Recombination Located over 2A-2B Junction Ribosome Frameshifting Region of Saffold Cardiovirus

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Processing
2.2. DNA Alignment
2.3. Recombination Analysis
2.4. Phylogenetic Trees and Likelihood Mapping
3. Results
3.1. Polyprotein Tree
3.2. Recombination Rates
3.3. Partitioned Trees
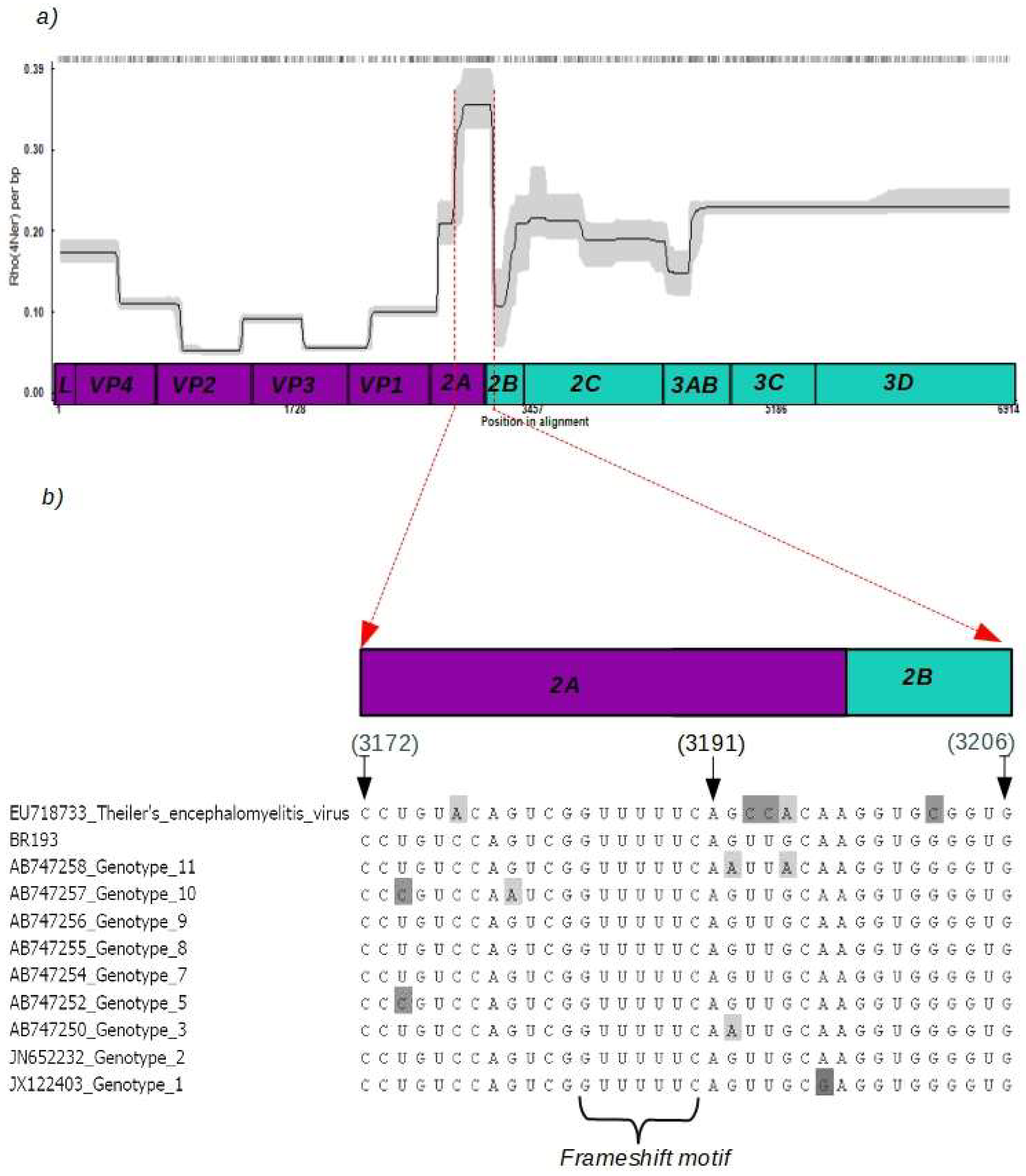
3.4. Mosaic Pattern
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zoll, J.; Erkens Hulshof, S.; Lanke, K.; Verduyn Lunel, F.; Melchers, W.J.; Schoondermark-van de Ven, E.; Roivainen, M.; Galama, J.M.; van Kuppeveld, F.J. Saffold virus, a human theiler’s-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 2009, 5, e1000416. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.S.; Lukashov, V.V.; Ganac, R.D.; Schnurr, D.P. Discovery of a novel human picornavirus in a stool sample from a pediatric patient presenting with fever of unknown origin. J. Clin. Microbiol. 2007, 45, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Baumgarte, S.; Luna, L.K.; Stocker, A.; Almeida, P.S.; Ribeiro, T.C.; Petersen, N.; Herzog, P.; Pedroso, C.; Brites, C.; et al. Genomic features and evolutionary constraints in saffold-like cardioviruses. J. Gen. Virol. 2010, 91, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Himeda, T.; Ohara, Y. Saffold virus, a novel human cardiovirus with unknown pathogenicity. J. Virol. 2012, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, T.; Aoki, Y.; Matoba, Y.; Tanaka, S.; Ikeda, T.; Matsuzaki, Y.; Mizuta, K. Detection of saffold viruses from children with acute respiratory infections in Yamagata, Japan, between 2008 and 2015. J. Med. Virol. 2018, 90, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Loughran, G.; Firth, A.E.; Atkins, J.F. Ribosomal frameshifting into an overlapping gene in the 2b-encoding region of the cardiovirus genome. Proc. Natl. Acad. Sci. USA 2011, 108, E1111–E1119. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Hosomi, T.; Nishimura, Y.; Alam, M.M.; Oka, T.; Zaidi, S.S.; Shimizu, H. Genetic diversity of circulating saffold viruses in Pakistan and Afghanistan. J. Gen. Virol. 2014, 95, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Xiao, Y.; Li, J.; Chen, L.; Zhang, J.; Vernet, G.; Wang, J. Multiple genomic recombination events in the evolution of saffold cardiovirus. PLoS ONE 2013, 8, e74947. [Google Scholar]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV virus taxonomy profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Blinkova, O.; Kapoor, A.; Victoria, J.; Jones, M.; Wolfe, N.; Naeem, A.; Shaukat, S.; Sharif, S.; Alam, M.M.; Angez, M.; et al. Cardioviruses are genetically diverse and cause common enteric infections in south Asian children. J. Virol. 2009, 83, 4631–4641. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Baumgarte, S.; Eschbach-Bludau, M.; Simon, A.; Kemen, C.; Bode, U.; Eis-Hubinger, A.M.; Madea, B.; Drosten, C. Human cardioviruses, meningitis, and sudden infant death syndrome in children. Emerg. Infect. Dis. 2011, 17, 2313–2315. [Google Scholar] [CrossRef] [PubMed]
- Martins Lde, O.; Leal, E.; Kishino, H. Phylogenetic detection of recombination with a bayesian prior on the distance between trees. PLoS ONE 2008, 3, e2651. [Google Scholar]
- Tan, S.Z.; Tan, M.Z.; Prabakaran, M. Saffold virus, an emerging human cardiovirus. Rev. Med. Virol. 2017, 27, e1908. [Google Scholar] [CrossRef] [PubMed]
- Charlys da Costa, A.; Theze, J.; Komninakis, S.C.V.; Sanz-Duro, R.L.; Felinto, M.R.L.; Moura, L.C.C.; Barroso, I.M.O.; Santos, L.E.C.; Nunes, M.A.L.; Moura, A.A.; et al. Spread of chikungunya virus east/central/south African genotype in northeast Brazil. Emerg. Infect. Dis. 2017, 23, 1742–1744. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. Jmodeltest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. Tree-puzzle: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.; Villanova, F.E.; Lin, W.; Hu, F.; Liu, Q.; Liu, Y.; Cui, S. Interclade recombination in porcine parvovirus strains. J. Gen. Virol. 2012, 93, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.A.; Lu, Q.B.; Wo, Y.; Zhao, J.; Huang, D.D.; Guo, C.T.; Xu, H.M.; Liu, E.M.; Liu, W.; Cao, W.C. Prevalence and genetic characteristics of saffold cardiovirus in China from 2009 to 2012. Sci. Rep. 2015, 5, 7704. [Google Scholar] [CrossRef] [PubMed]
- Abed, Y.; Boivin, G. New saffold cardioviruses in 3 children, Canada. Emerg. Infect. Dis. 2008, 14, 834–836. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Costa, A.C.; Luchs, A.; Milagres, F.A.d.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.C.A.B.S.; Brustulin, R.; Das Chagas, R.T.; Abrão, M.D.F.N.d.S.; Soares, C.V.d.D.A.; et al. Recombination Located over 2A-2B Junction Ribosome Frameshifting Region of Saffold Cardiovirus. Viruses 2018, 10, 520. https://doi.org/10.3390/v10100520
Da Costa AC, Luchs A, Milagres FAdP, Komninakis SV, Gill DE, Lobato MCABS, Brustulin R, Das Chagas RT, Abrão MDFNdS, Soares CVdDA, et al. Recombination Located over 2A-2B Junction Ribosome Frameshifting Region of Saffold Cardiovirus. Viruses. 2018; 10(10):520. https://doi.org/10.3390/v10100520
Chicago/Turabian StyleDa Costa, Antônio Charlys, Adriana Luchs, Flávio Augusto de Pádua Milagres, Shirley Vasconcelos Komninakis, Danielle Elise Gill, Márcia Cristina Alves Brito Sayão Lobato, Rafael Brustulin, Rogério Togisaki Das Chagas, Maria De Fátima Neves dos Santos Abrão, Cassia Vitória de Deus Alves Soares, and et al. 2018. "Recombination Located over 2A-2B Junction Ribosome Frameshifting Region of Saffold Cardiovirus" Viruses 10, no. 10: 520. https://doi.org/10.3390/v10100520
APA StyleDa Costa, A. C., Luchs, A., Milagres, F. A. d. P., Komninakis, S. V., Gill, D. E., Lobato, M. C. A. B. S., Brustulin, R., Das Chagas, R. T., Abrão, M. D. F. N. d. S., Soares, C. V. d. D. A., Deng, X., Sabino, E. C., Delwart, E., & Leal, É. (2018). Recombination Located over 2A-2B Junction Ribosome Frameshifting Region of Saffold Cardiovirus. Viruses, 10(10), 520. https://doi.org/10.3390/v10100520