Molecular Mechanisms of Hepatocarcinogenesis Following Sustained Virological Response in Patients with Chronic Hepatitis C Virus Infection

Abstract
:1. Introduction
1.1. Detection and Treatment of HCC
1.2. Chronic Hepatitis C Virus Infection
1.3. Differences between Treatment with DAA and IFN
2. Molecular Mechanisms of HCC
2.1. Fibrogenesis
2.2. Genetic Instability and Mutagenesis
2.3. Apoptosis and Cell Cycle Dysregulation
2.4. Viral Factors
2.5. Immune-Mediated Mechanisms
2.6. Host Factors
3. Experimental Systems
3.1. Cell Models
3.2. Animal Models
4. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of cancer incidence, mortality, and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Ribes, J.; Diaz, M.; Cleries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.P.; Klenerman, P.; Dusheiko, G.M. Hepatitis C. Lancet 2015, 385, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, G.L.; Alter, M.J.; McQuillan, G.M.; Margolis, H.S. The past incidence of hepatitis C virus infection: Implications for the future burden of chronic liver disease in the United States. Hepatology 2000, 31, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, I.M.; Davis, G.L.; El-Serag, H.; Negro, F.; Trepo, C. Prevalence and challenges of liver diseases in patients with chronic hepatitis C virus infection. Clin. Gastroenterol. Hepatol. 2010, 8, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Fuchs, B.C.; Bardeesy, N.; Baumert, T.F.; Chung, R.T. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 2014, 61, S79–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, T.; Giorgio, A.; Marin, G.; Salmi, A.; de Sio, I.; Bolondi, L.; Pompili, M.; Brunello, F.; Lazzaroni, S.; Torzilli, G.; et al. Hepatocellular carcinoma and cirrhosis in 746 patients: Long-term results of percutaneous ethanol injection. Radiology 1995, 197, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chuang, V.P.; Wallace, S. Hepatic artery embolization in the treatment of hepatic neoplasms. Radiology 1981, 140, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ando, E.; Tanaka, M.; Yamashita, F.; Kuromatsu, R.; Yutani, S.; Fukumori, K.; Sumie, S.; Yano, Y.; Okuda, K.; Sata, M. Hepatic arterial infusion chemotherapy for advanced hepatocellular carcinoma with portal vein tumor thrombosis: Analysis of 48 cases. Cancer 2002, 95, 588–595. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Frakes, J.M.; Abuodeh, Y.A.; Naghavi, A.O.; Echevarria, M.I.; Shridhar, R.; Friedman, M.; Kim, R.; El-Haddad, G.; Kis, B.; Biebel, B.; et al. Viral hepatitis associated hepatocellular carcinoma outcomes with yttrium-90 radioembolization. J. Gastrointest. Oncol. 2018, 9, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Abouchaleh, N.; Gabr, A.; Ali, R.; Al Asadi, A.; Mora, R.A.; Kallini, J.R.; Mouli, S.; Riaz, A.; Lewandowski, R.J.; Salem, R. (90)Y Radioembolization for Locally Advanced Hepatocellular Carcinoma with Portal Vein Thrombosis: Long-Term Outcomes in a 185-Patient Cohort. J. Nucl. Med. 2018, 59, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J. Viral Hepat. 1999, 6, 35–47. [Google Scholar]
- The Polaris Observatory HCV Collaborators. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.S.; Mehta, S.H.; Srikrishnan, A.K.; Solomon, S.; McFall, A.M.; Laeyendecker, O.; Celentano, D.D.; Iqbal, S.H.; Anand, S.; Vasudevan, C.K.; et al. Burden of hepatitis C virus disease and access to hepatitis C virus services in people who inject drugs in India: A cross-sectional study. Lancet Infect. Dis. 2015, 15, 36–45. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Shiratori, Y.; Moriyama, M.; Arakawa, Y.; Ide, T.; Sata, M.; Inoue, O.; Yano, M.; Tanaka, M.; Fujiyama, S.; et al. Interferon therapy reduces the risk for hepatocellular carcinoma: National surveillance program of cirrhotic and noncirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group. Inhibition of Hepatocarcinogenesis by Interferon Therapy. Ann. Intern. Med. 1999, 131, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, Y.; Ito, Y.; Yokosuka, O.; Imazeki, F.; Nakata, R.; Tanaka, N.; Arakawa, Y.; Hashimoto, E.; Hirota, K.; Yoshida, H.; et al. Antiviral therapy for cirrhotic hepatitis C: Association with reduced hepatocellular carcinoma development and improved survival. Ann. Intern. Med. 2005, 142, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Tateishi, R.; Arakawa, Y.; Sata, M.; Fujiyama, S.; Nishiguchi, S.; Ishibashi, H.; Yamada, G.; Yokosuka, O.; Shiratori, Y.; et al. Benefit of interferon therapy in hepatocellular carcinoma prevention for individual patients with chronic hepatitis C. Gut 2004, 53, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, A.J.; Wedemeyer, H.; Feld, J.J.; Dufour, J.F.; Zeuzem, S.; Hansen, B.E.; Janssen, H.L. Life expectancy in patients with chronic HCV infection and cirrhosis compared with a general population. JAMA 2014, 312, 1927–1928. [Google Scholar] [CrossRef] [PubMed]
- Rutter, K.; Stattermayer, A.F.; Beinhardt, S.; Scherzer, T.M.; Steindl-Munda, P.; Trauner, M.; Ferenci, P.; Hofer, H. Successful anti-viral treatment improves survival of patients with advanced liver disease due to chronic hepatitis C. Aliment. Pharmacol. Ther. 2015, 41, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Li, J.; Rupp, L.B.; Holmberg, S.D.; Moorman, A.C.; Spradling, P.R.; Teshale, E.H.; Zhou, Y.; Boscarino, J.A.; Schmidt, M.A.; et al. Hepatitis C treatment failure is associated with increased risk of hepatocellular carcinoma. J. Viral Hepat. 2016, 23, 718–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.A.; Friedman, S.L. Reversal, maintenance or progression: What happens to the liver after a virologic cure of hepatitis C? Antivir. Res. 2014, 107, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, R.T.; Baumert, T.F. Curing chronic hepatitis C—The arc of a medical triumph. N. Engl. J. Med. 2014, 370, 1576–1578. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Mariño, Z.; Perelló, C.; Iñarrairaegui, M.; Ribeiro, A.; Lens, S.; Díaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.C.M.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.H.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Kozbial, K.; Moser, S.; Schwarzer, R.; Laferl, H.; Al-Zoairy, R.; Stauber, R.; Stattermayer, A.F.; Beinhardt, S.; Graziadei, I.; Freissmuth, C.; et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. J. Hepatol. 2016, 65, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.-L.; Li, Z.-Q.; Liang, H.-X.; Xu, G.-H.; Li, C.-X.; Zhang, D.-W.; Li, W.; Sun, C.-Y.; Wang, F.-S.; Yu, Z.-J. Unexpected high incidence of hepatocellular carcinoma in patients with hepatitis C in the era of DAAs: Too alarming? J. Hepatol. 2016, 65, 1068–1069. [Google Scholar] [CrossRef] [PubMed]
- Meissner, E.G.; Wu, D.; Osinusi, A.; Bon, D.; Virtaneva, K.; Sturdevant, D.; Porcella, S.; Wang, H.; Herrmann, E.; McHutchison, J.; et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J. Clin. Investig. 2014, 124, 3352–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serti, E.; Chepa-Lotrea, X.; Kim, Y.J.; Keane, M.; Fryzek, N.; Liang, T.J.; Ghany, M.; Rehermann, B. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology 2015, 149, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.H.; Hsu, C.W.; Chang, M.L.; Chen, Y.C.; Lai, M.W.; Yeh, C.T. Peginterferon Is Superior to Nucleos(t)ide Analogues for Prevention of Hepatocellular Carcinoma in Chronic Hepatitis B. J. Infect. Dis. 2016, 213, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Chatterjee, A.; Chandra, P.K.; Chava, S.; Chen, W.; Tandon, A.; Dash, A.; Chedid, M.; Moehlen, M.W.; Regenstein, F.; et al. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol. Commun. 2017, 1, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serag, H.B.; Kanwal, F.; Richardson, P.; Kramer, J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology 2016, 64, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahon, P.; Layese, R.; Bourcier, V.; Cagnot, C.; Marcellin, P.; Guyader, D.; Pol, S.; Larrey, D.; De Lédinghen, V.; Ouzan, D.; et al. Incidence of Hepatocellular Carcinoma After Direct Antiviral Therapy for HCV in Patients With Cirrhosis Included in Surveillance Programs. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]
- Stanislas, P. Lack of evidence of an effect of direct-acting antivirals on the recurrence of hepatocellular carcinoma: Data from three ANRS cohorts. J. Hepatol. 2016, 65, 734–740. [Google Scholar]
- Nagata, H.; Nakagawa, M.; Asahina, Y.; Sato, A.; Asano, Y.; Tsunoda, T.; Miyoshi, M.; Kaneko, S.; Otani, S.; Kawai-Kitahata, F.; et al. Effect of interferon-based and -free therapy on early occurrence and recurrence of hepatocellular carcinoma in chronic hepatitis C. J. Hepatol. 2017, 67, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Kurosaki, M.; Komiyama, Y.; Takada, H.; Tamaki, N.; Watakabe, K.; Okada, M.; Wang, W.; Shimizu, T.; Kubota, Y.; et al. Wisteria floribunda agglutinin-positive Mac-2 binding protein predicts early occurrence of hepatocellular carcinoma after SVR by direct acting antivirals for HCV. Hepatol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, H.; Tamori, A.; Kubo, S.; Uchida-Kobayashi, S.; Takemura, S.; Tanaka, S.; Ohfuji, S.; Teranishi, Y.; Kozuka, R.; Kawamura, E.; et al. Stagnation of histopathological improvement is a predictor of hepatocellular carcinoma development after hepatitis C virus eradication. PLoS ONE 2018, 13, e0194163. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Zavaglia, C.; Okolicsanyi, S.; Cesarini, L.; Mazzarelli, C.; Pontecorvi, V.; Ciaccio, A.; Strazzabosco, M.; Belli, L.S. Is the risk of neoplastic recurrence increased after prescribing direct-acting antivirals for HCV patients whose HCC was previously cured? J. Hepatol. 2017, 66, 236–237. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, R.; Colombo, M. Should surveillance for liver cancer be modified in hepatitis C patients after treatment-related cirrhosis regression? Liver Int. 2016, 36, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Juhling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akuta, N.; Suzuki, F.; Hirakawa, M.; Kawamura, Y.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Saitoh, S.; et al. Amino acid substitutions in hepatitis C virus core region predict hepatocarcinogenesis following eradication of HCV RNA by antiviral therapy. J. Med. Virol. 2011, 83, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Hung, C.H.; Lu, S.N.; Wang, J.H.; Lee, C.M.; Chen, C.H.; Yen, M.F.; Lin, S.C.; Yen, Y.H.; Tsai, M.C.; et al. A novel predictive score for hepatocellular carcinoma development in patients with chronic hepatitis C after sustained response to pegylated interferon and ribavirin combination therapy. J. Antimicrob. Chemother. 2012, 67, 2766–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.F.; Yeh, M.L.; Tsai, P.C.; Hsieh, M.H.; Yang, H.L.; Hsieh, M.Y.; Yang, J.F.; Lin, Z.Y.; Chen, S.C.; Wang, L.Y.; et al. Baseline gamma-glutamyl transferase levels strongly correlate with hepatocellular carcinoma development in non-cirrhotic patients with successful hepatitis C virus eradication. J. Hepatol. 2014, 61, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Oze, T.; Hiramatsu, N.; Yakushijin, T.; Miyazaki, M.; Yamada, A.; Oshita, M.; Hagiwara, H.; Mita, E.; Ito, T.; Fukui, H.; et al. Post-treatment levels of alpha-fetoprotein predict incidence of hepatocellular carcinoma after interferon therapy. Clin. Gastroenterol. Hepatol. 2014, 12, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Chiba, T.; Suzuki, E.; Shinozaki, M.; Goto, N.; Kanogawa, N.; Motoyama, T.; Ogasawara, S.; Ooka, Y.; Tawada, A.; et al. Effect of previous interferon-based therapy on recurrence after curative treatment of hepatitis C virus-related hepatocellular carcinoma. Int. J. Med. Sci. 2014, 11, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Ohho, A.; Yamasaki, A.; Kurokawa, M.; Kotoh, K.; Kajiwara, E. Hepatocarcinogenesis in chronic hepatitis C patients achieving a sustained virological response to interferon: Significance of lifelong periodic cancer screening for improving outcomes. J. Gastroenterol. 2014, 49, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.F.; Yeh, M.L.; Yu, M.L.; Dai, C.Y.; Huang, C.F.; Huang, C.I.; Tsai, P.C.; Lin, P.C.; Chen, Y.L.; Chang, W.T.; et al. The tertiary prevention of hepatocellular carcinoma in chronic hepatitis C patients. J. Gastroenterol. Hepatol. 2015, 30, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Kumada, T.; Tada, T.; Kiriyama, S.; Tanikawa, M.; Hisanaga, Y.; Kanamori, A.; Kitabatake, S.; Ito, T. Risk factors of hepatocellular carcinoma development in non-cirrhotic patients with sustained virologic response for chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2015, 30, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Suzuki, F.; Fujiyama, S.; Kawamura, Y.; Sezaki, H.; Hosaka, T.; Akuta, N.; Suzuki, Y.; Saitoh, S.; Arase, Y.; et al. Sustained virologic response by direct antiviral agents reduces the incidence of hepatocellular carcinoma in patients with HCV infection. J. Med. Virol. 2017, 89, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, H.; Ikeda, K.; Sorin, Y.; Fujiyama, S.; Kawamura, Y.; Kobayashi, M.; Sezaki, H.; Hosaka, T.; Akuta, N.; Saitoh, S.; et al. Long-Term Outcomes of Hepatitis-C-Infected Patients Achieving a Sustained Virological Response and Undergoing Radical Treatment for Hepatocellular Carcinoma. Oncology 2016, 90, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Tateishi, R.; Nakagomi, R.; Fujiwara, N.; Sato, M.; Enooku, K.; Nakagawa, H.; Asaoka, Y.; Kondo, Y.; Shiina, S.; et al. The impact of direct-acting antivirals on early tumor recurrence after radiofrequency ablation in hepatitis C-related hepatocellular carcinoma. J. Hepatol. 2016, 65, 1272–1273. [Google Scholar] [CrossRef] [PubMed]
- Nagaoki, Y.; Aikata, H.; Nakano, N.; Shinohara, F.; Nakamura, Y.; Hatooka, M.; Morio, K.; Kan, H.; Fujino, H.; Kobayashi, T.; et al. Development of hepatocellular carcinoma in patients with hepatitis C virus infection who achieved sustained virological response following interferon therapy: A large-scale, long-term cohort study. J. Gastroenterol. Hepatol. 2016, 31, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Kumada, T.; Toyoda, H.; Kiriyama, S.; Tanikawa, M.; Hisanaga, Y.; Kanamori, A.; Kitabatake, S.; Yama, T.; Tanaka, J. Viral eradication reduces all-cause mortality in patients with chronic hepatitis C virus infection: A propensity score analysis. Liver Int. 2016, 36, 817–826. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.J.; Feld, J.J.; Hofer, H.; Almasio, P.L.; Calvaruso, V.; Fernandez-Rodriguez, C.M.; Aleman, S.; Ganne-Carrie, N.; D’Ambrosio, R.; Pol, S.; et al. Risk of cirrhosis-related complications in patients with advanced fibrosis following hepatitis C virus eradication. J. Hepatol. 2017, 66, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Yen, Y.H.; Yao, C.C.; Hung, C.H.; Chen, C.H.; Hu, T.H.; Lee, C.M.; Lu, S.N. Liver stiffness-based score in hepatoma risk assessment for chronic hepatitis C patients after successful antiviral therapy. Liver Int. 2016, 36, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Cabibbo, G.; Barbara, M.; Attardo, S.; Bucci, L.; Farinati, F.; Giannini, E.G.; Tovoli, F.; Ciccarese, F.; Rapaccini, G.L.; et al. Hepatocellular carcinoma recurrence in patients with curative resection or ablation: Impact of HCV eradication does not depend on the use of interferon. Aliment. Pharmacol. Ther. 2017, 45, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.; Vale, A.M.; Rodrigues, S.; Goncalves, R.; Albuquerque, A.; Pereira, P.; Lopes, S.; Silva, M.; Andrade, P.; Morais, R.; et al. High incidence of hepatocellular carcinoma following successful interferon-free antiviral therapy for hepatitis C associated cirrhosis. J. Hepatol. 2016, 65, 1070–1071. [Google Scholar] [CrossRef] [PubMed]
- Calleja, J.L.; Crespo, J.; Rincon, D.; Ruiz-Antoran, B.; Fernandez, I.; Perello, C.; Gea, F.; Lens, S.; Garcia-Samaniego, J.; Sacristan, B.; et al. Effectiveness, safety and clinical outcomes of direct-acting antiviral therapy in HCV genotype 1 infection: Results from a Spanish real-world cohort. J. Hepatol. 2017, 66, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Mettke, F.; Schlevogt, B.; Deterding, K.; Wranke, A.; Smith, A.; Port, K.; Manns, M.P.; Vogel, A.; Cornberg, M.; Wedemeyer, H. Interferon-free therapy of chronic hepatitis C with direct-acting antivirals does not change the short-term risk for de novo hepatocellular carcinoma in patients with liver cirrhosis. Aliment. Pharmacol. Ther. 2018, 47, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, V.; Cabibbo, G.; Cacciola, I.; Petta, S.; Madonia, S.; Bellia, A.; Tinè, F.; Distefano, M.; Licata, A.; Giannitrapani, L.; et al. Incidence of Hepatocellular Carcinoma in Patients With HCV-Associated Cirrhosis Treated With Direct-Acting Antiviral Agents. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 2007, 22, S108–S111. [Google Scholar] [CrossRef] [PubMed]
- Koskinas, J.; Petraki, K.; Kavantzas, N.; Rapti, I.; Kountouras, D.; Hadziyannis, S. Hepatic expression of the proliferative marker Ki-67 and p53 protein in HBV or HCV cirrhosis in relation to dysplastic liver cell changes and hepatocellular carcinoma. J. Viral Hepat. 2005, 12, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Alison, M.R.; Lovell, M.J. Liver cancer: The role of stem cells. Cell Prolif. 2005, 38, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S.; Yoshida, H.; Tateishi, R.; Masuzaki, R.; Ohki, T.; Hayakawa, Y.; Kinoshita, H.; Yamakado, M.; Kato, N.; et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: An analysis based on gender differences. Int. J. Cancer 2009, 125, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzanti, R.; Messerini, L.; Monsacchi, L.; Buzzelli, G.; Zignego, A.L.; Foschi, M.; Monti, M.; Laffi, G.; Morbidelli, L.; Fantappie, O.; et al. Chronic viral hepatitis induced by hepatitis C but not hepatitis B virus infection correlates with increased liver angiogenesis. Hepatology 1997, 25, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wei, X.; Chen, T.; Huang, C.; Liu, H.; Wang, Y. Characterization of fibrosis changes in chronic hepatitis C patients after virological cure: A systematic review with meta-analysis. J. Gastroenterol. Hepatol. 2017, 32, 548–557. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Karin, M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Apoptosis as a mechanism for liver disease progression. Semin. Liver Dis. 2010, 30, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Thimme, R.; Blum, H.E.; Zoulim, F. Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 2009, 51, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Kato, N.; Otsuka, M.; Goto, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J. Med. Virol. 2004, 72, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Hur, W.; Wang, J.S.; Jang, J.W.; Kim, C.W.; Bae, S.H.; Jang, S.K.; Yang, S.H.; Sung, Y.C.; Kwon, O.J.; et al. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-beta1. Exp. Mol. Med. 2005, 37, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Gregoire, D.; Floc’h, N.; Moreau, M.; Hernandez, C.; Simonin, Y.; Rosenberg, A.R.; Lassus, P.; Hibner, U. Hepatitis C viral protein NS5A induces EMT and participates in oncogenic transformation of primary hepatocyte precursors. J. Hepatol. 2012, 57, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Buonaguro, L.; Izzo, F.; Buonaguro, F.M. Molecular alterations in hepatocellular carcinoma associated with hepatitis B and hepatitis C infections. Oncotarget 2016, 7, 25087–25102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sghaier, I.; Mouelhi, L.; Rabia, N.A.; Alsaleh, B.R.; Ghazoueni, E.; Almawi, W.Y.; Loueslati, B.Y. Genetic variants in IL-6 and IL-10 genes and susceptibility to hepatocellular carcinoma in HCV infected patients. Cytokine 2017, 89, 62–67. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Moran, D.M.; Jin, X.; Koniaris, L.G. Molecular pathogenesis of hepatocellular carcinoma. J. Surg. Res. 2006, 136, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Jiang, Z.; Liu, J.; Haverty, P.M.; Guan, Y.; Stinson, J.; Yue, P.; Zhang, Y.; Pant, K.P.; Bhatt, D.; et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010, 465, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Is Cancer a Genetic Disease or a Metabolic Disease? EBioMedicine 2015, 2, 478–479. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Jeng, Y.M.; Chang, C.N.; Lee, H.J.; Hsu, H.C.; Lai, P.L.; Yuan, R.H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.S.; Park, S.H.; Jang, K.L. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012, 321, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukiyama-Kohara, K. Role of oxidative stress in hepatocarcinogenesis induced by hepatitis C virus. Int. J. Mol. Sci. 2012, 13, 15271–15278. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Mori, K.; Siddiqui, A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 2002, 76, 7453–7459. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.F.; Chen, S.Y.; Chen, J.Y.; Wu Lee, Y.H. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 2004, 23, 2472–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Cheng, Y.S.; Lai, M.M. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 2008, 370, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Nagano-Fujii, M.; Tanaka, M.; Nomura-Takigawa, Y.; Ikeda, M.; Kato, N.; Sada, K.; Hotta, H. NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 2006, 87, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Meyer, K.; Warner, R.; Basu, A.; Ray, R.B.; Ray, R. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 2006, 80, 4372–4379. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Majumder, M.; Steele, R.; Meyer, K.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein protects against TNF-alpha mediated apoptotic cell death. Virus Res. 2000, 67, 173–178. [Google Scholar] [CrossRef]
- Jiang, X.; Kanda, T.; Wu, S.; Nakamoto, S.; Wakita, T.; Shirasawa, H.; Yokosuka, O. Hepatitis C virus nonstructural protein 5A inhibits thapsigargin-induced apoptosis. PLoS ONE 2014, 9, e113499. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; Disson, O.; Lerat, H.; Antoine, E.; Biname, F.; Rosenberg, A.R.; Desagher, S.; Lassus, P.; Bioulac-Sage, P.; Hibner, U. Calpain activation by hepatitis C virus proteins inhibits the extrinsic apoptotic signaling pathway. Hepatology 2009, 50, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Liu, J.C.; McNamara, G.; Levine, A.; Duan, L.; Lai, M.M. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J. Virol. 2009, 83, 12590–12600. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of beta-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/beta-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Au, S.L.; Wong, C.C.; Lee, J.M.; Fan, D.N.; Tsang, F.H.; Ng, I.O.; Wong, C.M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Koike, K. Molecular basis of hepatitis C virus-associated hepatocarcinogenesis: Lessons from animal model studies. Clin. Gastroenterol. Hepatol. 2005, 3, S132–S135. [Google Scholar] [CrossRef]
- Bittar, C.; Shrivastava, S.; Bhanja Chowdhury, J.; Rahal, P.; Ray, R.B. Hepatitis C virus NS2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS ONE 2013, 8, e62581. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol. 2005, 76, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell Longev. 2016, 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Hara, Y.; Nishina, S. Mitochondrial reactive oxygen species as a mystery voice in hepatitis C. Hepatol. Res. 2014, 44, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Domitrovich, A.M.; Felmlee, D.J.; Siddiqui, A. Hepatitis C virus nonstructural proteins inhibit apolipoprotein B100 secretion. J. Biol. Chem. 2005, 280, 39802–39808. [Google Scholar] [CrossRef] [PubMed]
- Aytug, S.; Reich, D.; Sapiro, L.E.; Bernstein, D.; Begum, N. Impaired IRS-1/PI3-kinase signaling in patients with HCV: A mechanism for increased prevalence of type 2 diabetes. Hepatology 2003, 38, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Gonzalez-Peralta, R.P.; Qian, K.; Xu, Y.; Marousis, C.G.; Davis, G.L.; Lau, J.Y. Transforming growth factor-beta 1 in chronic hepatitis C. J. Viral Hepat. 1997, 4, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Pavio, N.; Battaglia, S.; Boucreux, D.; Arnulf, B.; Sobesky, R.; Hermine, O.; Brechot, C. Hepatitis C virus core variants isolated from liver tumor but not from adjacent non-tumor tissue interact with Smad3 and inhibit the TGF-beta pathway. Oncogene 2005, 24, 6119–6132. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hwang, S.B. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J. Biol. Chem. 2006, 281, 7468–7478. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Crotta, S.; Stilla, A.; Wack, A.; D’Andrea, A.; Nuti, S.; D’Oro, U.; Mosca, M.; Filliponi, F.; Brunetto, R.M.; Bonino, F.; et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 2002, 195, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, A. Aberrant DNA methylation in hepatocellular carcinoma tumor suppression (Review). Oncol. Lett. 2014, 8, 963–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, V.; Srinathan, S.; Pasini, E.; Angeli, M.; Chen, E.; Baciu, C.; Bhat, M. Epigenetic basis of hepatocellular carcinoma: A network-based integrative meta-analysis. World J. Hepatol. 2018, 10, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Ye, G.; Zhang, X.; Huang, T. p16 Methylation was associated with the development, age, hepatic viruses infection of hepatocellular carcinoma, and p16 expression had a poor survival: A systematic meta-analysis (PRISMA). Medicine 2017, 96, e8106. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.E.-R.N.; Nassar, A.A.-M.; El-Rouby, M.N.E.-D.; Shousha, H.I.; Barakat, A.B.; El-Desouky, E.D.; Zayed, N.A.; Ahmed, O.S.; Youssef, A.S.E.-D.; Kaseb, A.O.; et al. Disease Progression from Chronic Hepatitis C to Cirrhosis and Hepatocellular Carcinoma is Associated with Increasing DNA Promoter Methylation. Asian Pac. J. Cancer Prev. 2014, 14, 6721–6726. [Google Scholar] [CrossRef]
- Zekri, A.R.; Bahnasy, A.A.; Shoeab, F.E.; Mohamed, W.S.; El-Dahshan, D.H.; Ali, F.T.; Sabry, G.M.; Dasgupta, N.; Daoud, S.S. Methylation of multiple genes in hepatitis C virus associated hepatocellular carcinoma. J. Adv. Res. 2014, 5, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Couvert, P.; Carrie, A.; Paries, J.; Vaysse, J.; Miroglio, A.; Kerjean, A.; Nahon, P.; Chelly, J.; Trinchet, J.C.; Beaugrand, M.; et al. Liver insulin-like growth factor 2 methylation in hepatitis C virus cirrhosis and further occurrence of hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 5419–5427. [Google Scholar] [CrossRef] [PubMed]
- Rongrui, L.; Na, H.; Zongfang, L.; Fanpu, J.; Shiwen, J. Epigenetic Mechanism Involved in the HBV/HCV-Related Hepatocellular Carcinoma Tumorigenesis. Curr. Pharm. Des. 2014, 20, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Cevik, R.E.; Cesarec, M.; Da Silva Filipe, A.; Licastro, D.; McLauchlan, J.; Marcello, A. Hepatitis C Virus NS5A Targets Nucleosome Assembly Protein NAP1L1 To Control the Innate Cellular Response. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Hahn, Y.S.; Kwon, M.; Sadikot, R.T.; Blackwell, T.S.; Christman, J.W. Hepatitis C virus core protein suppresses NF-kappaB activation and cyclooxygenase-2 expression by direct interaction with IkappaB kinase beta. J. Virol. 2005, 79, 7648–7657. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Tovar, V.; Alsinet, C.; Villanueva, A.; Hoshida, Y.; Chiang, D.Y.; Sole, M.; Thung, S.; Moyano, S.; Toffanin, S.; Minguez, B.; et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J. Hepatol. 2010, 52, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.W.; Baek, W.K.; Suh, S.I.; Yang, S.H.; Chang, J.; Sung, Y.C.; Suh, M.H. Hepatitis C virus core protein promotes cell proliferation through the upregulation of cyclin E expression levels. Liver 2001, 21, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Giambartolomei, S.; Cupelli, F.; Merlo, P.; Fontemaggi, G.; Spaziani, A.; Balsano, C. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene 2003, 22, 2573–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, J.; Aoki, H.; Kajino, K.; Moriyama, M.; Arakawa, Y.; Hino, O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology 2000, 32, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. NS5A inhibitors in the treatment of hepatitis C. J. Hepatol. 2013, 59, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Honda, M.; Kaneko, S. Molecular mechanisms of hepatocarcinogenesis in chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2011, 26, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Staschke, K.A.; Tan, S.L. HCV NS5A: A Multifunctional Regulator of Cellular Pathways and Virus Replication. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Taylor & Francis: Abingdon-on-Thames, UK, 2006. [Google Scholar]
- Wu, S.C.; Chang, S.C.; Wu, H.Y.; Liao, P.J.; Chang, M.F. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J. Biol. Chem. 2008, 283, 29396–29404. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Choi, S.H.; Kang, S.M.; Kang, J.I.; Ahn, B.Y.; Kim, H.; Jung, G.; Choi, K.Y.; Hwang, S.B. Nonstructural 5A protein activates beta-catenin signaling cascades: Implication of hepatitis C virus-induced liver pathogenesis. J. Hepatol. 2009, 51, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Milward, A.; Mankouri, J.; Harris, M. Hepatitis C virus NS5A protein interacts with beta-catenin and stimulates its transcriptional activity in a phosphoinositide-3 kinase-dependent fashion. J. Gen. Virol. 2010, 91, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Zhang, L.; Yang, G.; Zhao, L.; Peng, F.; Tian, Y.; Xiao, X.; Chung, R.T.; Gong, G. Hepatitis C virus NS5A drives a PTEN-PI3K/Akt feedback loop to support cell survival. Liver Int. 2015, 35, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- El-Shamy, A.; Shindo, M.; Shoji, I.; Deng, L.; Okuno, T.; Hotta, H. Polymorphisms of the core, NS3, and NS5A proteins of hepatitis C virus genotype 1b associate with development of hepatocellular carcinoma. Hepatology 2013, 58, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Korba, B.; Shetty, K.; Medvedev, A.; Viswanathan, P.; Varghese, R.; Zhou, B.; Roy, R.; Makambi, K.; Ressom, H.; Loffredo, C.A. Hepatitis C virus Genotype 1a core gene nucleotide patterns associated with hepatocellular carcinoma risk. J. Gen. Virol. 2015, 96, 2928–2937. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, S.; Inoue, M.; Tanaka, Y.; Mizokami, M.; Iwasaki, M.; Tsugane, S.; Group, J.S. Impact of viral load of hepatitis C on the incidence of hepatocellular carcinoma: A population-based cohort study (JPHC Study). Cancer Lett. 2011, 300, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Bruno, S.; Mondelli, M.U.; Maisonneuve, P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: A meta-analysis. J. Hepatol. 2009, 50, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Harouaka, D.; Engle, R.E.; Wollenberg, K.; Diaz, G.; Tice, A.B.; Zamboni, F.; Govindarajan, S.; Alter, H.; Kleiner, D.E.; Farci, P. Diminished viral replication and compartmentalization of hepatitis C virus in hepatocellular carcinoma tissue. Proc. Natl. Acad. Sci. USA 2016, 113, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.D.; de Knegt, R.J.; Boonstra, A. The Path to Cancer and Back: Immune Modulation During Hepatitis C Virus Infection, Progression to Fibrosis and Cancer, and Unexpected Roles of New Antivirals. Transplantation 2017, 101, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Haybaeck, J.; Zeller, N.; Wolf, M.J.; Weber, A.; Wagner, U.; Kurrer, M.O.; Bremer, J.; Iezzi, G.; Graf, R.; Clavien, P.A.; et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell 2009, 16, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, M.; Sturm, N.; Decaens, T.; Bioulac-Sage, P.; Bancel, B.; Merle, P.; Tran Van Nhieu, J.; Slama, R.; Letoublon, C.; Zarski, J.P.; et al. Liver-infiltrating CD8(+) lymphocytes as prognostic factor for tumour recurrence in hepatitis C virus-related hepatocellular carcinoma. Liver Int. 2016, 36, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Fisicaro, P.; Tola, D.; Orlandini, A.; Sacchelli, L.; Mori, C.; Missale, G.; Ferrari, C. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 2006, 44, 126–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehermann, B.; Bertoletti, A. Immunological aspects of antiviral therapy of chronic hepatitis B virus and hepatitis C virus infections. Hepatology 2015, 61, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Sung, S.S.; Krueger, P.D.; Jo, Y.A.; Rosen, H.R.; Ziegler, S.F.; Hahn, Y.S. Hepatitis C virus promotes T-helper (Th)17 responses through thymic stromal lymphopoietin production by infected hepatocytes. Hepatology 2013, 57, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Rehermann, B. Immune responses to HCV and other hepatitis viruses. Immunity 2014, 40, 13–24. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Miki, D.; Ochi, H.; Hayes, C.N.; Abe, H.; Yoshima, T.; Aikata, H.; Ikeda, K.; Kumada, H.; Toyota, J.; Morizono, T.; et al. Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers. Nat. Genet. 2011, 43, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kato, N.; Urabe, Y.; Takahashi, A.; Muroyama, R.; Hosono, N.; Otsuka, M.; Tateishi, R.; Omata, M.; Nakagawa, H.; et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat. Genet. 2011, 43, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Bibert, S.; Dufour, J.F.; Cellerai, C.; Cerny, A.; Heim, M.H.; Kaiser, L.; Malinverni, R.; Mullhaupt, B.; Negro, F.; et al. Comparative genetic analyses point to HCP5 as susceptibility locus for HCV-associated hepatocellular carcinoma. J. Hepatol. 2013, 59, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Sawai, H.; Ikeo, K.; Ogawa, S.; Iio, E.; Isogawa, M.; Shimada, N.; Komori, A.; Toyoda, H.; Kumada, T.; et al. Genome-Wide Association Study Identifies TLL1 Variant Associated With Development of Hepatocellular Carcinoma After Eradication of Hepatitis C Virus Infection. Gastroenterology 2017, 152, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Maekawa, S.; Kadokura, M.; Sueki, R.; Komase, K.; Shindo, H.; Ohmori, T.; Kanayama, A.; Shindo, K.; Amemiya, F.; et al. Analysis of viral amino acids sequences and the IL28B SNP influencing the development of hepatocellular carcinoma in chronic hepatitis C. Hepatol. Int. 2012, 6, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Dragani, T.A. Risk of HCC: Genetic heterogeneity and complex genetics. J. Hepatol. 2010, 52, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Aroucha, D.C.; Carmo, R.F.; Vasconcelos, L.R.; Lima, R.E.; Mendonca, T.F.; Arnez, L.E.; Cavalcanti Mdo, S.; Muniz, M.T.; Aroucha, M.L.; Siqueira, E.R.; et al. TNF-alpha and IL-10 polymorphisms increase the risk to hepatocellular carcinoma in HCV infected individuals. J. Med. Virol. 2016, 88, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Labib, H.A.; Ahmed, H.S.; Shalaby, S.M.; Wahab, E.A.; Hamed, E.F. Genetic polymorphism of IL-23R influences susceptibility to HCV-related hepatocellular carcinoma. Cell. Immunol. 2015, 294, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Tseng, P.L.; Wu, Y.Y.; Hung, H.C.; Huang, C.M.; Lu, S.N.; Wang, J.H.; Lee, C.M.; Chen, C.H.; Tsai, M.C.; et al. A polymorphism in interferon L3 is an independent risk factor for development of hepatocellular carcinoma after treatment of hepatitis C virus infection. Clin. Gastroenterol. Hepatol. 2015, 13, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Balagopal, A.; Thomas, D.L.; Thio, C.L. IL28B and the control of hepatitis C virus infection. Gastroenterology 2010, 139, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, V.; Bronte, F.; Calvaruso, V.; Capra, M.; Borsellino, Z.; Maggio, A.; Renda, M.C.; Pitrolo, L.; Lo Pinto, M.C.; Rizzo, M.; et al. IL28B polymorphisms influence stage of fibrosis and spontaneous or interferon-induced viral clearance in thalassemia patients with hepatitis C virus infection. Haematologica 2012, 97, 679–686. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.P.; Horner, S.M.; Jarret, A.; Joslyn, R.C.; Bindewald, E.; Shapiro, B.A.; Delker, D.A.; Hagedorn, C.H.; Carrington, M.; Gale, M., Jr.; et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat. Immunol. 2014, 15, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, R.C.; Alves, D.; Guz, B.; Matos, C.; Viana, M.; Harriz, M.; Terrabuio, D.; Kondo, M.; Gampel, O.; Polletti, P. Advanced hepatocellular carcinoma. Review of targeted molecular drugs. Ann. Hepatol. 2011, 10, 21–27. [Google Scholar] [PubMed]
- Teoh, W.W.; Xie, M.; Vijayaraghavan, A.; Yaligar, J.; Tong, W.M.; Goh, L.K.; Sabapathy, K. Molecular characterization of hepatocarcinogenesis using mouse models. Dis. Model. Mech. 2015, 8, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, J.; Zhang, L.; Lv, S.; Zhang, C.; Jiang, S. Biomarkers for Hepatocellular Carcinoma. Biomark. Cancer 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Sangiovanni, A.; Sole, M.; Hur, C.; Andersson, K.L.; Chung, R.T.; Gould, J.; Kojima, K.; Gupta, S.; et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology 2013, 144, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Liang, K.H.; Chien, R.N.; Hu, T.H.; Lin, K.H.; Hsu, C.W.; Lin, C.L.; Pan, T.L.; Ke, P.Y.; Yeh, C.T. A Circulating MicroRNA Signature Capable of Assessing the Risk of Hepatocellular Carcinoma in Cirrhotic Patients. Sci. Rep. 2017, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Andreo, U.; Chung, H.Y.; Espiritu, C.; Branch, A.D.; Silva, J.M.; Rice, C.M. SEC14L2 enables pan-genotype HCV replication in cell culture. Nature 2015, 524, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.D.; Fuchs, B.C.; Hoshida, Y. In vitro modeling of hepatocellular carcinoma molecular subtypes for anti-cancer drug assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell, P.; Villanueva, A.; Friedman, S.L.; Koike, K.; Llovet, J.M. Experimental models of hepatocellular carcinoma. J. Hepatol. 2008, 48, 858–879. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.P.; Colaco, A.A.; Oliveira, P.A. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukh, J. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 2004, 39, 1469–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchmore, E.; Popper, H.; Peterson, D.A.; Miller, M.F.; Lieberman, H.M. Non-A, non-B hepatitis-related hepatocellular carcinoma in a chimpanzee. J. Med. Primatol. 1988, 17, 235–246. [Google Scholar] [PubMed]
- Mesalam, A.A.; Vercauteren, K.; Meuleman, P. Mouse Systems to Model Hepatitis C Virus Treatment and Associated Resistance. Viruses 2016, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chayama, K.; Hayes, C.N.; Hiraga, N.; Abe, H.; Tsuge, M.; Imamura, M. Animal model for study of human hepatitis viruses. J. Gastroenterol. Hepatol. 2011, 26, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Thorgeirsson, S.S. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol. Pathol. 2005, 33, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V.; Pinkert, C.A.; Milich, D.R.; Filippi, P.; McLachlan, A.; Palmiter, R.D.; Brinster, R.L. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science 1985, 230, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Ghebranious, N.; Sell, S. The mouse equivalent of the human p53ser249 mutation p53ser246 enhances aflatoxin hepatocarcinogenesis in hepatitis B surface antigen transgenic and p53 heterozygous null mice. Hepatology 1998, 27, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, Y.; Chen, J.; Cheng, G.; Xue, J. Transgenic mice expressing hepatitis B virus X protein are more susceptible to carcinogen induced hepatocarcinogenesis. Exp. Mol. Pathol. 2004, 76, 44–50. [Google Scholar] [CrossRef] [PubMed]
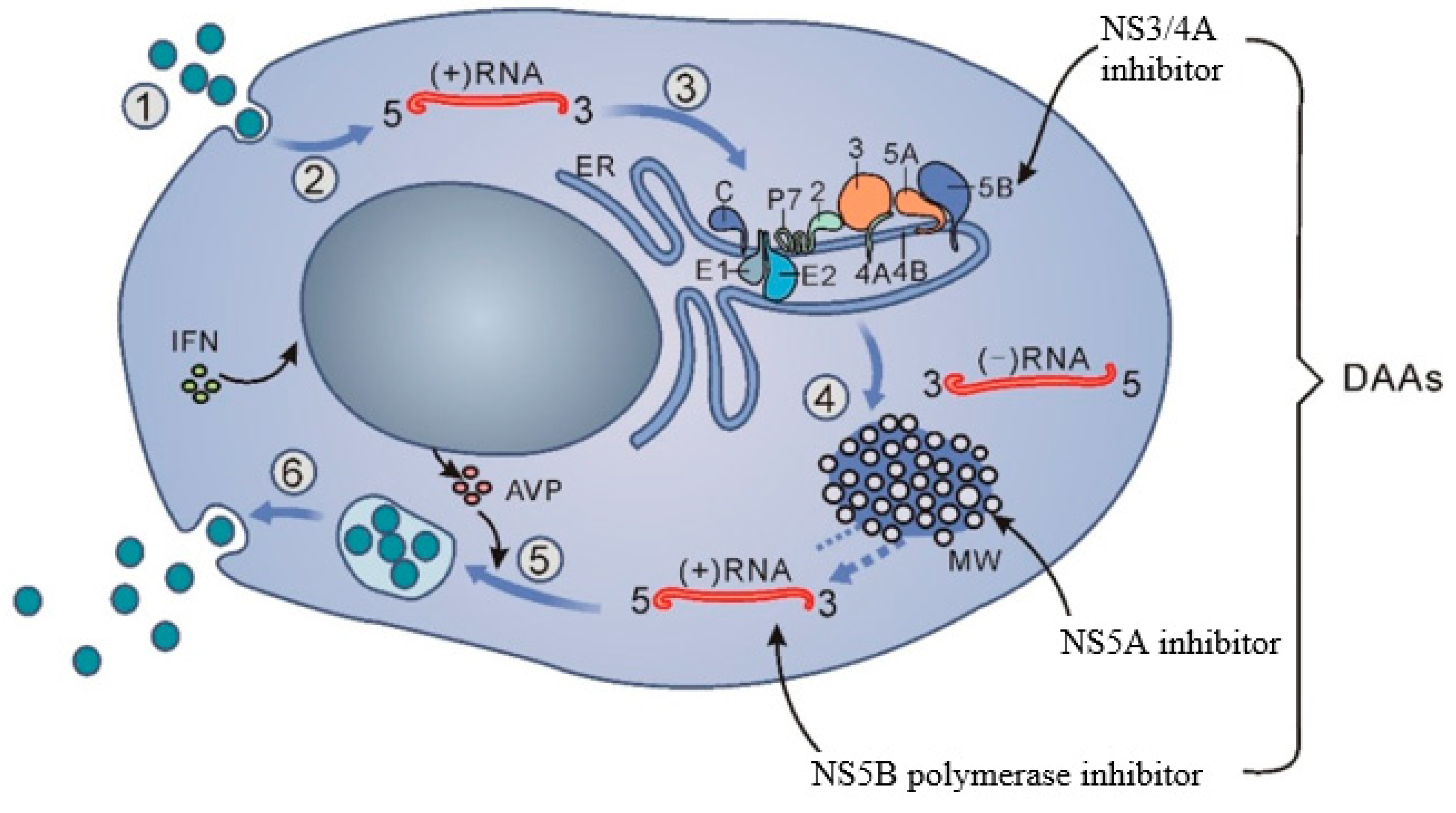

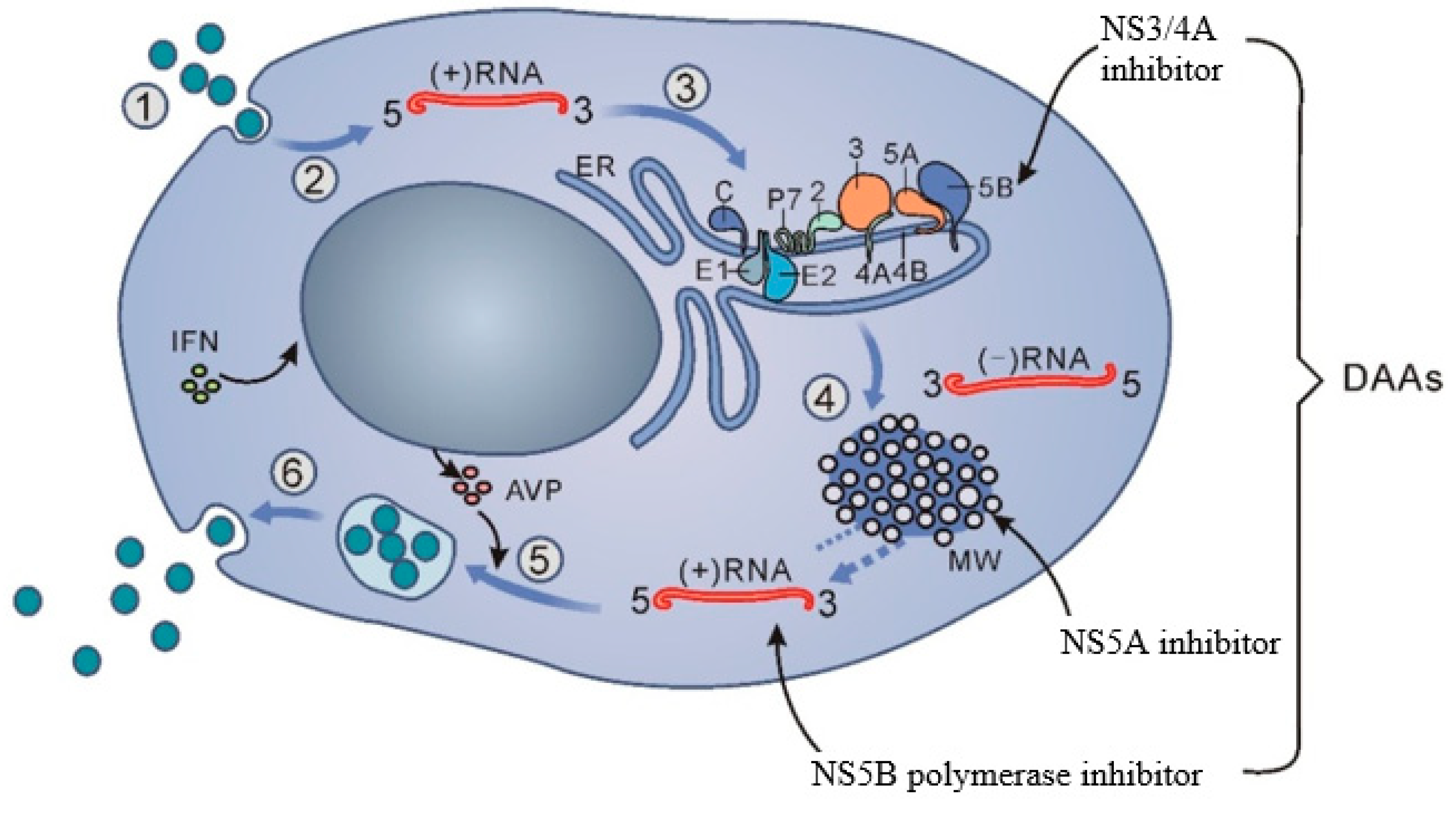{kind=link}
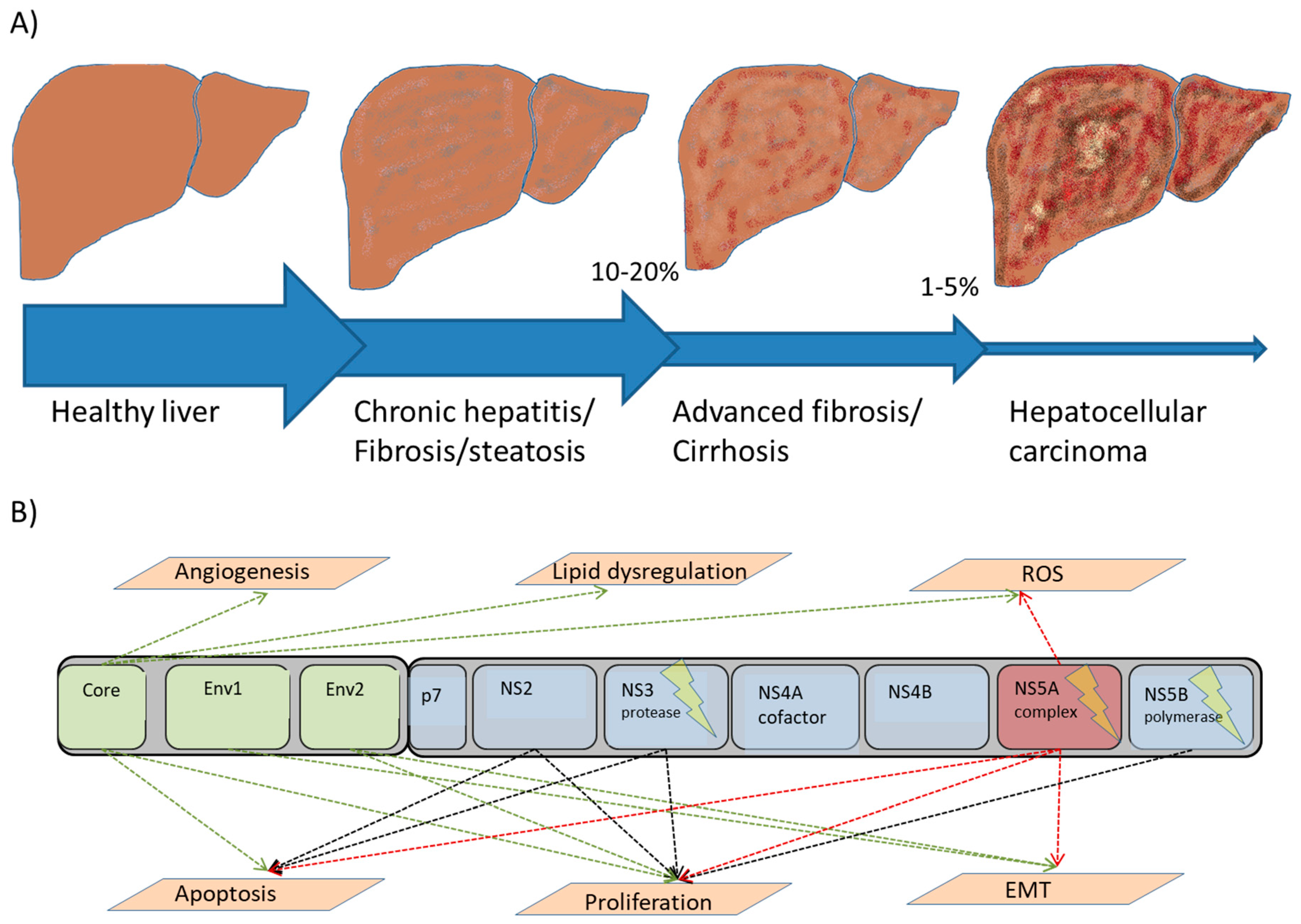
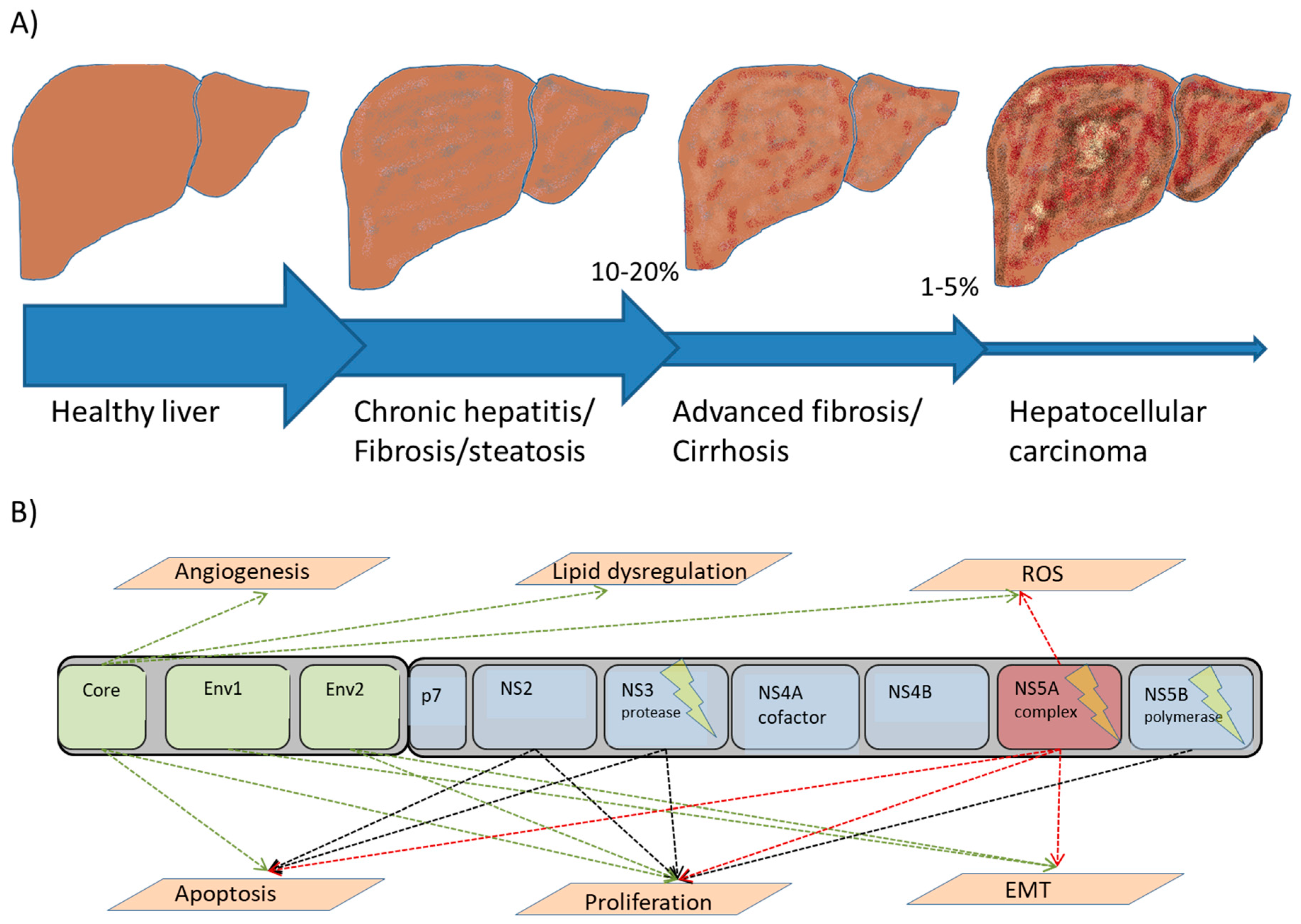{kind=link}
| Reference | Country | N | Follow-Up (Months) | Males (%) | Age | Cirrhosis (%) | Post-SVR HCC (%) * |
|---|---|---|---|---|---|---|---|
| Akuta (2011) [50] | Japan | 1273 | 1.1 | 61.5 | 53 | 8.6 | 3.2 |
| Chang (2012) [51] | Taiwan | 1271 | 3.4 | 75.9 | 55 | 27.9 | 1.2 |
| Huang (2014) [52] | Taiwan | 642 | 4.4 | 54.3 | 51 | 13.4 | 5.8 |
| Oze (2014) [53] | Japan | 1425 | 3.3 | 51.0 | 55 | 11.6 | 2.6 |
| Saito (2014) [54] | Japan | 14 | 3.9 | 92.9 | 72 | 85.7 | 18.0 |
| Yamashita (2014) [55] | Japan | 562 | 4.8 | 55.3 | 57 | 23.0 | 3.1 |
| Huang (2015) [56] | Taiwan | 56 | 4.4 | 64.3 | 62 | 37.5 | 43.2 |
| Toyoda (2015) [57] | Japan | 522 | 7.2 | 55.9 | 51 | 5.5 | 1.2 |
| El-Serag (2016) [40] | USA | 10,738 | 2.8 | 95.3 | 53 | 14.4 | 0.3 |
| Kobayashi (2016) [58] | Japan | 528 | 7.3 | 58.4 | 54 | 14.8 | 2.2 |
| Kunimoto (2016) [59] | Japan | 40 | 5.1 | 87.5 | 65 | 35.0 | 23.0 |
| Minami (2016) [60] | Japan | 38 | - | 71.0 | 66 | 0 | 52.9 |
| Nagaoki (2016) [61] | Japan | 1094 | 4.2 | 53.5 | 60 | 1.9 | 4.0 |
| Tada (2016) [62] | Japan | 587 | 14.0 | 55.2 | 50 | - | 4.4 |
| Tada (2016) [62] | Japan | 170 | 14.2 | 62.4 | 53 | - | 7.1 |
| van der Meer (2016) [63] | EU, Canada | 1000 | 5.7 | 68.0 | 53 | 85.0 | 7.6 |
| Wang (2016) [64] | Taiwan | 376 | 7.6 | 49.2 | 54 | 33.8 | 1.4 |
| Nagata (2017) [43] | Japan | 1145 | 6.8 | 54.0 | 59 | - | 2.6 |
| Kobayashi (2017) [58] | Japan | 77 | 4.0 | 44.2 | 63 | 3.0 | |
| Petta (2017) [65] | Italy | 57 | 2.8 | 72.0 | 62 | 0 | 15.0 |
| Motoyama (2018) [45] | Japan | 11 | 8.1 | 81.0 | 55 | 36.3 | - |
| Reference | Country | N | Follow-Up (Months) | Males (%) | Age | Cirrhosis (%) | Post-SVR HCC (%) * |
|---|---|---|---|---|---|---|---|
| ANRS (2016) [42] | France | 189 | 2.2 | 78.0 | 62 | 80.0 | 0.7 |
| ANRS (2016) [42] | France | 13 | 1.8 | 85.0 | 61 | 100.0 | 1.1 |
| ANRS (2016) [42] | France | 314 | - | 82.0 | 61 | 15.6 | 2.2 |
| Cardoso (2016) [66] | Portugal | 54 | 1.0 | 76.0 | 59 | - | 7.4 |
| Cheung (2016) [32] | UK | 317 | 1.3 | - | 54 | 80.1 | 5.4 |
| Conti (2016) [31] | Italy | 344 | 0.5 | 60.1 | 63 | 11.3 | 3.2 |
| Conti (2016) [31] | Italy | 59 | 0.5 | 67.8 | 72 | 16.9 | 28.8 |
| Kobayashi (2016) [58] | Japan | 77 | 4.0 | 44.2 | 63 | 29.9 | 3.0 |
| Kozbial (2016) [34] | Austria | 19 | - | 73.7 | - | 73.7 | 50.0 |
| Minami (2016) [60] | Japan | 27 | - | 67.0 | 71 | 0 | 29.8 |
| Petta (2016) [65] | Italy | 58 | 1.5 | 69.0 | 66 | 4.0 | 26.3 |
| Reig (2016) [30] | Spain | 58 | 0.5 | 69.0 | 66 | 8.6 | 27.6 |
| Calleja (2017) [67] | Spain | 1567 | 53.7 | 60 | 46.7 | 0.9 | |
| Nagata (2017) [43] | Japan | 752 | 1.8 | 45.0 | 69 | - | 3.3 |
| Kanwal (2017) [46] | US | 22,500 | 2.0 | 96.7 | 62 | 68.7 | 0.9 |
| Kobayashi (2017) [58] | Japan | 528 | 7.3 | 58.4 | 54 | 2.2 | |
| Mettke (2017) [68] | Germany | 158 | 1.2 | 55.0 | 59 | 100.0 | 2.9 |
| Petta (2017) [65] | Italy | 58 | 1.5 | 69.0 | 66 | - | 10.8 |
| Calvaruso (2018) [69] | Italy | 2249 | 1.1 | 56.9 | 65 | - | 3.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, C.N.; Zhang, P.; Zhang, Y.; Chayama, K. Molecular Mechanisms of Hepatocarcinogenesis Following Sustained Virological Response in Patients with Chronic Hepatitis C Virus Infection. Viruses 2018, 10, 531. https://doi.org/10.3390/v10100531
Hayes CN, Zhang P, Zhang Y, Chayama K. Molecular Mechanisms of Hepatocarcinogenesis Following Sustained Virological Response in Patients with Chronic Hepatitis C Virus Infection. Viruses. 2018; 10(10):531. https://doi.org/10.3390/v10100531
Chicago/Turabian StyleHayes, C. Nelson, Peiyi Zhang, Yizhou Zhang, and Kazuaki Chayama. 2018. "Molecular Mechanisms of Hepatocarcinogenesis Following Sustained Virological Response in Patients with Chronic Hepatitis C Virus Infection" Viruses 10, no. 10: 531. https://doi.org/10.3390/v10100531
APA StyleHayes, C. N., Zhang, P., Zhang, Y., & Chayama, K. (2018). Molecular Mechanisms of Hepatocarcinogenesis Following Sustained Virological Response in Patients with Chronic Hepatitis C Virus Infection. Viruses, 10(10), 531. https://doi.org/10.3390/v10100531