Viruses.STRING: A Virus-Host Protein-Protein Interaction Database

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Text Mining Evidence
2.2. Experimental Evidence
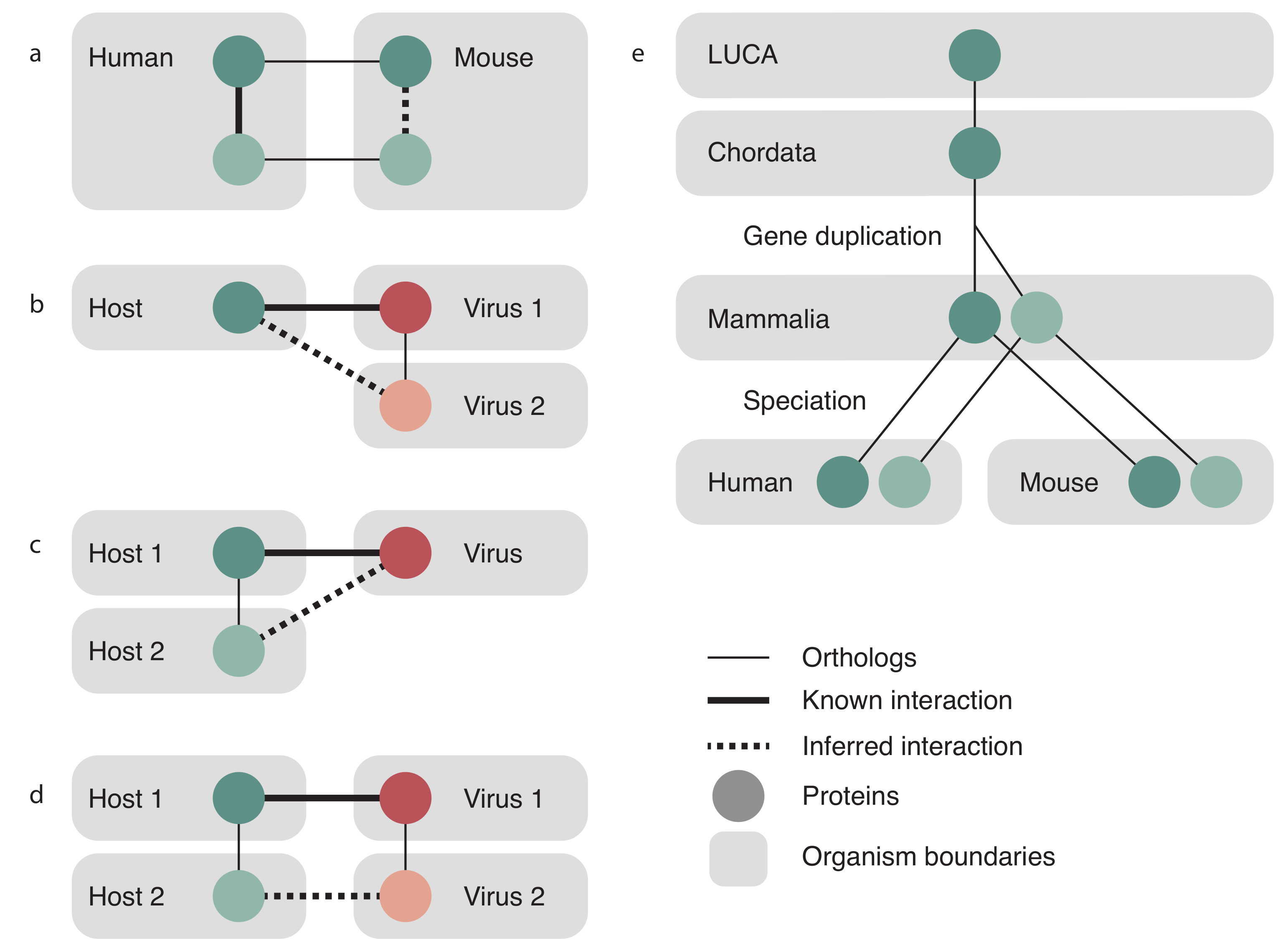
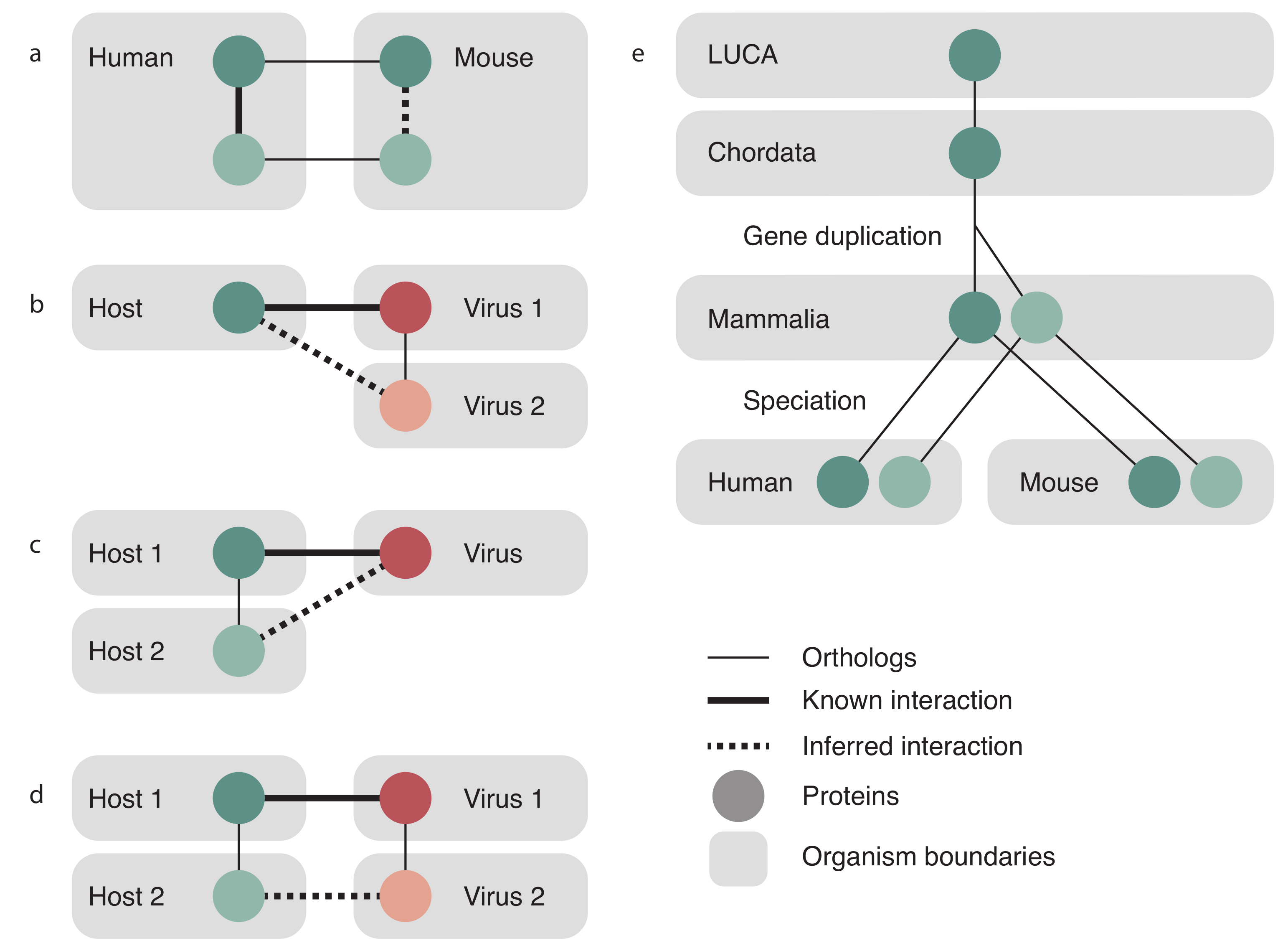
2.3. Transfer Evidence
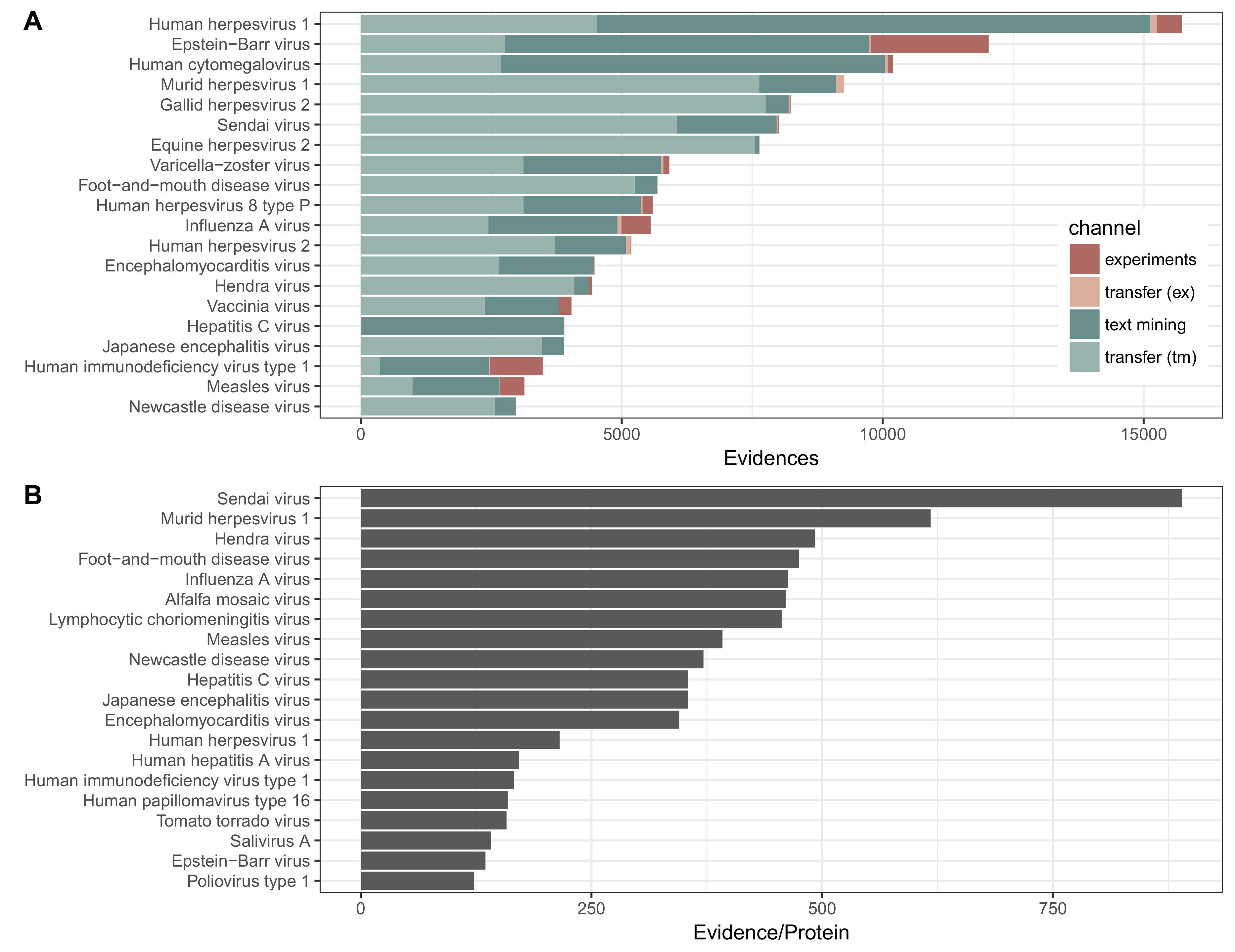
3. Results
3.1. Utility and Examples
3.1.1. Web Interface
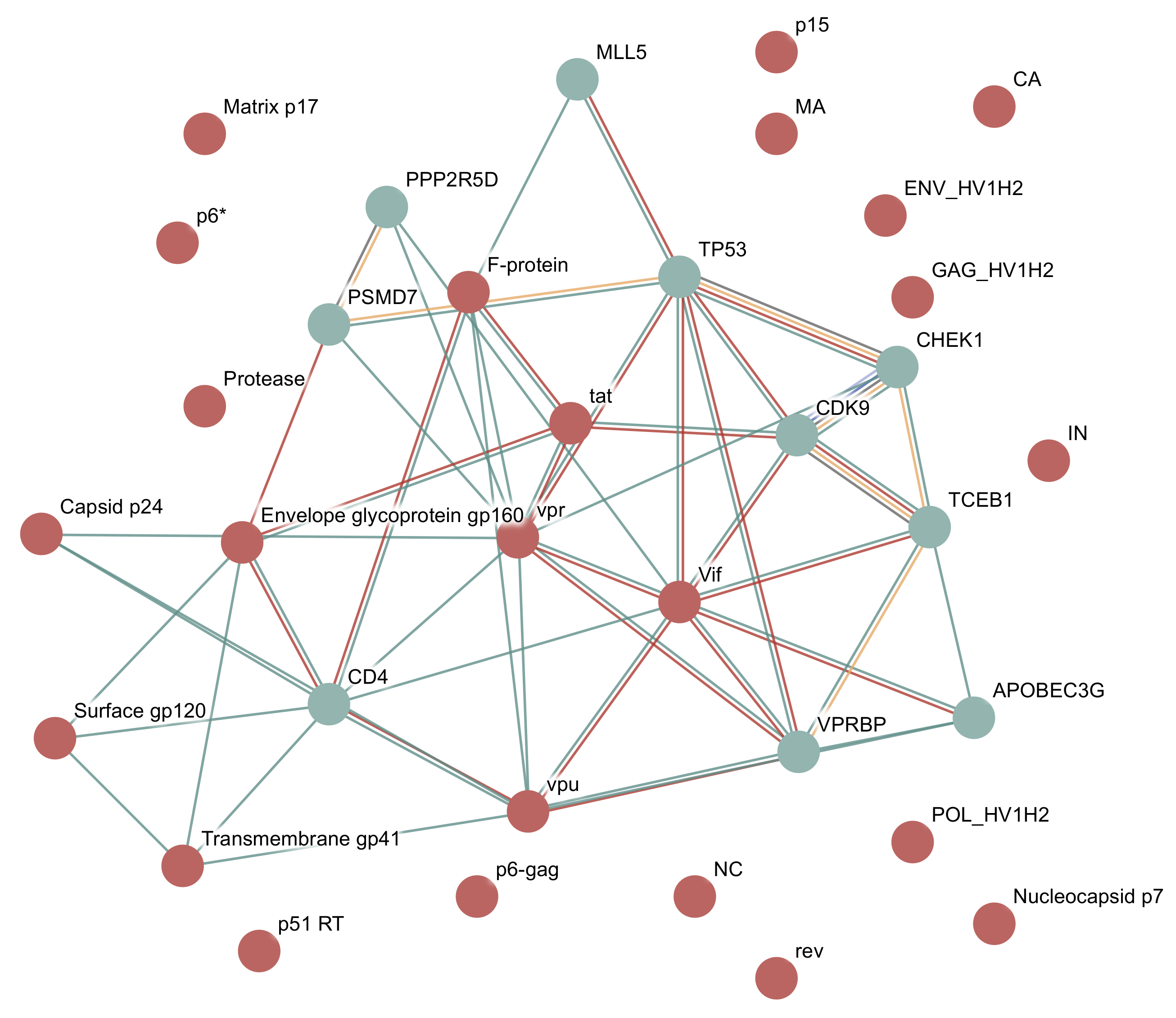
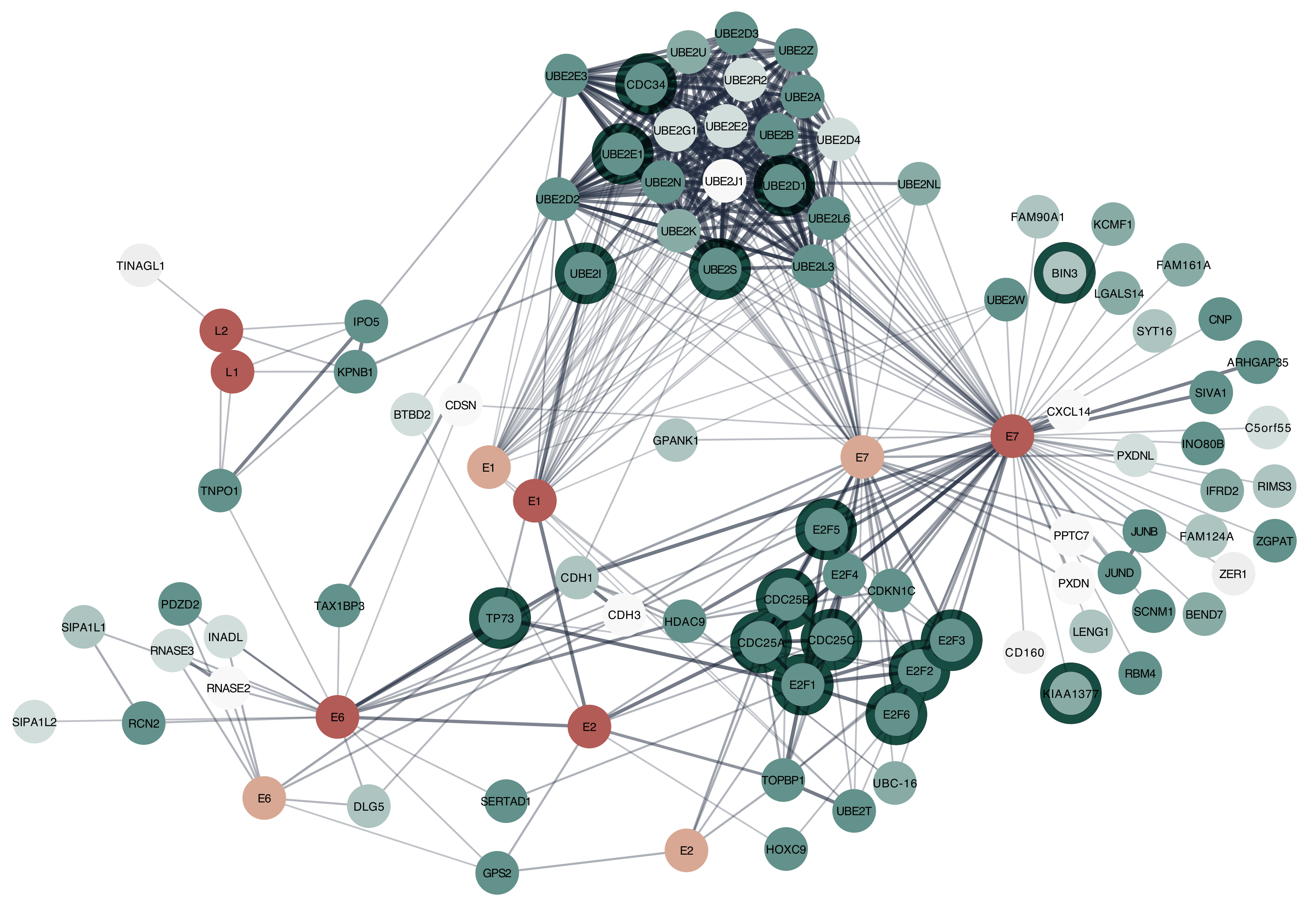
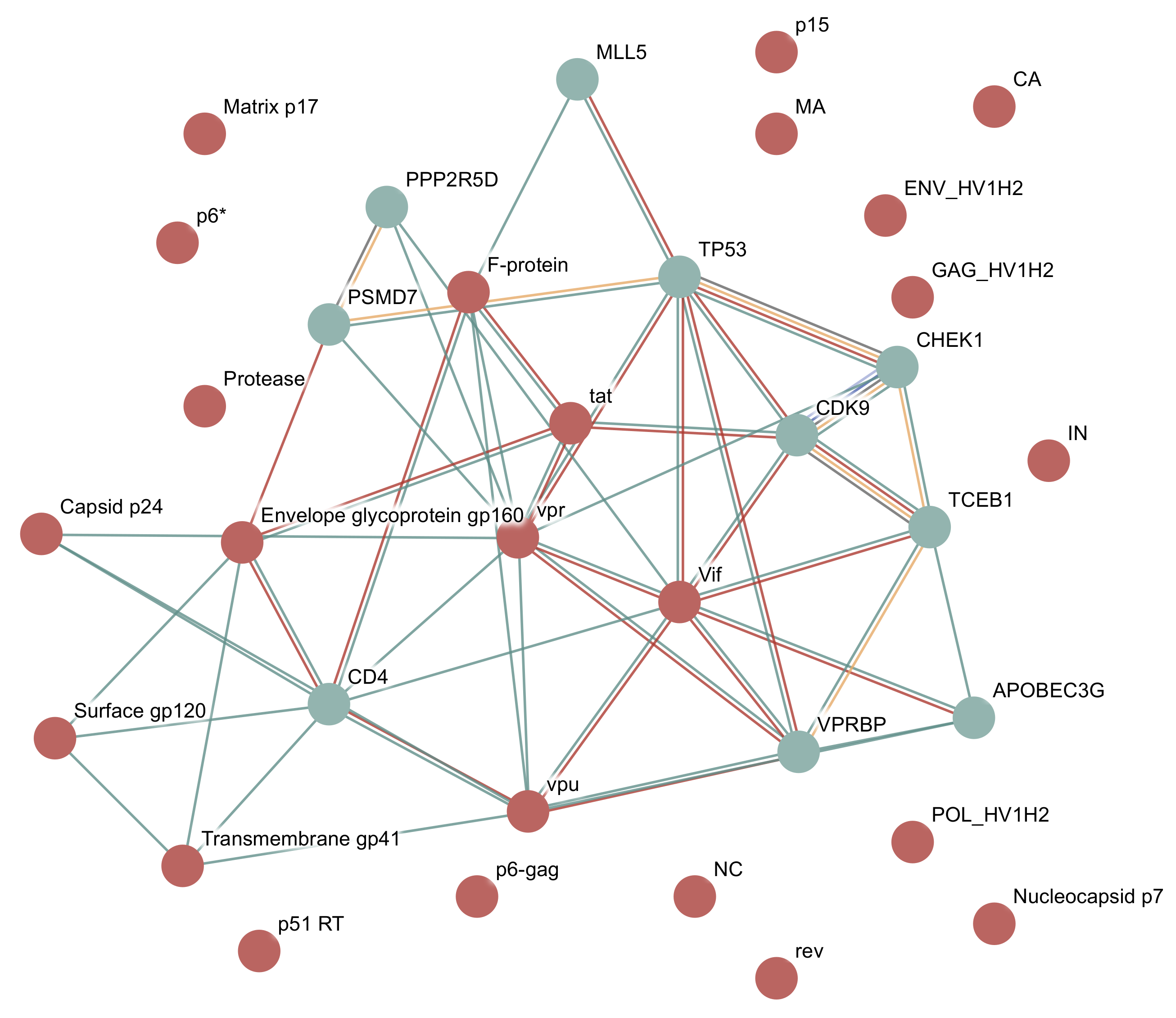
3.1.2. Example: HIV-1
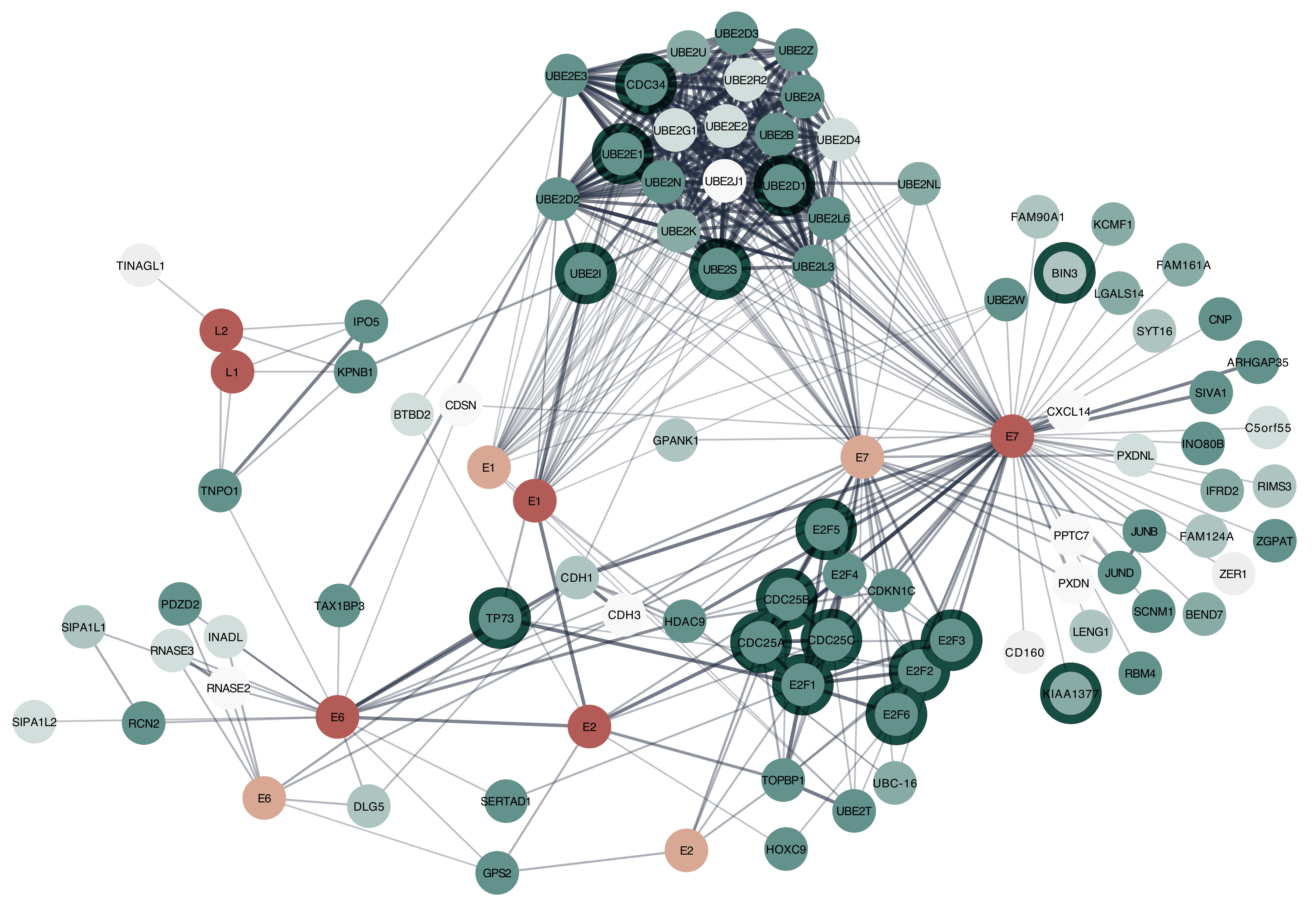
3.1.3. Cytoscape STRING App
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCMV | Human cytomegalovirus |
| HIV | Human immunodeficiency virus |
| MCMV | Murine cytomegalovirus |
| PPI | Protein–protein interaction |
References
- WHO. WHO Fact Sheets: Influenza, HCV, HPV; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Ceters for Disease Control and Prevention, Ebola (Ebola Virus Disease). Cost of the Ebola Epidemic. Available online: https://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/cost-of-ebola.html (accessed on 7 July 2017).
- Molinari, N.A.M.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, J.N.; Gage, K.L.; Khan, A.S. Potential influence of climate change on vector-borne and zoonotic diseases: A review and proposed research plan. Environ. Health Perspect. 2010, 118, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Morens, D.M. Zika Virus in the Americas—Yet Another Arbovirus Threat. N. Engl. J. Med. 2016, 374, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Crameri, G. Emerging zoonotic viral diseases. Rev. Sci. Tech. Off. Int. Epizoot. 2014, 33, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Maynard, N.D.; Gutschow, M.V.; Birch, E.W.; Covert, M.W. The virus as metabolic engineer. Biotechnol. J. 2010, 5, 686–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbahce, N.; Yan, H.; Dricot, A.; Padi, M.; Byrdsong, D.; Franchi, R.; Lee, D.S.; Rozenblatt-Rosen, O.; Mar, J.C.; Calderwood, M.A.; et al. Viral Perturbations of Host Networks Reflect Disease Etiology. PLoS Comput. Biol. 2012, 8, e1002531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Frentz, D.; Boucher, C.A.B.; Van De Vijver, D.A.M.C. Temporal changes in the epidemiology of transmission of drug-resistant HIV-1 across the world. AIDS Rev. 2012, 14, 17–27. [Google Scholar] [PubMed]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Herpesvirus Resistance to Antiviral Drugs. In Antimicrobial Drug Resistance; Number 18; Academic Press: New York, NY, USA, 2009; pp. 171–181. [Google Scholar]
- Murali, T.M.; Dyer, M.D.; Badger, D.; Tyler, B.M.; Katze, M.G. Network-based prediction and analysis of HIV dependency factors. PLoS Comput. Biol. 2011, 7, e1002164. [Google Scholar] [CrossRef] [PubMed]
- Ehreth, J. The global value of vaccination. Vaccine 2003, 21, 596–600. [Google Scholar] [CrossRef]
- Soema, P.C.; Kompier, R.; Amorij, J.P.; Kersten, G.F.A. Current and next generation influenza vaccines: Formulation and production strategies. Eur. J. Pharm. Biopharm. 2015, 94, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Moyle, P.M.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. ChemMedChem 2013, 8, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Calderone, A.; Licata, L.; Cesareni, G. VirusMentha: A new resource for virus–host protein interactions. Nucleic Acids Res. 2014, 43, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host-pathogen interactions. Database 2016, 2016, baw103. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z. PubMed and beyond: A survey of web tools for searching biomedical literature. Database 2011, 2011, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Attwood, T.; Agit, B.; Ellis, L. Longevity of Biological Databases. EMBnet J. 2015, 21, e803. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Pafilis, E.; Frankild, S.P.; Fanini, L.; Faulwetter, S.; Pavloudi, C.; Vasileiadou, A.; Arvanitidis, C.; Jensen, L.J. The SPECIES and ORGANISMS Resources for Fast and Accurate Identification of Taxonomic Names in Text. PLoS ONE 2013, 8, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 37, D5–D15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibbe, W.A.; Arze, C.; Felix, V.; Mitraka, E.; Bolton, E.; Fu, G.; Mungall, C.J.; Binder, J.X.; Malone, J.; Vasant, D.; et al. Disease Ontology 2015 update: An expanded and updated database of Human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2015, 43, D1071–D1078. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: New York, NY, USA, 2012. [Google Scholar]
- The UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef]
- Cook, H.V.; Berzins, R.; Rodriguez, C.L.; Cejuela, J.M.; Jensen, L.J. Creation and evaluation of a dictionary-based tagger for virus species and proteins. In Proceedings of the BioNLP 2017 Workshop, Association for Computational Linguistics, Vancouver, BC, Canada, 4 August 2017; pp. 91–98. [Google Scholar]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D15. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project - IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwínski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.; Soloyvov, A.; Ojeda-Flores, R.; Arrigio, N.C.; Islam, A.; Kahn, S.A.; et al. A Strategy to Estimate Unknown Viral Diversity in Mammals. mBio 2013, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Palasca, O.; Santos, A.; Stolte, C.; Gorodkin, J.; Jensen, L.J. TISSUES 2.0: An integrative web resource on mammalian tissue expression. Database 2018, 2018, bay003. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Henno, L.; Toots, M.; Ustav, M.; Ustav, M. The cell cycle timing of human papillomavirus DNA replication. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De Chassey, B.; Meyniel-Schicklin, L.; Vonderscher, J.; André, P.; Lotteau, V. Virus-host interactomics: New insights and opportunities for antiviral drug discovery. Genome Med. 2014, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, B.; Li, W.; Patil, G.; Liu, L.; Dorf, M.E.; Li, S. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat. Commun. 2017, 8, 13876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaballa, A.; Newton, G.L.; Antelmann, H.; Parsonage, D.; Upton, H.; Rawat, M.; Claiborne, A.; Fahey, R.C.; Helmann, J.D. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. USA 2010, 107, 6482–6486. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Genes | FDR p-Value | GO Term |
|---|---|---|
| 549 | 2.28E-104 | positive regulation of macromolecule metabolic process |
| 718 | 5.96E-93 | positive regulation of cellular process |
| 452 | 3.02E-91 | cell surface receptor signaling pathway |
| 758 | 7.87E-91 | protein binding |
| 779 | 1.23E-90 | positive regulation of biological process |
| 539 | 1.61E-87 | positive regulation of cellular metabolic process |
| 459 | 3.86E-84 | multi-organism process |
| 277 | 4.56E-82 | innate immune response |
| 468 | 5.27E-82 | carbohydrate derivative binding |
| 595 | 1.99E-81 | response to stress |
| 240 | 2.07E-81 | multi-organism cellular process |
| 254 | 2.4E-81 | regulation of immune response |
| 239 | 2.55E-81 | viral process |
| 583 | 4.17E-81 | regulation of response to stimulus |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cook, H.V.; Doncheva, N.T.; Szklarczyk, D.; Von Mering, C.; Jensen, L.J. Viruses.STRING: A Virus-Host Protein-Protein Interaction Database. Viruses 2018, 10, 519. https://doi.org/10.3390/v10100519
Cook HV, Doncheva NT, Szklarczyk D, Von Mering C, Jensen LJ. Viruses.STRING: A Virus-Host Protein-Protein Interaction Database. Viruses. 2018; 10(10):519. https://doi.org/10.3390/v10100519
Chicago/Turabian StyleCook, Helen Victoria, Nadezhda Tsankova Doncheva, Damian Szklarczyk, Christian Von Mering, and Lars Juhl Jensen. 2018. "Viruses.STRING: A Virus-Host Protein-Protein Interaction Database" Viruses 10, no. 10: 519. https://doi.org/10.3390/v10100519
APA StyleCook, H. V., Doncheva, N. T., Szklarczyk, D., Von Mering, C., & Jensen, L. J. (2018). Viruses.STRING: A Virus-Host Protein-Protein Interaction Database. Viruses, 10(10), 519. https://doi.org/10.3390/v10100519