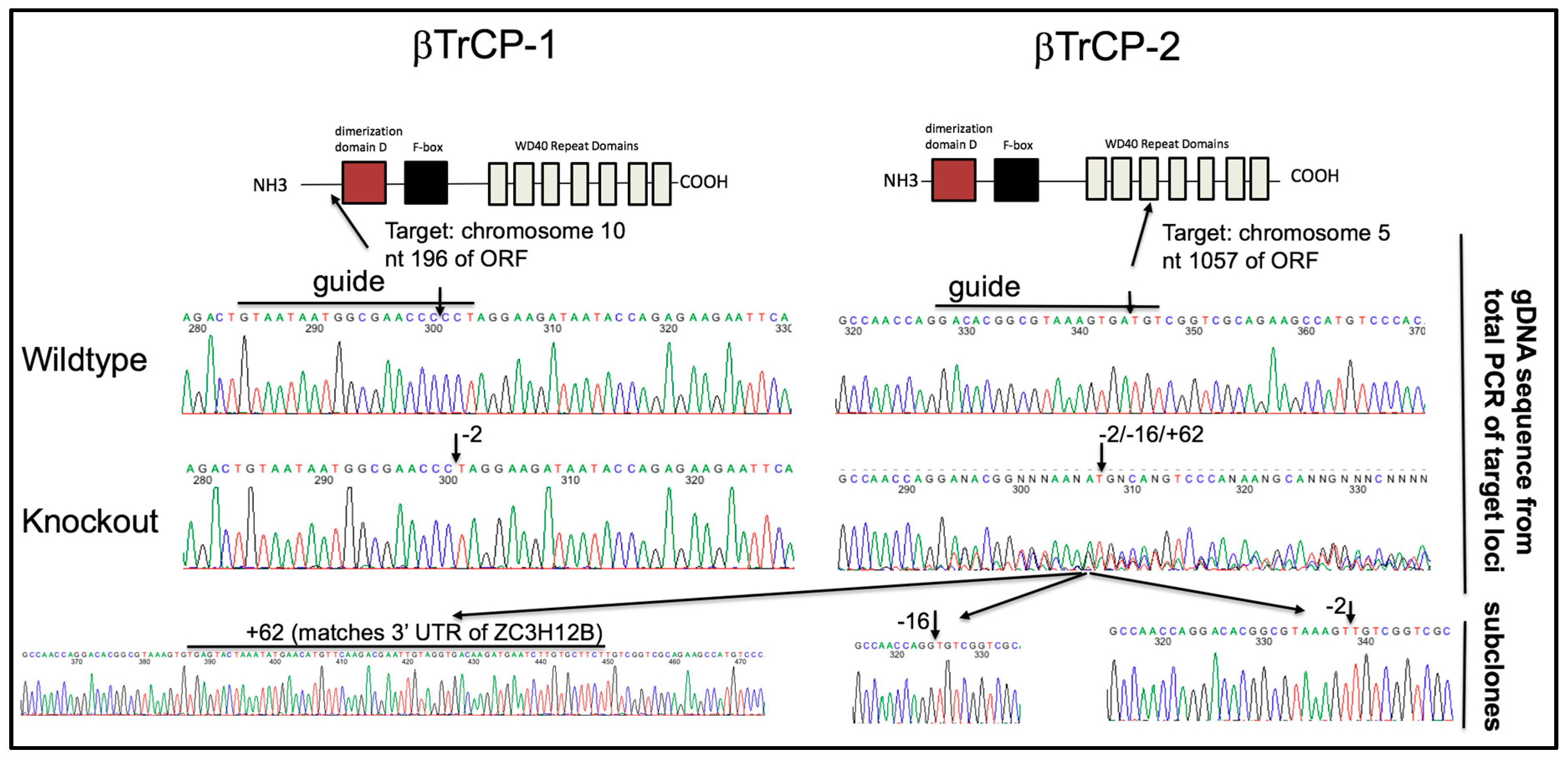
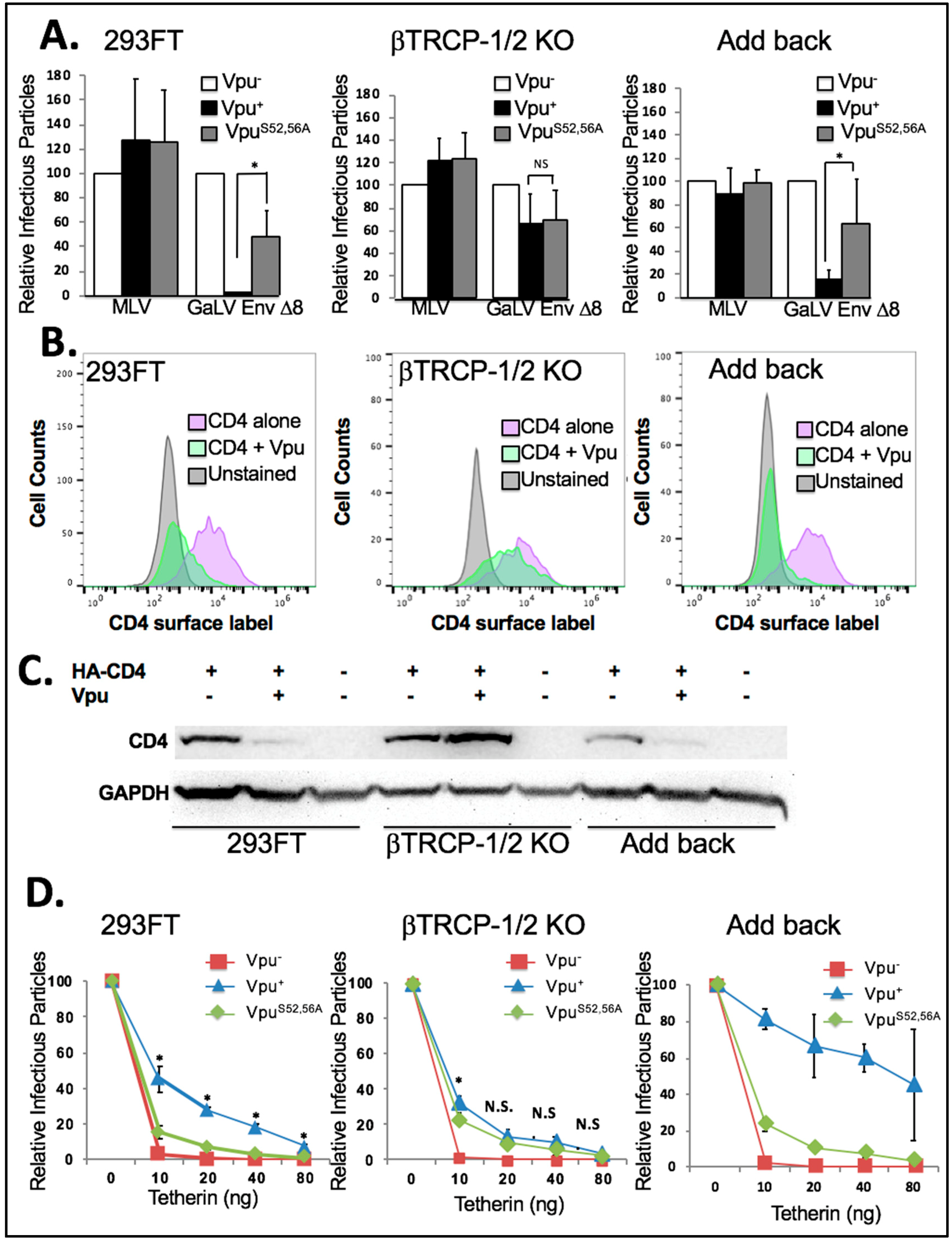
3.7. βTrCP Is Required for Vpu Activity
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Sugden, S.M.; Bego, M.G.; Pham, T.N.; Cohen, E.A. Remodeling of the host cell plasma membrane by HIV-1 Nef and Vpu: A strategy to ensure viral fitness and persistence. Viruses 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.; Strebel, K. HIV-1 Vpu targets cell surface markers CD4 and BST-2 through distinct mechanisms. Mol. Aspects Med. 2010, 31, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Schneider, T.; Henklein, P.; Hoffmann, K.; Berthold, E.; Hauser, H.; Pauli, G.; Porstmann, T. Human-immunodeficiency-virus-type-1-encoded Vpu protein is phosphorylated by casein kinase II. Eur. J. Biochem. 1992, 204, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Maldarelli, F.; Karczewski, M.K.; Willey, R.L.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces degradation of CD4 in vitro: The cytoplasmic domain of CD4 contributes to Vpu sensitivity. J. Virol. 1993, 67, 3877–3884. [Google Scholar] [PubMed]
- Schubert, U.; Strebel, K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 1994, 68, 2260–2271. [Google Scholar] [PubMed]
- Schubert, U.; Bour, S.; Ferrer-Montiel, A.V.; Montal, M.; Maldarell, F.; Strebel, K. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 1996, 70, 809–819. [Google Scholar] [PubMed]
- Schubert, U.; Anton, L.C.; Bacik, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-β Trcp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Butticaz, C.; Michielin, O.; Wyniger, J.; Telenti, A.; Rothenberger, S. Silencing of both β-Trcp1 and hos (β-Trcp2) is required to suppress human immunodeficiency virus type 1 Vpu-mediated CD4 down-modulation. J. Virol. 2007, 81, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of BST-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-Trcp and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Hatziioannou, T.; Bartlett, M.; Fofana, I.B.; Johnson, W.E.; Neil, S.J.; Bieniasz, P.D. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009, 5, e1000300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, A.J.; Miyagi, E.; Strebel, K. Differential effects of human immunodeficiency virus type 1 Vpu on the stability of BST-2/tetherin. J. Virol. 2011, 85, 2611–2619. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Fritz, J.V.; Bitzegeio, J.; Fackler, O.T.; Keppler, O.T. HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. MBio 2011, 2, e00036-00011. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βtrcp-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Tervo, H.M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. β-trcp is dispensable for vpu’s ability to overcome the CD317/tetherin-imposed restriction to HIV-1 release. Retrovirology 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J. Serine phosphorylation of HIV-1 Vpu and its binding to tetherin regulates interaction with clathrin adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Muller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 Vpu antagonizes CD317/tetherin by adaptor protein-1-mediated exclusion from virus assembly sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 vpu neutralizes the antiviral factor tetherin/BST-2 by binding it and directing its β-trcp2-dependent degradation. PLoS Pathog. 2009, 5, e1000574. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef] [PubMed]
- Stoneham, C.A.; Singh, R.; Jia, X.; Xiong, Y.; Guatelli, J. Endocytic activity of HIV-1 Vpu: Phosphoserine-dependent interactions with clathrin adaptors. Traffic 2017, 18, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.W.; DePaula-Silva, A.B.; Szaniawski, M.; Barker, E.; Bosque, A.; Planelles, V. HIV-1 vpu utilizes both cullin-ring ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology 2015, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.; Stoneham, C.; Lewinski, M.K.; Mukim, A.; Deshmukh, S.; Vollbrecht, T.; Spina, C.A.; Guatelli, J. Pharmacologic inhibition of Nedd8 activation enzyme exposes CD4-induced epitopes within Env on cells expressing HIV-1. J. Virol. 2016, 90, 2486–2502. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The escrt-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Paquay, C.; Roy, B.B.; Bego, M.G.; Mercier, J.; Cohen, E.A. Hiv-1 vpu antagonizes bst-2 by interfering mainly with the trafficking of newly synthesized bst-2 to the cell surface. Traffic 2011, 12, 1714–1729. [Google Scholar] [CrossRef] [PubMed]
- Gustin, J.K.; Douglas, J.L.; Bai, Y.; Moses, A.V. Ubiquitination of BST-2 protein by HIV-1 Vpu protein does not require lysine, serine, or threonine residues within the BST-2 cytoplasmic domain. J. Biol. Chem. 2012, 287, 14837–14850. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Neil, S.J. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 2012, 8, e1002609. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Kuruppu, N.D.; Felton, K.L.; D’Souza, D.; Freed, E.O. In cos cells vpu can both stabilize tetherin expression and counteract its antiviral activity. PLoS ONE 2014, 9, e111628. [Google Scholar] [CrossRef] [PubMed]
- Christodoulopoulos, I.; Droniou-Bonzom, M.E.; Oldenburg, J.E.; Cannon, P.M. Vpu-dependent block to incorporation of galv Env into lentiviral vectors. Retrovirology 2010, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.M.; Lyddon, T.D.; Cannon, P.M.; Johnson, M.C. Pseudotyping incompatibility between HIV-1 and gibbon ape leukemia virus Env is modulated by Vpu. J. Virol. 2010, 84, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.M.; Janaka, S.K.; Stephens, E.B.; Johnson, M.C. Vpu downmodulates two distinct targets, tetherin and gibbon ape leukemia virus envelope, through shared features in the vpu cytoplasmic tail. PLoS ONE 2012, 7, e51741. [Google Scholar] [CrossRef] [PubMed]
- Janaka, S.K.; Lucas, T.M.; Johnson, M.C. Sequences in gibbon ape leukemia virus envelope that confer sensitivity to HIV-1 accessory protein Vpu. J. Virol. 2011, 85, 11945–11954. [Google Scholar] [CrossRef] [PubMed]
- Janaka, S.K.; Faurot, J.; Johnson, M.C. Functional complementation of a model target to study Vpu sensitivity. PLoS ONE 2013, in press. [Google Scholar] [CrossRef] [PubMed]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Ingmundson, A.; Horner, S.M.; Cicchetti, G.; Allen, P.G.; Pypaert, M.; Cunningham, J.M.; Mothes, W. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 2003, 4, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, L.M.; Pacyniak, E.; Flick, M.; Hout, D.R.; Gomez, M.L.; Nerrienet, E.; Ayouba, A.; Santiago, M.L.; Hahn, B.H.; Stephens, E.B. Vpu-mediated CD4 down-regulation and degradation is conserved among highly divergent siv(CPZ) strains. Virology 2005, 335, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular cullin-ring ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Rajan, D.; Banning, C.; Wimmer, P.; Koppensteiner, H.; Iwanski, A.; Specht, A.; Sauter, D.; Dobner, T.; Kirchhoff, F. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology 2010, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.C.; Chinnasamy, N.; Morgan, R.A.; Varmus, H.E. Development of an avian leukosis-sarcoma virus subgroup a pseudotyped lentiviral vector. J. Virol. 2001, 75, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.D.; Garten, W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 1992, 360, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Arnason, T.; Ellison, M.J. Stress resistance in saccharomyces cerevisiae is strongly correlated with assembly of a novel type of multiubiquitin chain. Mol. Cell Biol. 1994, 14, 7876–7883. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.; Ardila-Osorio, H.; Fournier, M.; Brice, A.; Corti, O. Biochemical analysis of parkinson’s disease-causing variants of parkin, an e3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 2006, 15, 2059–2075. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for lewy body formation. J. Neurosci. 2005, 25, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, K.; Matsumoto, M.; Oyamada, K.; Nakayama, K.I. Proteome-wide identification of ubiquitylation sites by conjugation of engineered lysine-less ubiquitin. J. Proteome Res. 2012, 11, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Patel, S.V.; Snyder, P.M. Nedd4–2 catalyzes ubiquitination and degradation of cell surface Enac. J. Biol. Chem. 2007, 282, 20207–20212. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of Nedd8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Merlet, J.; Burger, J.; Gomes, J.E.; Pintard, L. Regulation of cullin-ring E3 ubiquitin-ligases by neddylation and dimerization. Cell. Mol. Life Sci. 2009, 66, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.E.; Cyburt, D.; Lucas, T.M.; Gregory, D.A.; Lyddon, T.D.; Johnson, M.C. βTrCP is Required for HIV-1 Vpu Modulation of CD4, GaLV Env, and BST-2/Tetherin. Viruses 2018, 10, 573. https://doi.org/10.3390/v10100573
Song YE, Cyburt D, Lucas TM, Gregory DA, Lyddon TD, Johnson MC. βTrCP is Required for HIV-1 Vpu Modulation of CD4, GaLV Env, and BST-2/Tetherin. Viruses. 2018; 10(10):573. https://doi.org/10.3390/v10100573
Chicago/Turabian StyleSong, Yul Eum, Daniel Cyburt, Tiffany M. Lucas, Devon A. Gregory, Terri D. Lyddon, and Marc C. Johnson. 2018. "βTrCP is Required for HIV-1 Vpu Modulation of CD4, GaLV Env, and BST-2/Tetherin" Viruses 10, no. 10: 573. https://doi.org/10.3390/v10100573
APA StyleSong, Y. E., Cyburt, D., Lucas, T. M., Gregory, D. A., Lyddon, T. D., & Johnson, M. C. (2018). βTrCP is Required for HIV-1 Vpu Modulation of CD4, GaLV Env, and BST-2/Tetherin. Viruses, 10(10), 573. https://doi.org/10.3390/v10100573