Comparative Analysis of 37 Acinetobacter Bacteriophages

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
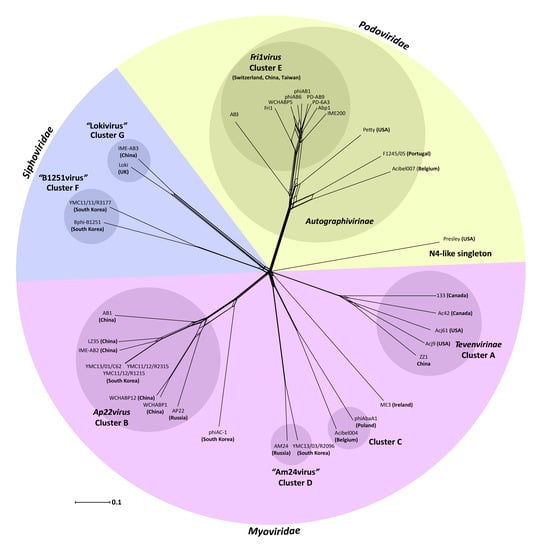
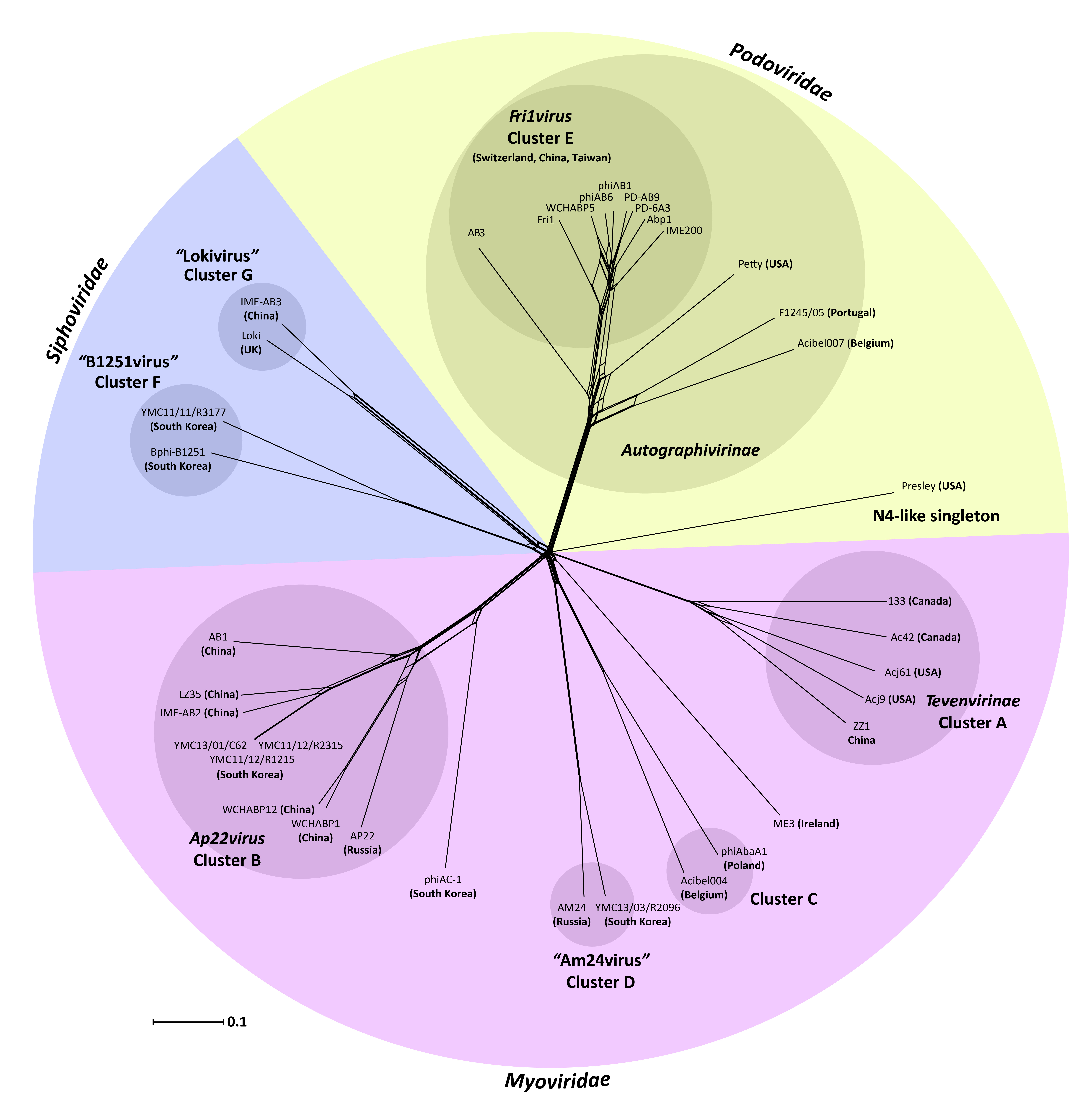
3.1. Acinetobacter Phages with Whole Genome Sequences
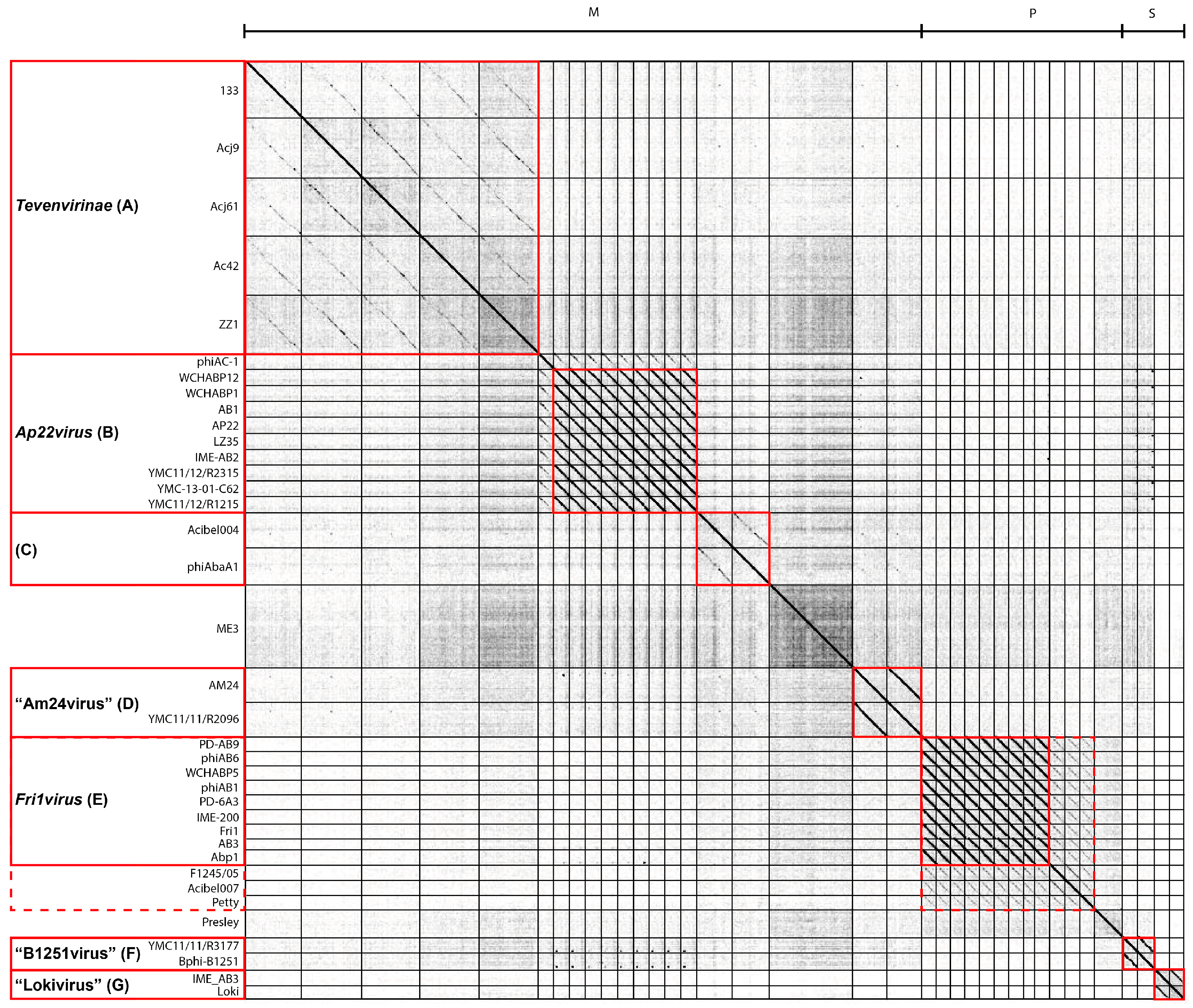
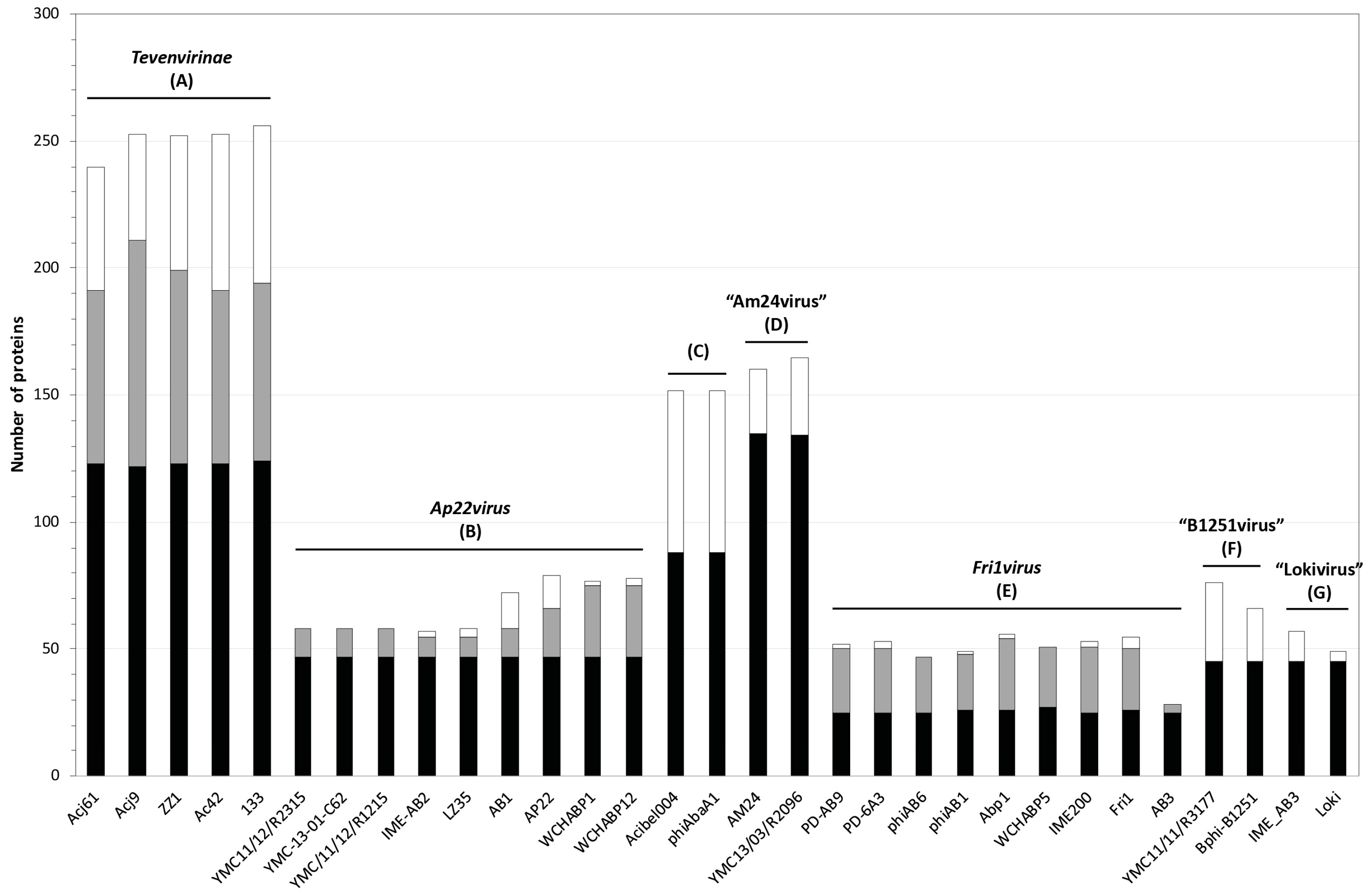
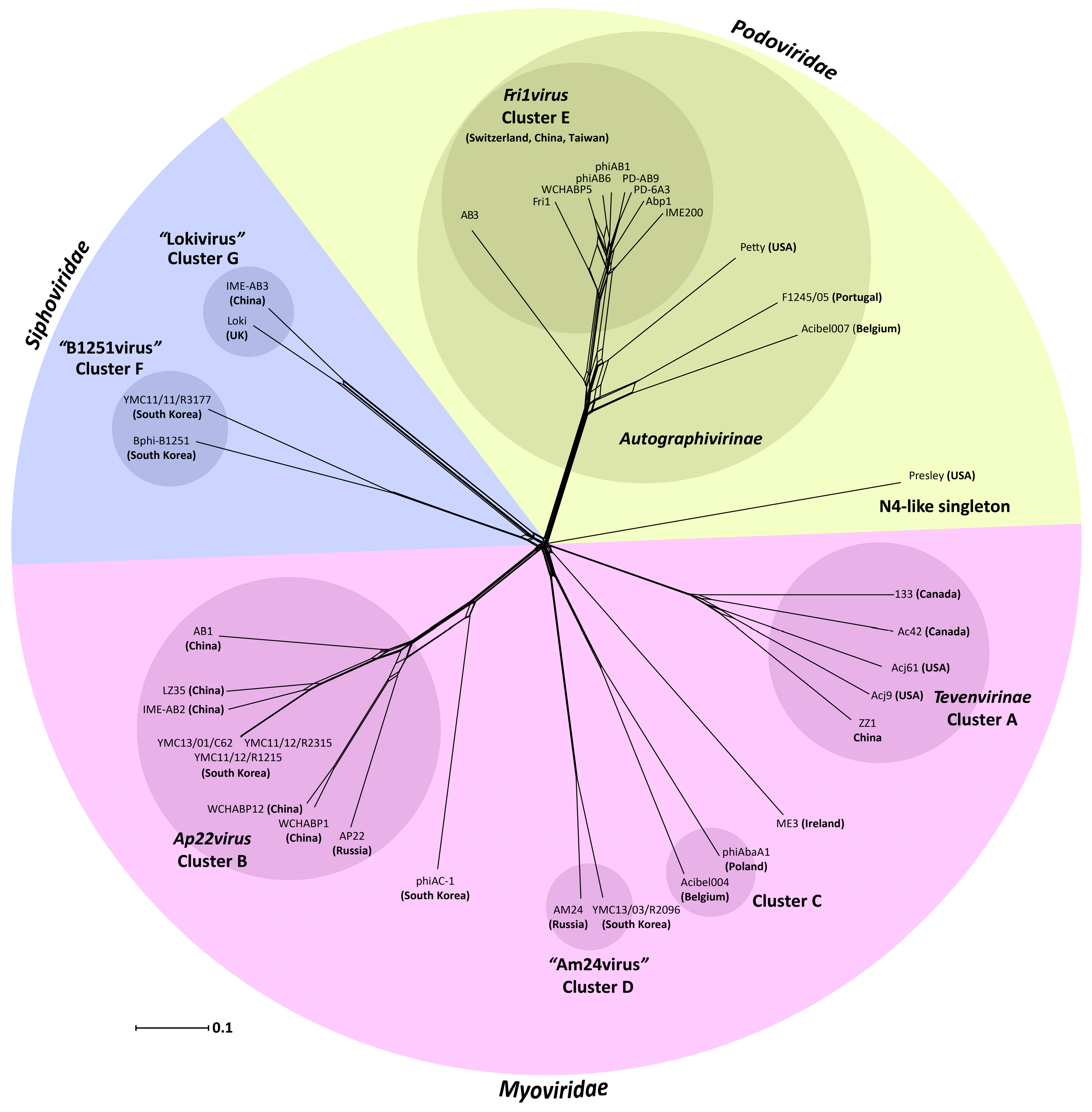
3.2. Whole Genome and Proteome Comparisons of the Acinetobacter Phages
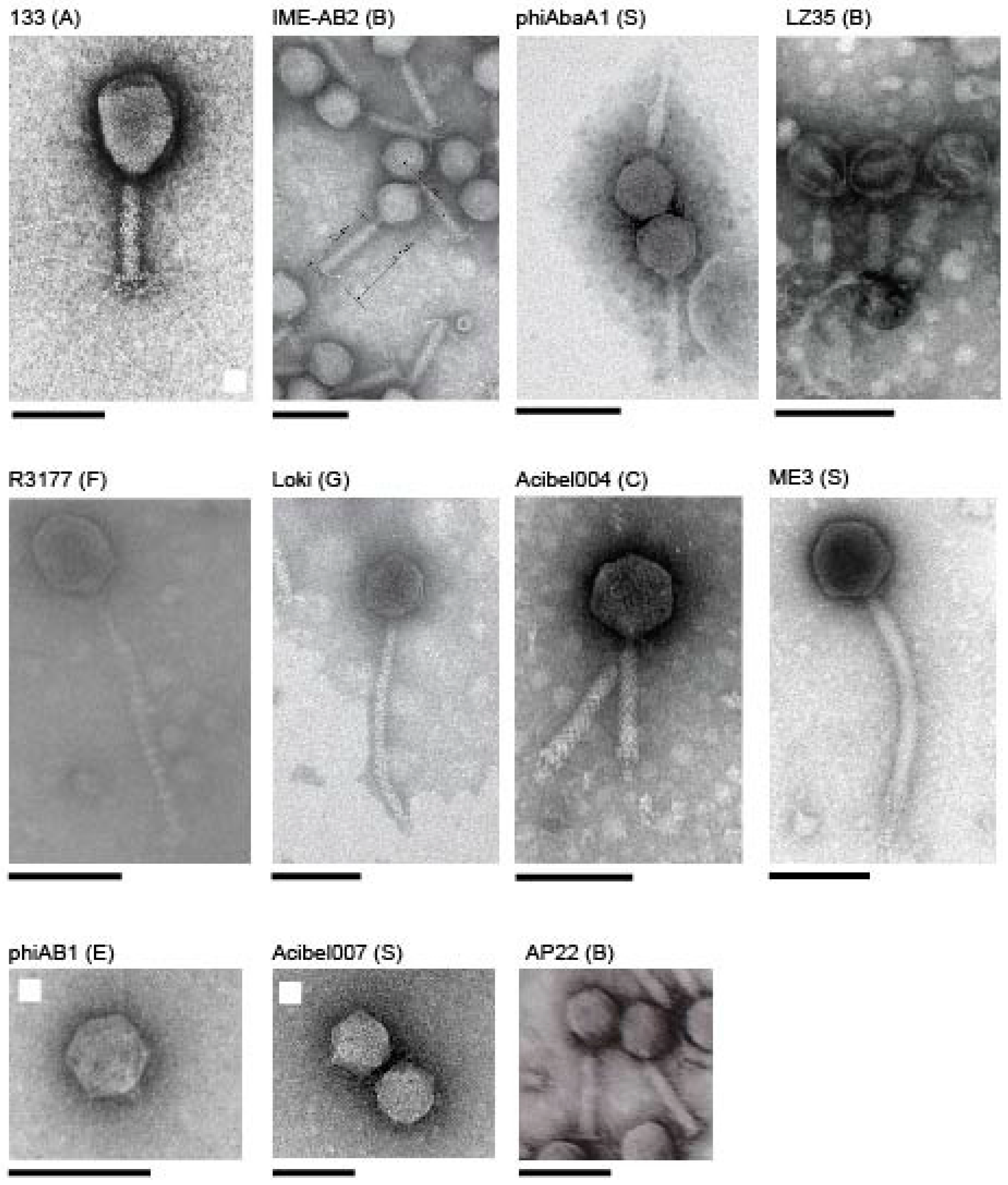
3.2.1. Cluster A: The T4-Like Acinetobacter Phages
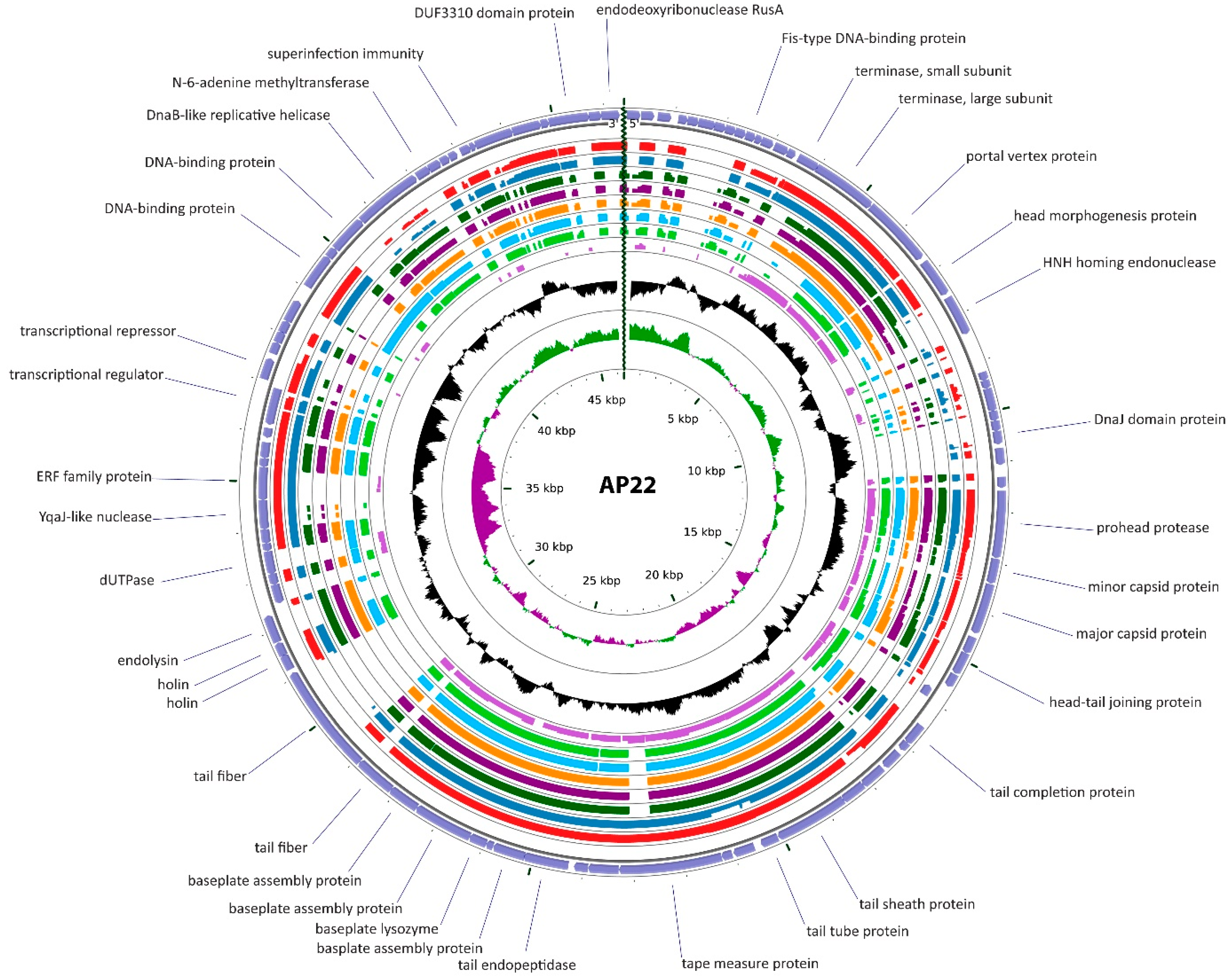
3.2.2. Cluster B: The AP22-Like Acinetobacter Myoviruses (Ap22virus)
3.2.3. Cluster C: Myoviruses Acibel004 and PhiAbaA1
3.2.4. Cluster D: Myoviruses AM24 and YMC13/03/R2096
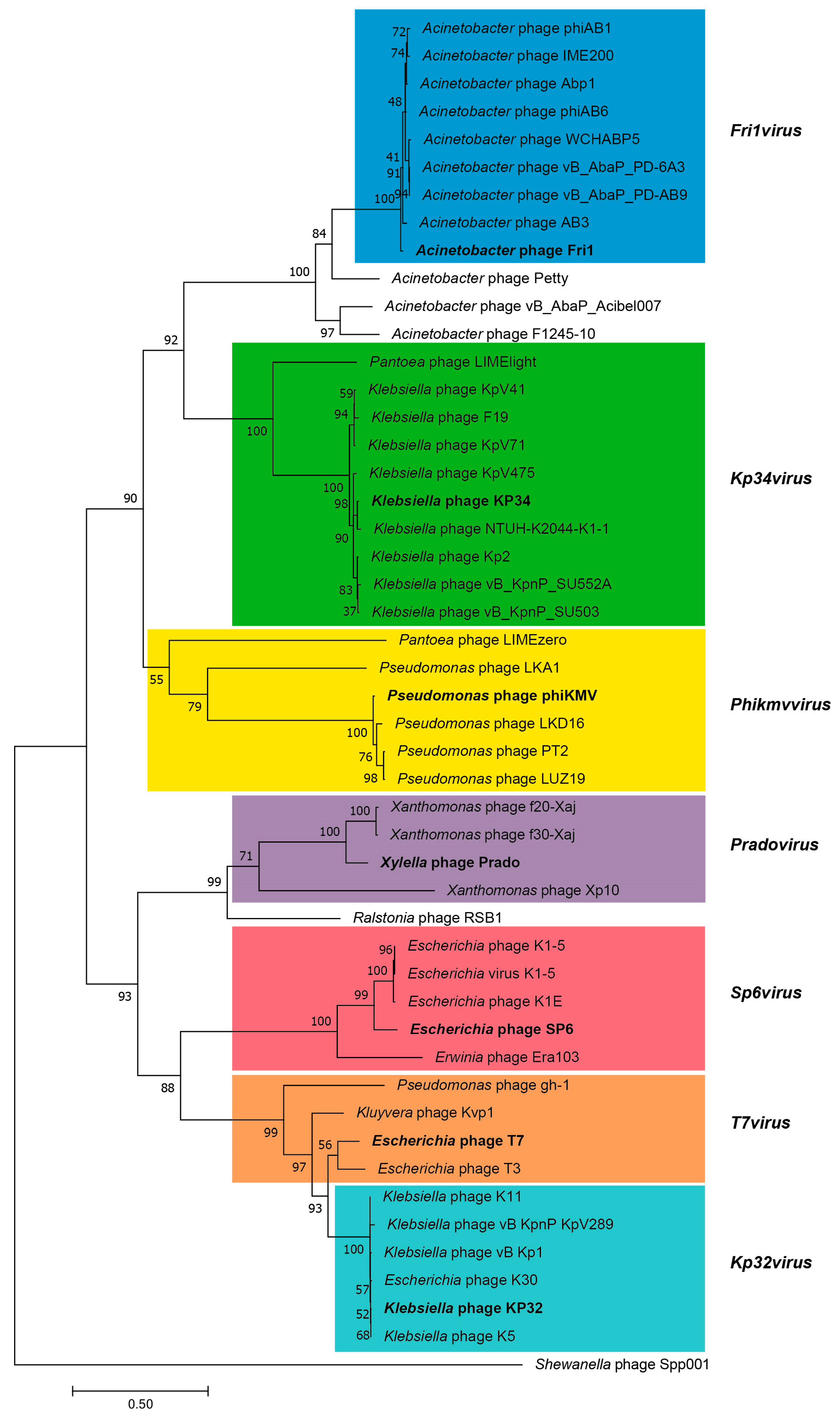
3.2.5. Cluster E: Acinetobacter Phages of the Subfamily Autographivirinae; Genus Fri1virus
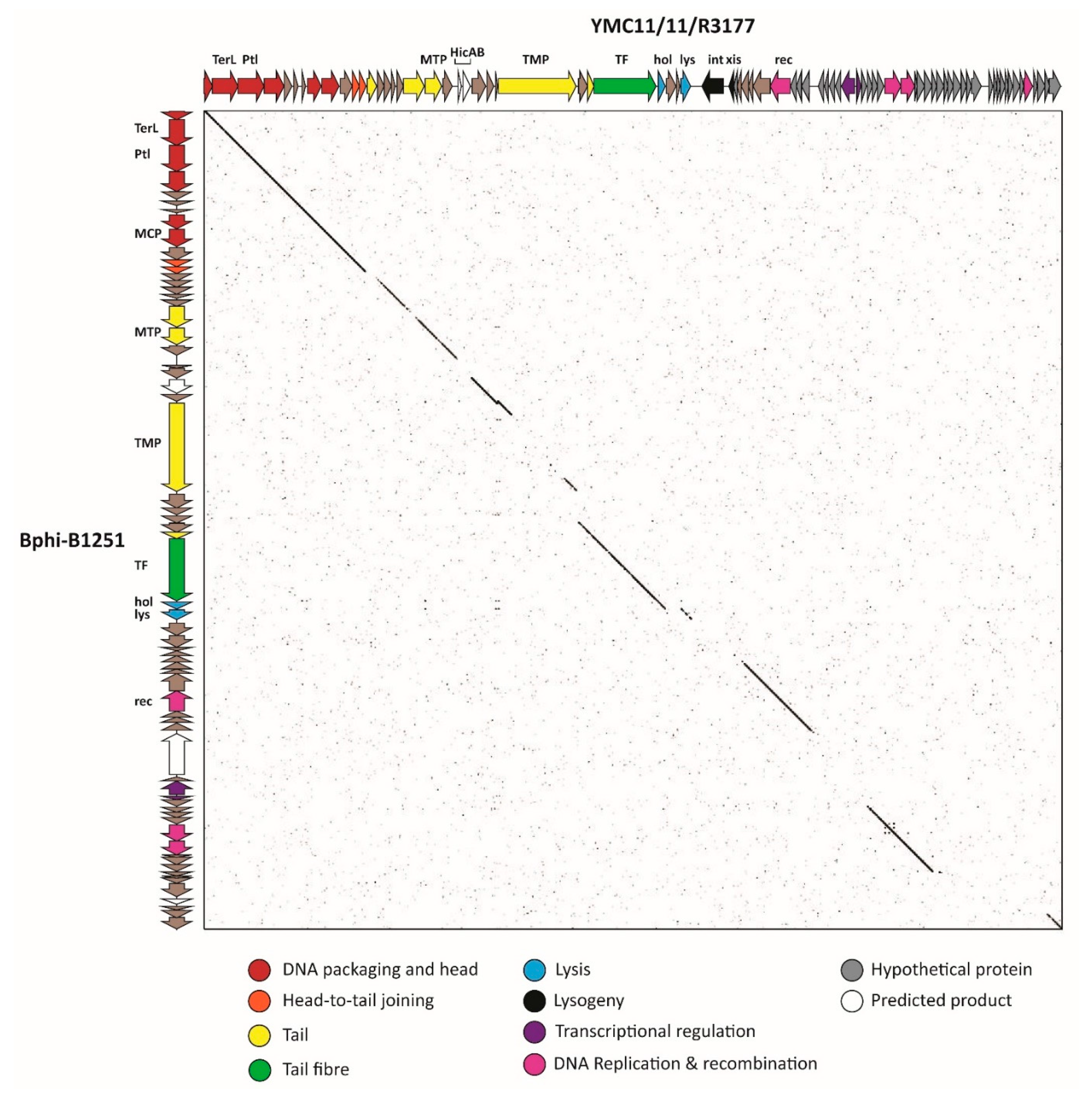
3.2.6. Cluster F: The 531-Like Acinetobacter Siphoviruses
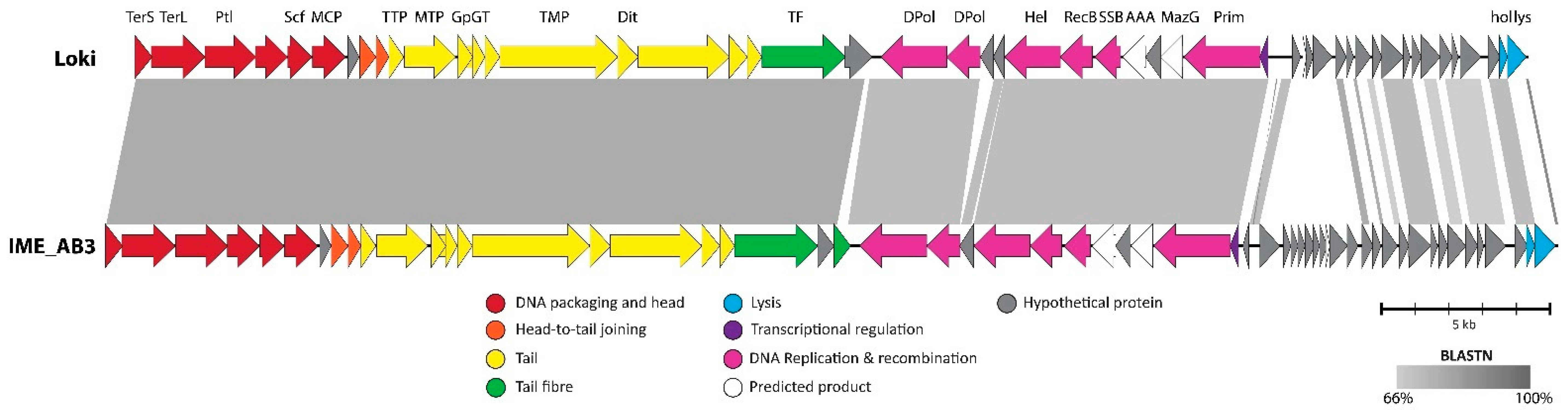
3.2.7. Cluster G: The Loki-Like Acinetobacter Siphoviruses
3.3. The Singleton Acinetobacter Phages
3.3.1. Singleton vB_AbaM_ME3
3.3.2. Acinetobacter Phage Presley Is an N4-Like Singleton
3.3.3. Leviviridae
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bergh, O.; Borsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Dark matter of the biosphere: The amazing world of bacteriophage diversity. J. Virol. 2015, 89, 8107–8110. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Jensen, G.L.; Burnett, S.H.; Breakwell, D.P. Genomic comparison of 93 Bacillus phages reveals 12 clusters, 14 singletons and remarkable diversity. BMC Genom. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J. Bacteriol. 2006, 188, 1184–1187. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R., Jr.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 2015, 4, e06416. [Google Scholar] [CrossRef] [PubMed]
- Moreno Switt, A.I.; Sulakvelidze, A.; Wiedmann, M.; Kropinski, A.M.; Wishart, D.S.; Poppe, C.; Liang, Y. Salmonella phages and prophages: Genomics, taxonomy, and applied aspects. In Salmonella: Methods and Protocols; Schatten, H., Eisenstark, A., Eds.; Springer: New York, NY, USA, 2015; pp. 237–287. [Google Scholar]
- Deveau, H.; Labrie, S.J.; Chopin, M.-C.; Moineau, S. Biodiversity and classification of lactococcal phages. Appl. Environ. Microbiol. 2006, 72, 4338–4346. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2014, 468–470, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Solonenko, S.A.; Ignacio-Espinoza, J.C.; LaButti, K.; Copeland, A.; Sudek, S.; Maitland, A.; Chittick, L.; dos Santos, F.; Weitz, J.S.; et al. Genomic differentiation among wild cyanophages despite widespread horizontal gene transfer. BMC Genom. 2016, 17, 930. [Google Scholar] [CrossRef] [PubMed]
- Millard, A.D.; Zwirglmaier, K.; Downey, M.J.; Mann, N.H.; Scanlan, D.J. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: Implications for mechanisms of cyanophage evolution. Environ. Microbiol. 2009, 11, 2370–2387. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, G.P.; Rørbo, I.N.; Castillo, D.; Mauritzen, J.J.; Jørgensen, J.; Kokkari, C.; Zhang, F.; Katharios, P.; Middelboe, M. Stumbling across the same phage: Comparative genomics of widespread temperate phages infecting the fish pathogen Vibrio anguillarum. Viruses 2017, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, K.; Solonenko, N.; Shah, M.; Corrier, K.; Riemann, L.; VerBerkmoes, N.C.; Sullivan, M.B. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12798–12803. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.D.; McAllister, T.A.; Nash, J.H.E.; Kropinski, A.M.; Stanford, K. Four Escherichia coli O157:H7 phages: A new bacteriophage genus and taxonomic classification of T1-like phages. PLoS ONE 2014, 9, e100426. [Google Scholar] [CrossRef] [PubMed]
- Anany, H.; Moreno Switt, A.I.; de Lappe, N.; Ackermann, H.-W.; Reynolds, D.; Kropinski, A.; Wiedmann, M.; Griffiths, M.; Tremblay, D.; Moineau, S.; et al. A proposed new bacteriophage subfamily: “Jerseyvirinae”. Arch. Virol. 2015, 160, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Maciejewska, B.; Latka, A.; Majkowska-Skrobek, G.; Hellstrand, M.; Melefors, Ö.; Wang, J.-T.; Kropinski, A.; Drulis-Kawa, Z.; Nilsson, A. A suggested new bacteriophage genus, “kp34likevirus”, within the Autographivirinae subfamily of Podoviridae. Viruses 2015, 7, 1804–1822. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Bacterial ‘immunity’ against bacteriophages. Bacteriophage 2012, 2, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Seed, K.D. Battling phages: How bacteria defend against viral attack. PLoS Pathog. 2015, 11, e1004847. [Google Scholar] [CrossRef] [PubMed]
- Towner, K.J. The genus Acinetobacter. In The Prokaryotes, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 6, pp. 746–758. [Google Scholar]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [PubMed]
- List of prokaryotic names with standing in nomenclature. Available online: http://www.bacterio.net/acinetobacter.html (accessed on 4 September 2017).
- Bouvet, P.J.M.; Grimont, P.A.D. Taxonomy of the genus Acinetobacter with the recognition of acinetobacter baumannii sp. Nov., Acinetobacter haemolyticus sp. Nov., Acinetobacter johnsonii sp. Nov., and Acinetobacter junii sp. Nov. and emended descriptions of Acinetobacter calcoaceticus and Acinetobacter lwoffii. Int. J. Syst. Bacteriol. 1986, 36, 228–240. [Google Scholar] [CrossRef]
- Dijkshoorn, L.; van Harsselaar, B.; Tjernberg, I.; Bouvet, P.J.M.; Vaneechoutte, M. Evaluation of amplified ribosomal DNA restriction analysis for identification of Acinetobacter genomic species. Syst. Appl. Microbiol. 1998, 21, 33–39. [Google Scholar] [CrossRef]
- Diancourt, L.; Passet, V.; Nemec, A.; Dijkshoorn, L.; Brisse, S. The population structure of Acinetobacter baumannii: Expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS ONE 2010, 5, e10034. [Google Scholar] [CrossRef] [PubMed]
- Bartual, S.G.; Seifert, H.; Hippler, C.; Luzon, M.A.D.; Wisplinghoff, H.; Rodríguez-Valera, F. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J. Clin. Microbiol. 2005, 43, 4382–4390. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Z.M.; Halachev, M.R.; Loman, N.J.; Constantinidou, C.; Pallen, M.J. Defining bacterial species in the genomic era: Insights from the genus Acinetobacter. BMC Microbiol. 2012, 12, 302. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Brochu, G.; Emadi Konjin, H.P. Classification of Acinetobacter phages. Arch. Virol. 1994, 135, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Blouse, L.; Twarog, R. Properties of four Herellea phages. Can. J. Microbiol. 1966, 12, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Actis, L.; Pachón, J. Acinetobacter baumannii: Human infections, factors contributing to pathogenesis and animal models. FEMS Microbiol. Rev. 2013, 37, 130–155. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery and Development of New Antibiotics; WHO Press: Geneva, Switzerland, 2017; p. 7. [Google Scholar]
- Bergogne-Bérézin, E.; Towner, K.J. Acinetobacter spp. As nosocomial pathogens: Microbiological, clinical, and epidemiological features. Clin. Microbiol. Rev. 1996, 9, 148–165. [Google Scholar] [PubMed]
- Turton, J.F.; Shah, J.; Ozongwu, C.; Pike, R. Incidence of Acinetobacter species other than A. baumannii among clinical isolates of Acinetobacter: Evidence for emerging species. J. Clin. Microbiol. 2010, 48, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.; Land, M.; Larimer, F.; Hauser, L. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with glimmer. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Borodovsky, M. Genemark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic virus orthologous groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Naville, M.; Ghuillot-Gaudeffroy, A.; Marchais, A.; Gautheret, D. ARNold: A web tool for the prediction of Rho-independent transcription terminators. RNA Biol. 2011, 8, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Kingsford, C.; Ayanbule, K.; Salzberg, S. Rapid, accurate, computational discovery of rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007, 8, R22. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-se: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindin, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Phylogeny.fr format conversion. Available online: http://phylogeny.lirmm.fr/phylo_cgi/data_converter.cgi (accessed on 21 July 2017).
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Söding, J. Protein homology detection by HMM–HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. Coregenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.; Arantes, A.; Stothard, P. Comparing thousands of circular genomes using the CGView comparison tool. BMC Genom. 2012, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Mi, Z.; Huang, Y.; Yuan, X.; Niu, W.; Wang, Y.; Hua, Y.; Fan, H.; Bai, C.; Tong, Y. Characterization, sequencing and comparative genomic analysis of vB_AbaM-IME-AB2, a novel lytic bacteriophage that infects multidrug-resistant Acinetobacter baumannii clinical isolates. BMC Microbiol. 2014, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Vandenheuvel, D.; Kropinski, A.M.; Mast, J.; de Vos, D.; Verbeken, G.; Noben, J.-P.; Lavigne, R.; Vaneechoutte, M.; Pirnay, J.-P. Characterization of newly isolated lytic bacteriophages active against Acinetobacter baumannii. PLoS ONE 2014, 9, e104853. [Google Scholar] [CrossRef] [PubMed]
- Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q, Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2012. [Google Scholar]
- Klovins, J.; Overbeek, G.P.; van den Worm, S.H.E.; Ackermann, H.-W.; van Duin, J. Nucleotide sequence of a ssRNA phage from Acinetobacter: Kinship to coliphages. J. Gen. Virol. 2002, 83, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.; Ratnayaka, S.; Nolan, J.; Miller, E.; Karam, J. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 2010, 7, 292. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, Z.-J.; Wang, S.-W.; Wang, S.-M.; Huang, D.-H.; Li, Y.-H.; Ma, Y.-Y.; Wang, J.; Liu, F.; Chen, X.-D.; et al. Isolation and characterization of ZZ1, a novel lytic phage that infects Acinetobacter baumannii clinical isolates. BMC Microbiol. 2012, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, Z.-J.; Wang, S.-W.; Wang, S.-M.; Chen, S.-J.; Huang, D.-H.; Zhang, G.; Li, Y.-H.; Wang, X.-T.; Wang, J.; et al. Genome organisation of the Acinetobacter lytic phage ZZ1 and comparison with other T4-like Acinetobacter phages. BMC Genom. 2014, 15, 793. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.V.; Myakinina, V.P.; Platonov, M.E.; Volozhantsev, N.V. Molecular genetic characterization of multidrug-resistant Acinetobacter baumannii strains and assessment of their sensitivity to phage AP22. Mol. Genet. Microbiol. Virol. 2012, 27, 154–159. [Google Scholar] [CrossRef]
- Dubrovin, E.V.; Popova, A.V.; Kraevskiy, S.V.; Ignatov, S.G.; Ignatyuk, T.E.; Yaminsky, I.V.; Volozhantsev, N.V. Atomic force microscopy analysis of the Acinetobacter baumannii bacteriophage AP22 lytic cycle. PLoS ONE 2012, 7, e47348. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, B.; Song, Z.; Song, Y.; Yang, Y.; Ma, P.; Wang, H.; Ying, J.; Ren, P.; Yang, L.; et al. Bioinformatic analysis of the Acinetobacter baumannii phage AB1 genome. Gene 2012, 507, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liang, L.; Lin, S.; Jia, S. Isolation and characterization of a virulent bacteriophage AB1 of Acinetobacter baumannii. BMC Microbiol. 2010, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Ryu, C.-M.; Lee, J.-Y.; Park, J.-H.; Yong, D.; Lee, K. In vivo application of bacteriophage as a potential therapeutic agent to control OXA-66-like carbapenemases-producing Acinetobacter baumannii strains belonging to ST357. Appl. Environ. Microbiol. 2016, 82, 4200–4208. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; D’Souza, R.; Pinto, N.; Ryu, C.-M.; Park, J.; Yong, D.; Lee, K. Characterization and complete genome sequence analysis of two myoviral bacteriophages infecting clinical carbapenem-resistant Acinetobacter baumannii isolates. J. Appl. Microbiol. 2016, 121, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Huang, H.; Wu, X.; Hao, Y.; Sun, Y. Complete genome sequence of lytic bacteriophage LZ35 infecting Acinetobacter baumannii isolates. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; O’Sullivan, L.; Elbreki, M.; Neve, H.; McAuliffe, O.; Ross, R.P.; Hill, C.; O’Mahony, J.; Coffey, A. Genome sequence of jumbo phage vB_AbaM_ME3 of Acinetobacter baumanni. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, C.; Choresca, C.H.; Shin, S.P.; Han, J.E.; Jun, J.W.; Heo, S.-J.; Kang, D.-H.; Park, S.C. Complete genome sequence of bacteriophage phiAC-1 infecting Acinetobacter soli strain KZ-1. J. Virol. 2012, 86, 13131–13132. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Le, S.; Peng, Y.; Zhao, Y.; Yin, S.; Zhang, L.; Yao, X.; Tan, Y.; Li, M.; Hu, F. Characterization and genome sequencing of phage Abp1, a new phiKMV-like virus infecting multidrug-resistant Acinetobacter baumannii. Curr. Microbiol. 2013, 66, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-C.; Lin, N.-T.; Hu, A.; Lin, Y.-S.; Chen, L.-K.; Lai, M.-J. Genomic analysis of bacteriophage ϕAB1, a ϕKMV-like virus infecting multidrug-resistant Acinetobacter baumannii. Genomics 2011, 97, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Li, X.-J. Bioinformatic analysis of phage AB3, a phiKMV-like virus infecting Acinetobacter baumannii. Genet. Mol. Res. 2015, 14, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.J.; Chang, K.C.; Huang, S.W.; Luo, C.H.; Chiou, P.Y.; Wu, C.C.; Lin, N.T. The tail associated protein of Acinetobacter baumannii phage phiAB6 is the host specificity determinant possessing exopolysaccharide depolymerase activity. PLoS ONE 2016, 11, e0153361. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Tu, I.F.; Yang, F.L.; Ko, T.P.; Liao, J.H.; Lin, N.T.; Wu, C.Y.; Ren, C.T.; Wang, A.H.; Chang, C.M.; et al. Structural basis for fragmenting the exopolysaccharide of Acinetobacter baumannii by bacteriophage phiAB6 tailspike protein. Sci. Rep. 2017, 7, 42711. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.J.; Leandro, C.; Mottola, C.; Barbosa, R.; Silva, F.A.; Oliveira, M.; Vilela, C.L.; Melo-Cristino, J.; Górski, A.; Pimentel, M.; et al. In vitro design of a novel lytic bacteriophage cocktail with therapeutic potential against organisms causing diabetic foot infections. J. Med. Microbiol. 2014, 63, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.J.; Leandro, C.; Corte-Real, S.; Barbosa, R.; Cavaco-Silva, P.; Melo-Cristino, J.; Górski, A.; Garcia, M. Wound healing potential of topical bacteriophage therapy on diabetic cutaneous wounds. Wound Repair Regen. 2013, 21, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Mumm, I.P.; Wood, T.L.; Chamakura, K.R.; Kuty Everett, G.F. Complete genome of Acinetobacter baumannii podophage Petty. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Farmer, N.G.; Wood, T.L.; Chamakura, K.R.; Kuty Everett, G.F. Complete genome of Acinetobacter baumannii N4-like podophage Presley. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; D’Souza, R.; Pinto, N.; Ryu, C.-M.; Park, J.-H.; Yong, D.; Lee, K. Complete genome sequence of the siphoviral bacteriophage βϕ-R3177, which lyses an OXA-66-producing carbapenem-resistant Acinetobacter baumannii isolate. Arch. Virol. 2015, 160, 3157–3160. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Kim, J.-W.; Yong, D.; Lee, K.; Chong, Y. Complete genome sequence of the podoviral bacteriophage YMC/09/02/B1251 ABA BP, which causes the lysis of an oxa-23-producing carbapenem-resistant Acinetobacter baumannii isolate from a septic patient. J. Virol. 2012, 86, 12437–12438. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Wand, M.E.; Briers, Y.; Lavigne, R.; Sutton, J.M.; Reynolds, D.M. Characterisation and genome sequence of the lytic Acinetobacter baumannii bacteriophage vB_AbaS_Loki. PLoS ONE 2017, 12, e0172303. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Krisch, H.M. A catalogue of T4-type bacteriophages. Arch. Virol. 1997, 142, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.-T.; Chiou, P.-Y.; Chang, K.-C.; Chen, L.-K.; Lai, M.-J. Isolation and characterization of ϕAB2: A novel bacteriophage of Acinetobacter baumannii. Res. Microbiol. 2010, 161, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Haggård-Ljungquist, E.; Halling, C.; Calendar, R. DNA sequences of the tail fiber genes of bacteriophage P2: Evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J. Bacteriol. 1992, 174, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Stummeyer, K.; Schwarzer, D.; Claus, H.; Vogel, U.; Gerardy-Schahn, R.; Mühlenhoff, M. Evolution of bacteriophages infecting encapsulated bacteria: Lessons from Escherichia coli K1-specific phages. Mol. Microbiol. 2006, 60, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.M. Transcriptional control in the prereplicative phase of T4 development. Virol. J. 2010, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- Geiduschek, E.P.; Kassavetis, G.A. Transcription of the T4 late genes. Virol. J. 2010, 7, 288. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-N.; Tseng, T.-T.; Lin, J.-W.; Fu, Y.-C.; Weng, S.-F.; Tseng, Y.-H. Lytic myophage Abp53 encodes several proteins similar to those encoded by host Acinetobacter baumannii and phage phiKO2. Appl. Environ. Microbiol. 2011, 77, 6755–6762. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M. Accurate description of phages and their genomes—Genet. Mol. Res. 14 (1): 190–198 “Bioinformatic analysis of phage AB3, a phiKMV-like virus infecting Acinetobacter baumannii”. Genet. Mol. Res. 2015, 14, 15092–15093. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.; Stothard, P.; Dennis, J. Comparative analysis of two phenotypically-similar but genomically-distinct Burkholderia cenocepacia-specific bacteriophages. BMC Genom. 2012, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, J.; Klumpp, J.; Moreno Switt, A.I.; Yagubi, A.; Ackermann, H.-W.; Wiedmann, M.; Svircev, A.; Nash, J.H.E.; Kropinski, A.M. Taxonomic reassessment of N4-like viruses using comparative genomics and proteomics suggests a new subfamily—“Enquartavirinae”. Arch. Virol. 2015, 160, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Brabban, A.; Rogge, L.; Lewis, M.S.; Pickard, D.; Goulding, D.; Dougan, G.; Noben, J.-P.; Kropinski, A.; Kutter, E.; et al. Molecular and physiological analysis of three Pseudomonas aeruginosa phages belonging to the “N4-like viruses”. Virology 2010, 405, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Bollback, P.J.; Huelsenbeck, P.J. Phylogeny, genome evolution, and host specificity of single-stranded RNA bacteriophage (family Leviviridae). J. Mol. Evol. 2001, 52, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Turner, D. University of the West of England, Bristol, United Kingdom. A preliminary bioinformatics analysis of completely sequenced Acinetobacter genomes for the presence of prophage. 2017. [Google Scholar]
- Renda, B.A.; Chan, C.; Parent, K.N.; Barrick, J.E. Emergence of a competence reducing filamentous phage from the genome of Acinetobacter baylyi ADP1. J. Bacteriol. 2016, 198, 3209–3219. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Cury, J.; Yoon, E.-J.; Krizova, L.; Cerqueira, G.C.; Murphy, C.; Feldgarden, M.; Wortman, J.; Clermont, D.; Lambert, T.; et al. The genomic diversification of the whole Acinetobacter genus: Origins, mechanisms, and consequences. Genome Biol. Evol. 2014, 6, 2866–2882. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Molecular genetics of mycobacteriophages. Microbiol. Spectr. 2014, 2, 1–36. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Host | Country | Genome Size | No. of ORFs # | % G+C | Accession | Coding% | Genes/kbp | tRNAs | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Family: Leviviridae; Genus: Levivirus | ||||||||||
| AP205 | Acinetobacter sp. (HER 1424) | Canada | 4268 | 4 | 43.86 | AF334111 | 90.9 | 0.937 | 0 | [61] |
| Family: Myoviridae | ||||||||||
| Subfamily: Tevenvirinae (Cluster A) | ||||||||||
| 133 | A. johnsonii (HER 1423) | Canada | 159,801 | 257 (257) | 39.67 | HM114315 | 95.7 | 1.608 | 16 | [62] |
| Acj9 | A. johnsonii | USA | 169,947 | 253 (253) | 40.03 | HM004124 | 93.3 | 1.488 | 19 | [62] |
| Acj61 | A. johnsonii | USA | 164,093 | 241 (241) | 39.01 | GU911519 | 92.5 | 1.468 | 12 | [62] |
| Ac42 | Acinetobacter sp. (HER 1422) | Canada | 167,716 | 255 (255) | 36.37 | HM032710 | 94.6 | 1.52 | 6 | [62] |
| ZZ1 | A. baumannii AB09V | China | 166,682 | 256 (256) | 34.41 | HQ698922 | 93.6 | 1.535 | 8 | [63,64] |
| Genus: Ap22virus (Cluster B) | ||||||||||
| AP22 | A. baumannii 1053 | Russia | 46,387 | 89 (90) | 37.74 | HE806280 | 91.9 | 1.918 | 0 | [65,66] |
| AB1 | A. baumannii KD311 | China | 45,159 | 88 (88) | 37.69 | HM368260 | 93.8 | 1.948 | 0 | [67,68] |
| IME-AB2 | A. baumannii MDR-AB2 | China | 43,665 | 82 (82) | 37.5 | JX976549 | 93.4 | 1.877 | 0 | [58] |
| YMC-13-01-C62 | A. baumannii YMC/13/01/C62 | South Korea | 44,844 | 84 (87) | 37.6 | KJ817802 | 94.3 | 1.94 | 0 | [69] |
| YMC11/12/R2315 | Acinetobacter sp. | South Korea | 44,846 | 85 (87) | 37.59 | KP861229 | 94.0 | 1.94 | 0 | [70] |
| YMC11/12/R1215 | Acinetobacter sp. | South Korea | 44,866 | 85 (87) | 37.59 | KP861231 | 94.3 | 1.94 | 0 | [70] |
| WCHABP1 | A. baumannii | China | 45,888 | 89 (91) | 37.63 | KY829116 | 93.9 | 1.939 | 0 | Unpublished |
| WCHABP12 | A. baumannii | China | 45,415 | 88 (91) | 37.58 | KY670595 | 94.1 | 1.937 | 0 | Unpublished |
| LZ35 | A. baumannii | China | 44,885 | 83 (85) | 38.36 | KU510289 | 92.6 | 1.782 | 0 | [71] |
| Unclassified myoviruses (Cluster C) | ||||||||||
| Acibel004 | A. baumannii 070517/0072 | Belgium | 99,730 | 156 (156) | 37.27 | KJ473422 | 89.2 | 1.564 | 22 | [59] |
| vB_AbaM_phiAbaA1 | A. baumannii Acb8/09 | Poland | 104,906 | 165 (165) | 37.77 | KJ628499 | 90.7 | 1.572 | 13 | Unpublished |
| Proposed genus: “Am24virus” (Cluster D) | ||||||||||
| AM24 | A. baumannii | Russia | 97,139 | 146 (168) | 37.25 | KY000079 | 80.4 | 1.503 | 17 | Unpublished |
| YMC13/03/R2096 | A. baumannii YMC13/03/R2096 | South Korea | 98,170 | 162 (170) | 37.04 | KM67266 | 87.3 | 1.731 | 17 | Unpublished |
| Singleton myoviruses | ||||||||||
| vB_AbaM_ME3 | A. baumannii DSM 30007 | Ireland | 234,900 | 326 (326) | 30.76 | KU935715 | 94.1 | 1.387 | 4 | [72] |
| phiAC-1 | A. soli KZ-1 | South Korea | 43,216 | 82 (82) | 38.48 | JX560521 | 93.1 | 1.897 | 0 | [73] |
| Family: Podoviridae | ||||||||||
| Subfamily: Autographivirinae; Genus: Fri1virus (Cluster E) | ||||||||||
| Fri1 | A. baumannii 28 | Switzerland | 41,805 | 54 (55) | 39.29 | KR149290 | 92.5 | 1.291 | 0 | Unpublished |
| Abp1 | A. baumanni AB1 | China | 42,185 | 57 (57) | 39.15 | JX658790 | 93.4 | 1.351 | 0 | [74] |
| phiAB1 | A. baumannii M68316 | Taiwan | 41,526 | 46 (49) | 39.09 | HQ186308 | 91.8 | 1.179 | 0 | [75] |
| AB3 * | A. baumannii | China | 31,185 | 27 (29) | 39.18 | KC311669 | 96.4 | 0.929 | 0 | [76] |
| WCHABP5 | A. baumannii WCHAB1334 | China | 40,409 | 47 (52) | 39.38 | KY888680 | 88.2 | 1.163 | 0 | Unpublished |
| phiAB6 | A. baumannii 54149 | Taiwan | 40,570 | 45 (47) | 39.47 | KT339321 | 90 | 1.109 | 0 | [77,78] |
| IME200 | A. baumannii | China | 41,243 | 52 (53) | 39.73 | KT804908 | 88.6 | 1.26 | 0 | Unpublished |
| vB_AbaP_PD-6A3 | A. baumannii | China | 41,563 | 48 (53) | 39.92 | KT388102 | 92.8 | 1.154 | 0 | Unpublished |
| vB_AbaP_PD-AB9 | A. baumannii | China | 40,938 | 48 (52) | 39.34 | KT388103 | 92.9 | 1.172 | 0 | Unpublished |
| Unclassified members of Subfamily Autographvirinae | ||||||||||
| F1245/05 | Acinetobacter sp. | Portugal | 43,016 | 0 (53) | 40.46 | HH777814 | 94.3 | 1.255 | 0 | [79,80] |
| Acibel007 | A. baumannii 070517/0072 | Belgium | 42,654 | 53 (53) | 41.7 | KJ473423 | 93.9 | 1.242 | 0 | [59] |
| Petty | A. baumannii AU0783 | USA | 40,739 | 45 (45) | 42.19 | KF669656 | 91.5 | 1.104 | 0 | [81] |
| Unclassified N4-like virus | ||||||||||
| Presley | A. baumannii M2 | USA | 77,792 | 95 (95) | 37.77 | KF669658 | 95.4 | 1.221 | 0 | [82] |
| Family: Siphoviridae | ||||||||||
| Proposed genus “B1251virus” (Cluster F) | ||||||||||
| YMC11/11/R3177 | Acinetobacter sp. | South Korea | 47,575 | 80 (81) | 39.83 | KP861230 | 90.7 | 1.681 | 0 | [83] |
| Bphi-B1251 | A. baumannii YMC/09/02/B1251 | South Korea | 45,364 | 62 (67) | 39.05 | JX403940 | 93.5 | 1.454 | 0 | [84] |
| Proposed genus: “Lokivirus” (Cluster G) | ||||||||||
| IME-AB3 | A. baumannii | China | 43,050 | 57 (58) | 45.48 | KF811200 | 97.1 | 1.347 | 0 | Unpublished |
| vB_AbaS_Loki | A. baumannii ATCC 17978 | UK | 41,308 | 51 (51) | 44.35 | LN890663 | 96.4 | 1.234 | 0 | [85] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turner, D.; Ackermann, H.-W.; Kropinski, A.M.; Lavigne, R.; Sutton, J.M.; Reynolds, D.M. Comparative Analysis of 37 Acinetobacter Bacteriophages. Viruses 2018, 10, 5. https://doi.org/10.3390/v10010005
Turner D, Ackermann H-W, Kropinski AM, Lavigne R, Sutton JM, Reynolds DM. Comparative Analysis of 37 Acinetobacter Bacteriophages. Viruses. 2018; 10(1):5. https://doi.org/10.3390/v10010005
Chicago/Turabian StyleTurner, Dann, Hans-Wolfgang Ackermann, Andrew M. Kropinski, Rob Lavigne, J. Mark Sutton, and Darren M. Reynolds. 2018. "Comparative Analysis of 37 Acinetobacter Bacteriophages" Viruses 10, no. 1: 5. https://doi.org/10.3390/v10010005
APA StyleTurner, D., Ackermann, H.-W., Kropinski, A. M., Lavigne, R., Sutton, J. M., & Reynolds, D. M. (2018). Comparative Analysis of 37 Acinetobacter Bacteriophages. Viruses, 10(1), 5. https://doi.org/10.3390/v10010005