RNA Interference Therapies for an HIV-1 Functional Cure


Abstract
1. Introduction
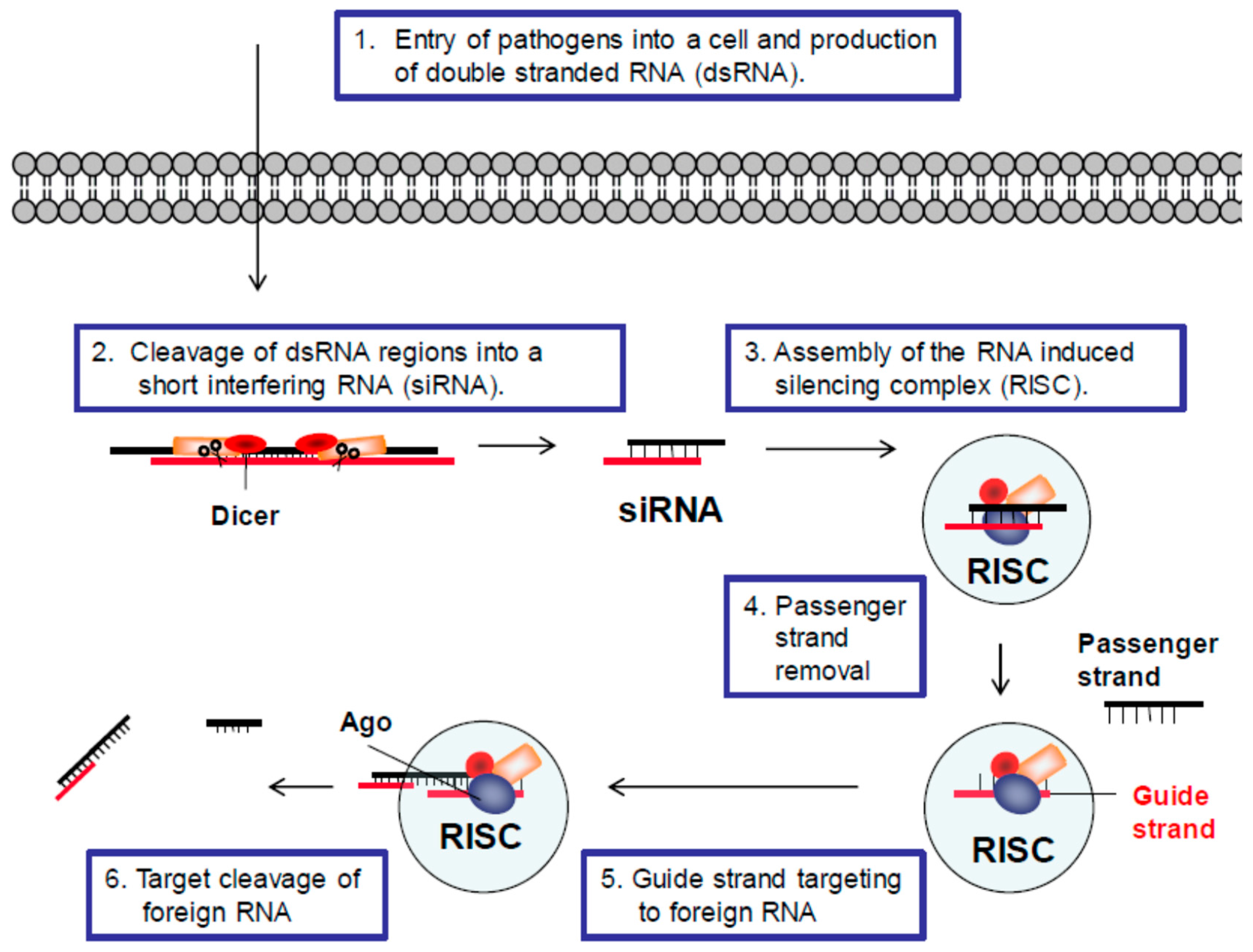
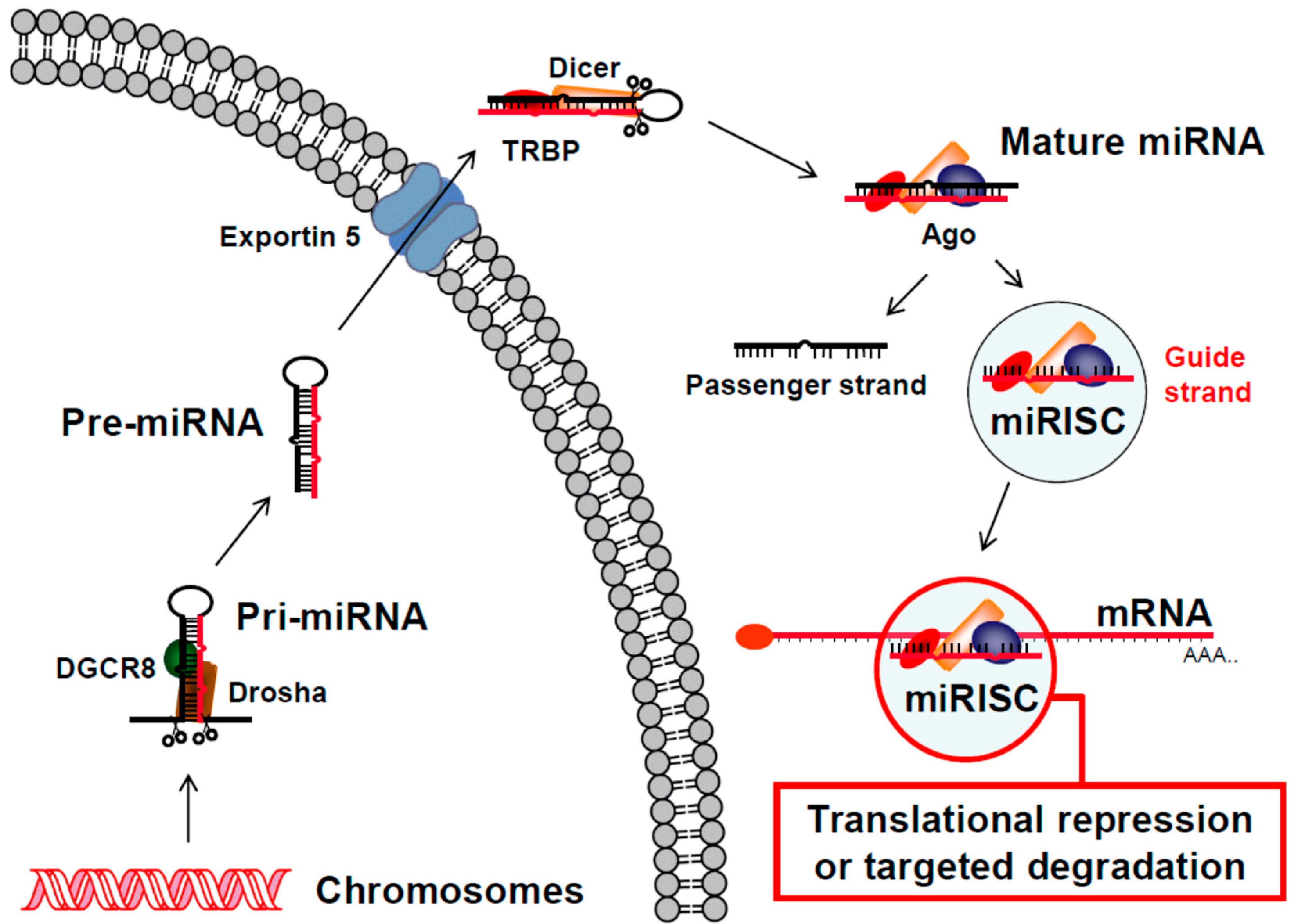
2. RNA Interference (RNAi) Pathways
2.1. RNAi Defense Mechanism
2.2. RNAi Post-Transcriptional Gene Regulation Mechanism
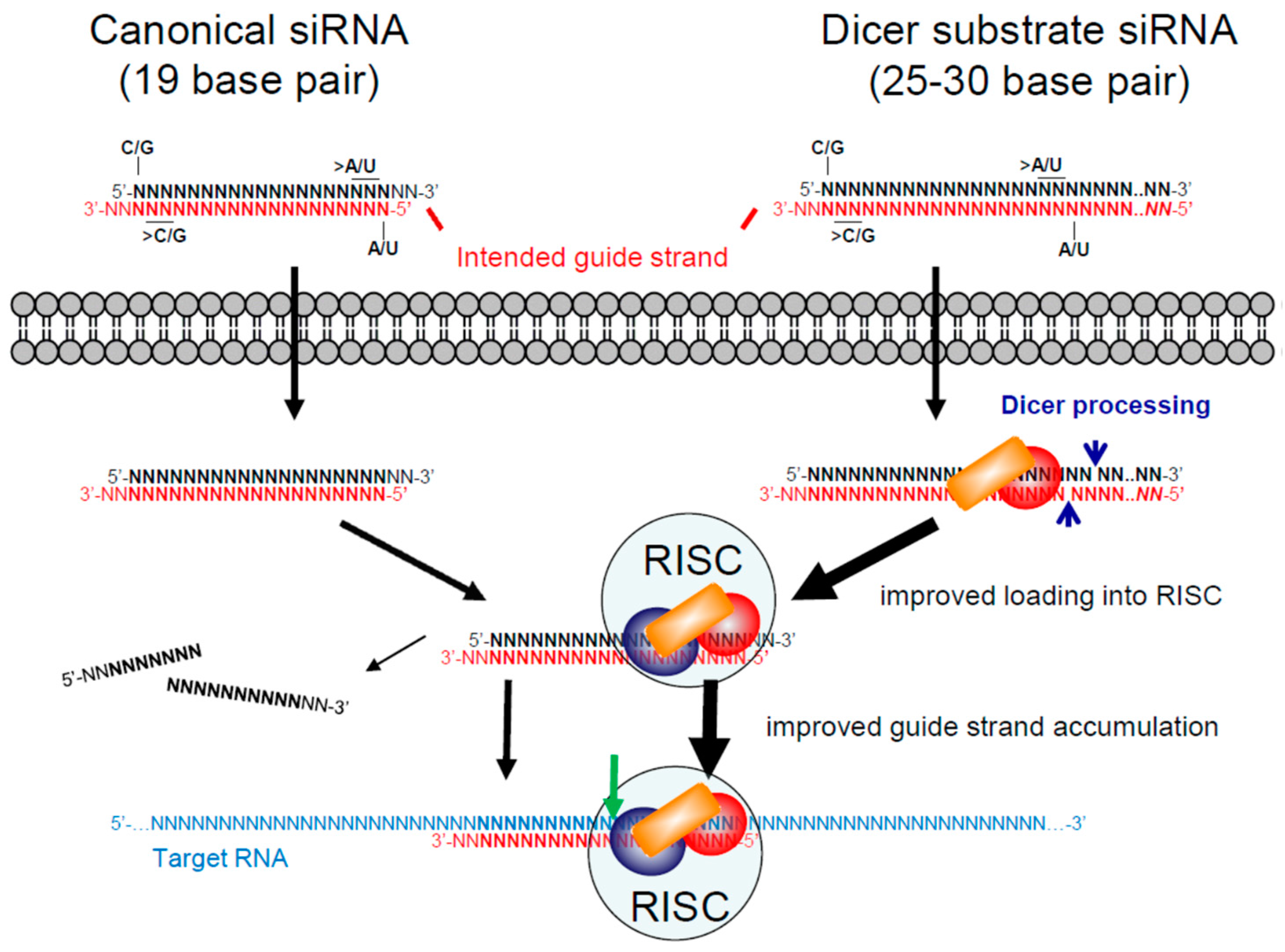
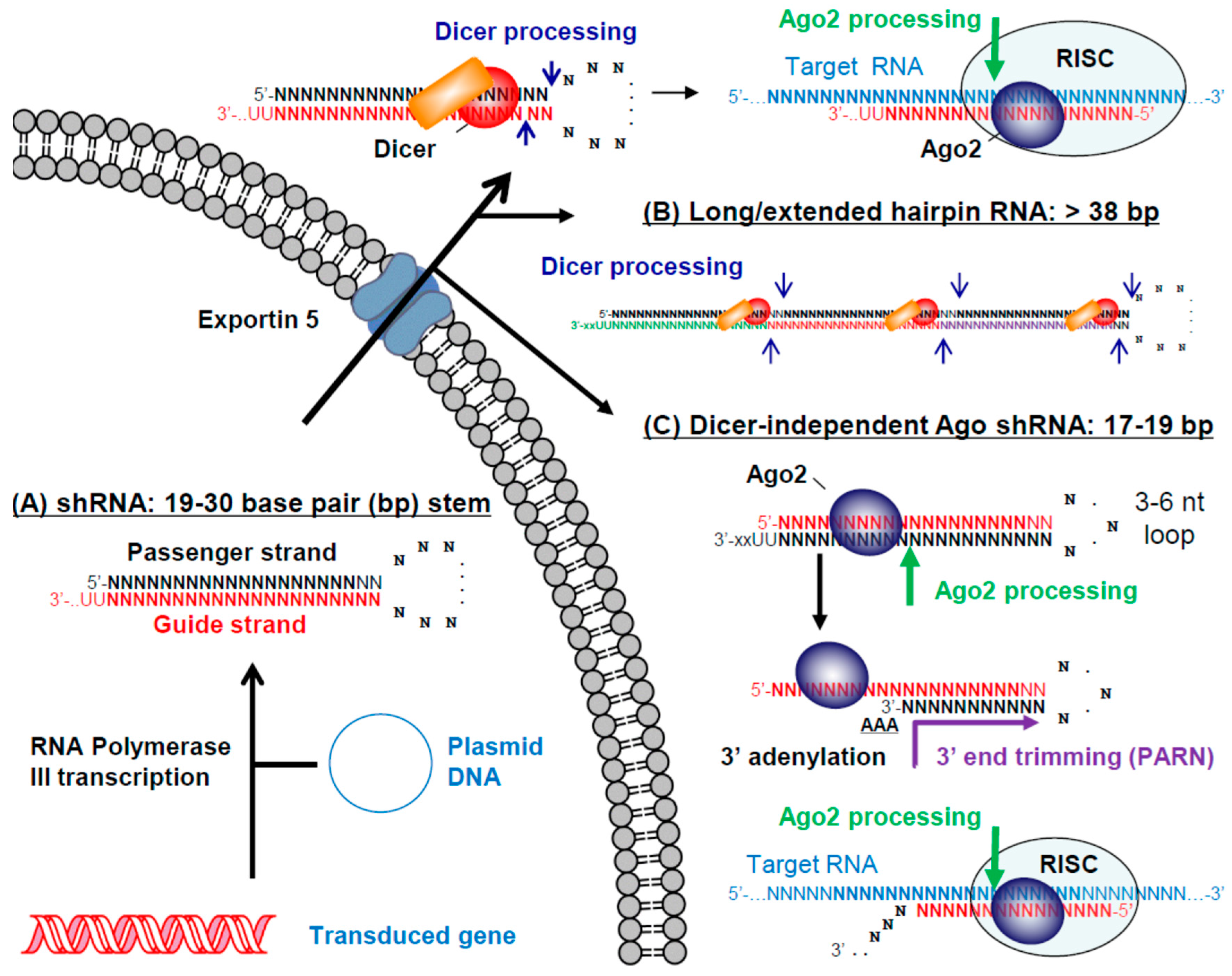
3. RNAi Therapeutics
3.1. Small/Short Interfering RNAs (siRNAs)
3.2. Small/Short Hairpin RNAs (shRNAs)
4. RNAi Therapies for HIV-1
4.1. Anti-HIV-1 siRNAs
4.2. Anti-HIV-1 shRNAs
4.2.1. shRNAs Targeting Human Genes
4.2.2. shRNAs Targeting HIV-1 RNA
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Clercq, E. Antiretroviral drugs. Curr. Opin. Pharmacol. 2010, 10, 507–615. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.T.; Grant, P.M.; Zolopa, A.R. Evolution of HIV treatment guidelines in high- and low-income countries: Converging recommendations. Antivir. Res. 2014, 103, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Tsibris, A.M.; Hirsch, M.S. Antiretroviral therapy in the clinic. J. Virol. 2010, 84, 5458–5464. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [PubMed]
- Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; Mascio, M.D.; Katlama, C.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the effort to cure HIV infection: Going beyond N = 1. J. Clin. Investig. 2016, 126, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Gay, H.; Ziemniak, C.; Sanborn, K.B.; Somasundaran, M.; Rainwater-Lovett, K.; Mellors, J.W.; Rosenbloom, D.; Persaud, D. Viremic relapse after HIV-1 remission in a perinatally infected child. N. Engl. J. Med. 2015, 372, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Persaud, D.; Gay, H.; Ziemniak, C.; Chen, Y.H.; Piatak, M., Jr.; Chun, T.W.; Strain, M.; Richman, D.; Luzuriaga, K. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N. Engl. J. Med. 2013, 369, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Frange, P.; Faye, A.; Avettand-Fenoel, V.; Bellaton, E.; Descamps, D.; Angin, M.; David, A.; Caillat-Zucman, S.; Peytavin, G.; Dollfus, C.; et al. HIV-1 virological remission lasting more than 12 years after interruption of early antiretroviral therapy in a perinatally infected teenager enrolled in the French ANRS EPF-CO10 paediatric cohort: A case report. Lancet HIV 2016, 3, e49–e54. [Google Scholar] [CrossRef]
- Rouzioux, C.; Hocqueloux, L.; Saez-Cirion, A. Posttreatment controllers: What do they tell us? Curr. Opin. HIV AIDS 2015, 10, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Saez-Cirion, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Valente, S.T. Didehydro-Cortistatin A: A new player in HIV-therapy? Expert Rev. Anti-Infect. Ther. 2016, 14, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed]
- Leal, L.; Lucero, C.; Gatell, J.M.; Gallart, T.; Plana, M.; Garcia, F. New challenges in therapeutic vaccines against HIV infection. Expert Rev. Vaccines 2017, 16, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G. Stem cell transplantation in strategies for curing HIV/AIDS. AIDS Res. Ther. 2016, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Baltimore, D. Gene therapy. Intracellular immunization. Nature 1988, 335, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Woffendin, C.; Ranga, U.; Yang, Z.; Xu, L.; Nabel, G.J. Expression of a protective gene-prolongs survival of T cells in human immunodeficiency virus-infected patients. Proc. Natl. Acad. Sci. USA 1996, 93, 2889–2894. [Google Scholar] [CrossRef] [PubMed]
- Ranga, U.; Woffendin, C.; Verma, S.; Xu, L.; June, C.H.; Bishop, D.K.; Nabel, G.J. Enhanced T cell engraftment after retroviral delivery of an antiviral gene in HIV-infected individuals. Proc. Natl. Acad. Sci. USA 1998, 95, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Wong-Staal, F.; Poeschla, E.M.; Looney, D.J. A controlled, Phase 1 clinical trial to evaluate the safety and effects in HIV-1 infected humans of autologous lymphocytes transduced with a ribozyme that cleaves HIV-1 RNA. Hum. Gene Ther. 1998, 9, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Mitsuyasu, R.T.; Symonds, G.; Rosenblatt, J.D.; Zack, J.; Sun, L.Q.; Miller, M.; Ely, J.; Gerlach, W. A phase I trial of autologous CD34+ hematopoietic progenitor cells transduced with an anti-HIV ribozyme. Hum. Gene Ther. 1999, 10, 2255–2270. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar] [PubMed]
- Podsakoff, G.M.; Engel, B.C.; Carbonaro, D.A.; Choi, C.; Smogorzewska, E.M.; Bauer, G.; Selander, D.; Csik, S.; Wilson, K.; Betts, M.R.; et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34(+) cells. Mol. Ther. 2005, 12, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Walker, R.; Carter, C.S.; Natarajan, V.; Tavel, J.A.; Bechtel, C.; Herpin, B.; Muul, L.; Zheng, Z.; Jagannatha, S.; et al. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum. Gene Ther. 2005, 16, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.M.; de Witte, M.; Malech, H.; Morgan, R.A.; Phang, S.; Carter, C.; Leitman, S.F.; Childs, R.; Barrett, A.J.; Little, R.; et al. Nonmyeloablative conditioning followed by transplantation of genetically modified HLA-matched peripheral blood progenitor cells for hematologic malignancies in patients with acquired immunodeficiency syndrome. Blood 2002, 99, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, J.; Washington, K.; Uchida, N.; Phang, O.; Kang, E.M.; Hsieh, M.M.; Tisdale, J.F. Long-term vector integration site analysis following retroviral mediated gene transfer to hematopoietic stem cells for the treatment of HIV infection. PLoS ONE 2009, 4, e4211. [Google Scholar] [CrossRef] [PubMed]
- Van Lunzen, J.; Glaunsinger, T.; Stahmer, I.; von Baehr, V.; Baum, C.; Schilz, A.; Kuehlcke, K.; Naundorf, S.; Martinius, H.; Hermann, F.; et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 2007, 15, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Cannon, P.M.; Holmes, M.C.; Li, L.; Rao, A.; Wang, J.; Lee, G.; Gregory, P.D.; Kim, K.A.; Hayward, S.B.; et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther. Methods Clin. Dev. 2016, 3, 16067. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Mitsuyasu, R.T.; Rosenblatt, J.D.; Ngok, F.K.; Bakker, A.; Cole, S.; Chorn, N.; Lin, L.S.; Bristol, G.; Boyd, M.P.; et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: Myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum. Gene Ther. 2004, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Michienzi, A.; Castanotto, D.; Lee, N.; Li, S.; Zaia, J.A.; Rossi, J.J. RNA-mediated inhibition of HIV in a gene therapy setting. Ann. N. Y. Acad. Sci. 2003, 1002, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R.; et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Binder-Scholl, G.; Mukherjee, R.; Brady, T.; Rebello, T.; Humeau, L.; Kalos, M.; Papasavvas, E.; Montaner, L.J.; et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 2013, 121, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E.; Chen, R.X.; McGee, J.; Nacey, C.; Pollard, R.B.; Abedi, M.; Bauer, G.; Nolta, J.A.; Anderson, J.S. Generation of an HIV-1-resistant immune system with CD34(+) hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J. Virol. 2012, 86, 5719–5729. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Akkina, R. Human immunodeficiency virus type 1 restriction by human-rhesus chimeric tripartite motif 5α (TRIM 5α) in CD34(+) cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum. Gene Ther. 2008, 19, 217–228. [Google Scholar] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Bone Marrow Gene Therapy for HIV/AIDS. Viruses 2015, 7, 3910–3936. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, H.; Gao, Y.; Chen, Z.; Xie, L.; Liu, Y.; Liu, Y.; Wang, X.; Li, H.; Lai, W.; et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol. Ther. 2017, 25, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.M.; Sinck, L.; Ward, N.J.; Melendez-Pena, C.E.; Scarborough, R.J.; Azar, I.; Rance, E.; Daher, A.; Pang, K.M.; Rossi, J.J.; et al. HIV-1 RRE RNA acts as an RNA silencing suppressor by competing with TRBP-bound siRNAs. RNA Biol. 2015, 12, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Scherer, L.J.; Gu, A.; Gardner, A.M.; Torres-Coronado, M.; Epps, E.W.; Digiusto, D.L.; Rossi, J.J. Optimized lentiviral vectors for HIV gene therapy: Multiplexed expression of small RNAs and inclusion of MGMT(P140K) drug resistance gene. Mol. Ther. 2014, 22, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Shum, K.T.; Zhou, J.; Rossi, J.J. Aptamer-based therapeutics: New approaches to combat human viral diseases. Pharmaceuticals (Basel) 2013, 6, 1507–1542. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.J.; Burke, D.H. Screening inhibitory potential of anti-HIV RT RNA aptamers. Methods Mol. Biol. 2014, 1103, 11–29. [Google Scholar] [PubMed]
- Duclair, S.; Gautam, A.; Ellington, A.; Prasad, V.R. High-affinity RNA Aptamers Against the HIV-1 Protease Inhibit Both In Vitro Protease Activity and Late Events of Viral Replication. Mol. Ther. Nucleic Acids 2015, 4, e228. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.M.; Castanotto, D.; Li, H.; Scherer, L.; Rossi, J.J. Incorporation of aptamers in the terminal loop of shRNAs yields an effective and novel combinatorial targeting strategy. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, D.; Duclair, S.; Datta, S.A.; Ellington, A.; Rein, A.; Prasad, V.R. RNA aptamers directed to human immunodeficiency virus type 1 Gag polyprotein bind to the matrix and nucleocapsid domains and inhibit virus production. J. Virol. 2011, 85, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.J.; Gatignol, A. HIV and Ribozymes. Adv. Exp. Med. Biol. 2015, 848, 97–116. [Google Scholar] [PubMed]
- Miyagishi, M.; Hayashi, M.; Taira, K. Comparison of the suppressive effects of antisense oligonucleotides and siRNAs directed against the same targets in mammalian cells. Antisense Nucleic Acid Drug Dev. 2003, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.-W.; Voinnet, O. Antiviral Immunity Directed by Small RNAs. Cell 2007, 130, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.M.; Gatignol, A. The multiple functions of TRBP, at the hub of cell responses to viruses, stress, and cancer. Microbiol. Mol. Biol. Rev. 2012, 76, 652–666. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- TenOever, B.R. Questioning antiviral RNAi in mammals. Nat. Microbiol. 2017, 2, 17052. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R.; Cherry, S.; tenOever, B.R. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell Host Microbe 2013, 14, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, J.; Han, Y.; Fan, X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013, 342, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; Ciaudo, C.; Marchais, A.; Li, Y.; Jay, F.; Ding, S.W.; Voinnet, O. Antiviral RNA interference in mammalian cells. Science 2013, 342, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Sagan, S.M.; Sarnow, P. Molecular biology. RNAi, Antiviral after all. Science 2013, 342, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Basavappa, M.; Lu, J.; Dong, S.; Cronkite, D.A.; Prior, J.T.; Reinecker, H.C.; Hertzog, P.; Han, Y.; Li, W.X.; et al. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat. Microbiol. 2016, 2, 16250. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; van der Veen, A.G.; Deddouche-Grass, S.; Rogers, N.C.; Merits, A.; Reis, E.S.C. Inactivation of the type I interferon pathway reveals long double-stranded RNA-mediated RNA interference in mammalian cells. EMBO J. 2016, 35, 2505–2518. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.M.; Melendez-Peña, C.E.; Scarborough, R.J.; Daher, A.; Christensen, H.S.; El Far, M.; Purcell, D.F.; Lainé, S.; Gatignol, A. Characterization of the TRBP domain required for dicer interaction and function in RNA interference. BMC Mol. Biol. 2009, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Plante, I.; Landry, P.; Barat, C.; Janelle, M.E.; Flamand, L.; Tremblay, M.J.; Provost, P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008, 36, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Suhr, O.B.; Dyck, P.J.; Litchy, W.J.; Leahy, R.G.; Chen, J.; Gollob, J.; Coelho, T. Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017, 17, 181. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Ui-Tei, K.; Zenno, S.; Miyata, Y.; Saigo, K. Sensitive assay of RNA interference in Drosophila and Chinese hamster cultured cells using firefly luciferase gene as target. FEBS Lett. 2000, 479, 79–82. [Google Scholar] [CrossRef]
- Caplen, N.J.; Fleenor, J.; Fire, A.; Morgan, R.A. dsRNA-mediated gene silencing in cultured Drosophila cells: A tissue culture model for the analysis of RNA interference. Gene 2000, 252, 95–105. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Parrish, S.; Fleenor, J.; Xu, S.; Mello, C.; Fire, A. Functional anatomy of a dsRNA trigger: Differential requirement for the two trigger strands in RNA interference. Mol. Cell 2000, 6, 1077–1087. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Caplen, N.J.; Parrish, S.; Imani, F.; Fire, A.; Morgan, R.A. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl. Acad. Sci. USA 2001, 98, 9742–9747. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Petri, S.; Meister, G. siRNA design principles and off-target effects. Methods Mol. Biol. 2013, 986, 59–71. [Google Scholar] [PubMed]
- Kim, D.H.; Behlke, M.A.; Rose, S.D.; Chang, M.S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.D.; Kim, D.H.; Amarzguioui, M.; Heidel, J.D.; Collingwood, M.A.; Davis, M.E.; Rossi, J.J.; Behlke, M.A. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005, 33, 4140–4156. [Google Scholar] [CrossRef] [PubMed]
- Amarzguioui, M.; Lundberg, P.; Cantin, E.; Hagstrom, J.; Behlke, M.A.; Rossi, J.J. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat. Protoc. 2006, 1, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Snead, N.M.; Wu, X.; Li, A.; Cui, Q.; Sakurai, K.; Burnett, J.C.; Rossi, J.J. Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res. 2013, 41, 6209–6221. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Barros, S.; Duncan, R.; Shaikh, S.; Cantley, W.; Dell, A.; Bulgakova, E.; O’Shea, J.; Taneja, N.; Kuchimanchi, S.; et al. Comprehensive evaluation of canonical versus Dicer-substrate siRNA in vitro and in vivo. RNA 2012, 18, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.J.; Levesque, M.V.; Boudrias-Dalle, E.; Chute, I.C.; Daniels, S.M.; Ouellette, R.J.; Perreault, J.P.; Gatignol, A. A Conserved Target Site in HIV-1 Gag RNA is Accessible to Inhibition by Both an HDV Ribozyme and a Short Hairpin RNA. Mol. Ther. Nucleic Acids 2014, 3, e178. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.J.; Lévesque, M.V.; Perreault, J.P.; Gatignol, A. Design and Evaluation of Clinically Relevant SOFA-HDV Ribozymes Targeting HIV RNA. Methods Mol. Biol. 2014, 1103, 31–43. [Google Scholar] [PubMed]
- Scarborough, R.J.; Adams, K.L.; Del Corpo, O.; Daher, A.; Gatignol, A. Evaluation of the Efficacy and Toxicity of RNAs Targeting HIV-1 Production for Use in Gene or Drug Therapy. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.J.; Adams, K.L.; Daher, A.; Gatignol, A. Effective Inhibition of HIV-1 Production by Short Hairpin RNAs and Small Interfering RNAs Targeting a Highly Conserved Site in HIV-1 Gag RNA Is Optimized by Evaluating Alternative Length Formats. Antimicrob. Agents Chemother. 2015, 59, 5297–5305. [Google Scholar] [CrossRef] [PubMed]
- Snead, N.M.; Rossi, J.J. RNA interference trigger variants: Getting the most out of RNA for RNA interference-based therapeutics. Nucleic Acid Ther. 2012, 22, 139–146. [Google Scholar] [PubMed]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, L.S.; Smith, C.A. Short hairpin RNA-mediated gene silencing. Methods Mol. Biol. 2013, 942, 205–232. [Google Scholar] [PubMed]
- Ma, H.; Wu, Y.; Dang, Y.; Choi, J.-G.; Zhang, J.; Wu, H. Pol III Promoters to Express Small RNAs: Delineation of Transcription Initiation. Mol. Ther. Nucleic Acids 2014, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Harwig, A.; Berkhout, B.; Herrera-Carrillo, E. Mutation of nucleotides around the +1 position of type 3 polymerase III promoters: The effect on transcriptional activity and start site usage. Transcription 2017, 8, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. Delineation of the Exact Transcription Termination Signal for Type 3 Polymerase III. Mol. Ther. Nucleic Acids 2018, 10, 36–44. [Google Scholar] [CrossRef]
- Yu, J.Y.; Taylor, J.; DeRuiter, S.L.; Vojtek, A.B.; Turner, D.L. Simultaneous inhibition of GSK3α and GSK3β using hairpin siRNA expression vectors. Mol. Ther. 2003, 7, 228–236. [Google Scholar] [CrossRef]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Miyagishi, M.; Sumimoto, H.; Miyoshi, H.; Kawakami, Y.; Taira, K. Optimization of an siRNA-expression system with an improved hairpin and its significant suppressive effects in mammalian cells. J. Gene Med. 2004, 6, 715–723. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, G.J.; Yu, Y.H.; Lomas, M.; Fanning, G.C. The effects of stem length and core placement on shRNA activity. BMC Mol. Biol. 2011, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lin, X.; Khvorova, A.; Fesik, S.W.; Shen, Y. Defining the optimal parameters for hairpin-based knockdown constructs. RNA 2007, 13, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, R.L.; Monteys, A.M.; Davidson, B.L. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA 2008, 14, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Haasnoot, J.; ter Brake, O.; Berkhout, B.; Konstantinova, P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008, 36, 2811–2824. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.L.; Ahmed, I.; Steel, L.F. RNA polymerase III can drive polycistronic expression of functional interfering RNAs designed to resemble microRNAs. Nucleic Acids Res. 2009, 37, e127. [Google Scholar] [CrossRef] [PubMed]
- Steel, L.F.; Sanghvi, V.R. Polycistronic expression of interfering RNAs from RNA polymerase III promoters. Methods Mol. Biol. 2012, 815, 347–359. [Google Scholar] [PubMed]
- Saayman, S.; Arbuthnot, P.; Weinberg, M.S. Deriving four functional anti-HIV siRNAs from a single Pol III-generated transcript comprising two adjacent long hairpin RNA precursors. Nucleic Acids Res. 2010, 38, 6652–6663. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Haasnoot, J.; Berkhout, B. Design of extended short hairpin RNAs for HIV-1 inhibition. Nucleic Acids Res. 2007, 35, 5683–5693. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Dicer-independent processing of small RNA duplexes: Mechanistic insights and applications. Nucleic Acids Res. 2017, 45, 10369–10379. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Schopman, N.C.; Berkhout, B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013, 41, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Herrera-Carrillo, E.; Jongejan, A.; van Kampen, A.H.; Berkhout, B. Deep Sequence Analysis of AgoshRNA Processing Reveals 3′ A Addition and Trimming. Mol. Ther. Nucleic Acids 2015, 4, e247. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Harwig, A.; Berkhout, B. Silencing of HIV-1 by AgoshRNA molecules. Gene Ther. 2017, 24, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Bobbin, M.L.; Burnett, J.C.; Rossi, J.J. RNA interference approaches for treatment of HIV-1 infection. Genome Med. 2015, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.N.; Wu, H.; Shankar, P. Recent advances in RNAi-based strategies for therapy and prevention of HIV-1/AIDS. Adv. Drug Deliv. Rev. 2016, 103, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Mizrahy, S.; Hazan-Halevy, I.; Dammes, N.; Landesman-Milo, D.; Peer, D. Current Progress in Non-viral RNAi-Based Delivery Strategies to Lymphocytes. Mol. Ther. 2017, 25, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J. Future anti-HBV strategies. Liver Int. 2017, 37, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Thi, E.P.; Mire, C.E.; Lee, A.C.; Geisbert, J.B.; Zhou, J.Z.; Agans, K.N.; Snead, N.M.; Deer, D.J.; Barnard, T.R.; Fenton, K.A.; et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature 2015, 521, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, J.; Zamora, M.R.; Hodges, T.; Musk, A.W.; Sommerwerk, U.; Dilling, D.; Arcasoy, S.; DeVincenzo, J.; Karsten, V.; Shah, S.; et al. ALN-RSV01 for prevention of bronchiolitis obliterans syndrome after respiratory syncytial virus infection in lung transplant recipients. J. Heart Lung Transplant. 2016, 35, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; de Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Berkhout, B. Toward a durable treatment of HIV-1 infection using RNA interference. Prog. Mol. Biol. Transl. Sci. 2011, 102, 141–163. [Google Scholar] [PubMed]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antivir. Res. 2011, 89, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Von Eije, K.J.; ter Brake, O.; Berkhout, B. Stringent testing identifies highly potent and escape-proof anti-HIV short hairpin RNAs. J. Gene Med. 2009, 11, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Von Eije, K.J.; ter Brake, O.; Berkhout, B. Human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J. Virol. 2008, 82, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Centlivre, M.; Legrand, N.; Klamer, S.; Liu, Y.P.; Jasmijn von Eije, K.; Bohne, M.; Rijnstra, E.S.; Weijer, K.; Blom, B.; Voermans, C.; et al. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol. Ther. Nucleic Acids 2013, 2, e120. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Nohtomi, K.; Onogi, T.; Uenishi, R.; Ui-Tei, K.; Saigo, K.; Takebe, Y. Optimal design and validation of antiviral siRNA for targeting HIV-1. Retrovirology 2007, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Ui-Tei, K.; Naito, Y.; Takahashi, F.; Haraguchi, T.; Ohki-Hamazaki, H.; Juni, A.; Ueda, R.; Saigo, K. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004, 32, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Amarzguioui, M.; Prydz, H. An algorithm for selection of functional siRNA sequences. Biochem. Biophys. Res. Commun. 2004, 316, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, G.J.; Groneman, J.L.; Yu, Y.H.; Jaramillo, A.; Shen, S.; Applegate, T.L. 96 shRNAs designed for maximal coverage of HIV-1 variants. Retrovirology 2009, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Ringpis, G.E.; Shimizu, S.; Arokium, H.; Camba-Colon, J.; Carroll, M.V.; Cortado, R.; Xie, Y.; Kim, P.Y.; Sahakyan, A.; Lowe, E.L.; et al. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PLoS ONE 2012, 7, e53492. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Knoepfel, S.A.; Watts, J.M.; ter Brake, O.; Berkhout, B.; Weeks, K.M. SHAPE-directed discovery of potent shRNA inhibitors of HIV-1. Mol. Ther. 2012, 20, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Gene therapy strategies to block HIV-1 replication by RNA interference. Adv. Exp. Med. Biol. 2015, 848, 71–95. [Google Scholar] [PubMed]
- Westerhout, E.M.; Ooms, M.; Vink, M.; Das, A.T.; Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005, 33, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hutter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef] [PubMed]
- Younan, P.; Kowalski, J.; Kiem, H.P. Genetically modified hematopoietic stem cell transplantation for HIV-1-infected patients: Can we achieve a cure? Mol. Ther. 2014, 22, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Jerome, K.R.; Deeks, S.G.; McCune, J.M. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell 2012, 10, 137–147. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Stan, R.; Krishnan, A.; Li, H.; Rossi, J.J.; Zaia, J.A. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: Considerations for proof of concept studies and translation to standard medical practice. Viruses 2013, 5, 2898–2919. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiviral Gene(s) | Ex Vivo Manipulation | Status | Year | Reference |
|---|---|---|---|---|
| Proteins and peptides | ||||
| Dominant negative mutant HIV-1 Rev protein (Rev M10) | Gold particle transfected CD4+ T cells | Completed | 1996 | [22] |
| Murine retrovirus transduced CD4+ T cells | Completed | 1998 | [23] | |
| Murine retrovirus transduced HSCs | Completed | 2005 | [27] | |
| Dominant negative HIV-1 Rev protein | Murine retrovirus transduced syngeneic CD4+ T cells | Completed | 2005 | [28] |
| Murine retrovirus transduced HSCs | Completed | 2002, 2009 | [29,30] | |
| gp41 peptide, fusion inhibitor (C46) | Murine retrovirus transduced CD4+ T cells | Completed | 2007 | [31] |
| Zinc finger nuclease targeting the CCR5 gene (SB-728) | Transient expression in CD4+ T cells | NCT02225665, 01543152, 02388594 | 2014 | [32] |
| Transient expression in HSCs | NCT02500849 | 2016 | [33] | |
| CRISPR/Cas9 targeting the CCR5 gene | Transient expression in HSCs | NCT103164135 | - | - |
| RNA | ||||
| Ribozyme targeting HIV-1 tat/vpr RNA (Rz2, OZ-1) | Murine retrovirus transduced CD4+ T cells | Completed | 1998 | [24] |
| Murine retrovirus transduced syngeneic CD4+ T cells | Completed | 2005 | [34] | |
| Murine retrovirus transduced HSCs | Completed | 1999, 2004 | [25,35] | |
| Murine retrovirus transduced HSCs, Phase II | Completed | 2009 | [36] | |
| Ribozymes targeting HIV-1 RNA | Murine retrovirus transduced HSCs | Completed | 2003 | [37] |
| Antisense RNA targeting HIV-1 env RNA (VRX496) | Lentivirus (HIV) transduced CD4+ T cells | Completed | 2006 | [38] |
| Lentivirus (HIV) transduced CD4+ T cells | NCT00295477, 00131560 | 2013 | [39] | |
| RRE-decoy targeting HIV-1 Rev | Murine retrovirus transduced HSCs | Completed | 1999 | [26] |
| Combinations | ||||
| 1. shRNA: tat/rev 2. ribozyme: CCR5 3. TAR-decoy: Tat | Lentivirus (HIV) transduced HSCs | NCT01961063, 02337985, 00569985 | 2010 | [40] |
| 1. shRNA: CCR5 2. gp41 peptide (C46) | Lentivirus (HIV) transduced HSCs and CD4+ T cells | NCT01734850, 02390297 | - | - |
| 1. shRNA: CCR5 2. TRIM5α-HRH 3. TAR decoy: Tat | Lentivirus (HIV) transduced HSCs | NCT02797470 | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarborough, R.J.; Gatignol, A. RNA Interference Therapies for an HIV-1 Functional Cure. Viruses 2018, 10, 8. https://doi.org/10.3390/v10010008
Scarborough RJ, Gatignol A. RNA Interference Therapies for an HIV-1 Functional Cure. Viruses. 2018; 10(1):8. https://doi.org/10.3390/v10010008
Chicago/Turabian StyleScarborough, Robert J., and Anne Gatignol. 2018. "RNA Interference Therapies for an HIV-1 Functional Cure" Viruses 10, no. 1: 8. https://doi.org/10.3390/v10010008
APA StyleScarborough, R. J., & Gatignol, A. (2018). RNA Interference Therapies for an HIV-1 Functional Cure. Viruses, 10(1), 8. https://doi.org/10.3390/v10010008