Interferons: Reprogramming the Metabolic Network against Viral Infection

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
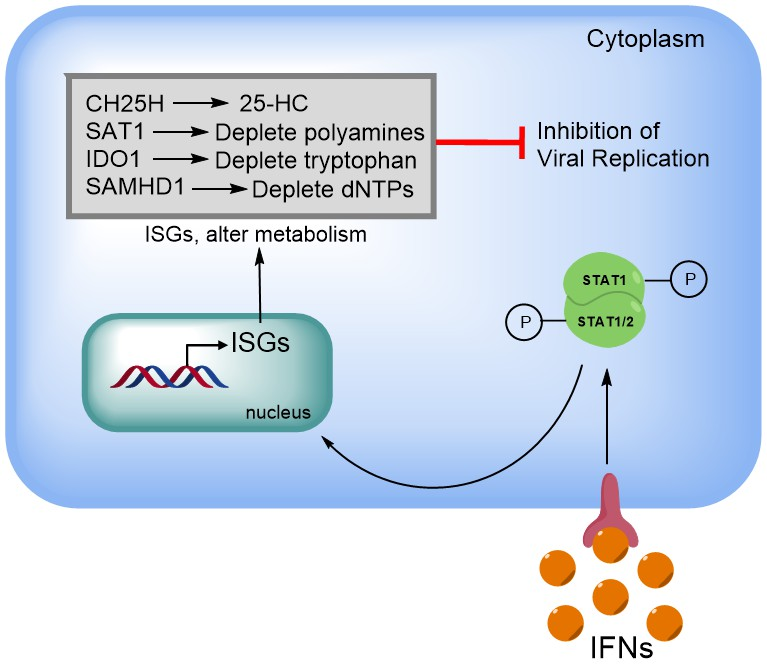
2. Viral Rewiring of Central Carbon Metabolism
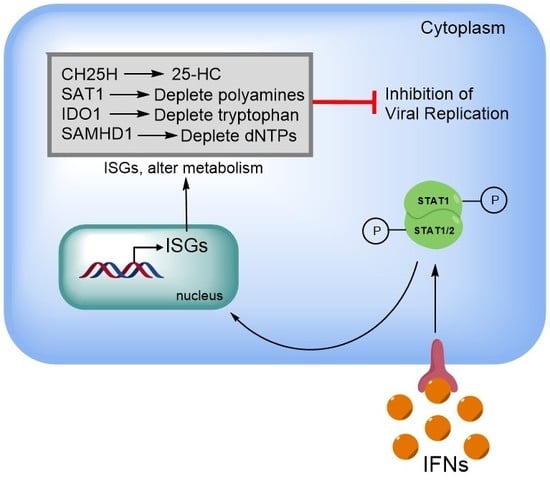
3. Diverse Antiviral Mechanisms by Interferons and Their Stimulated Genes
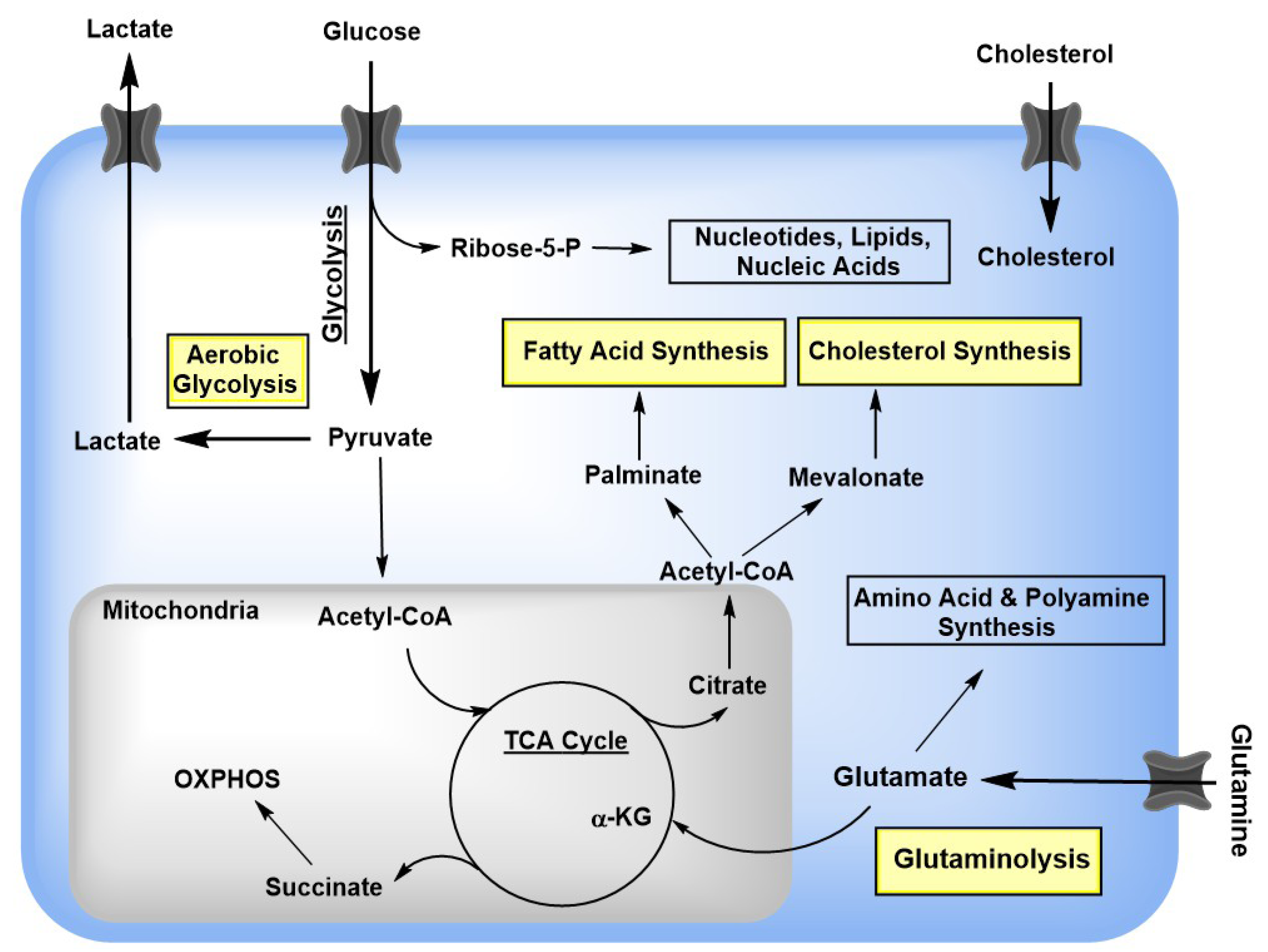
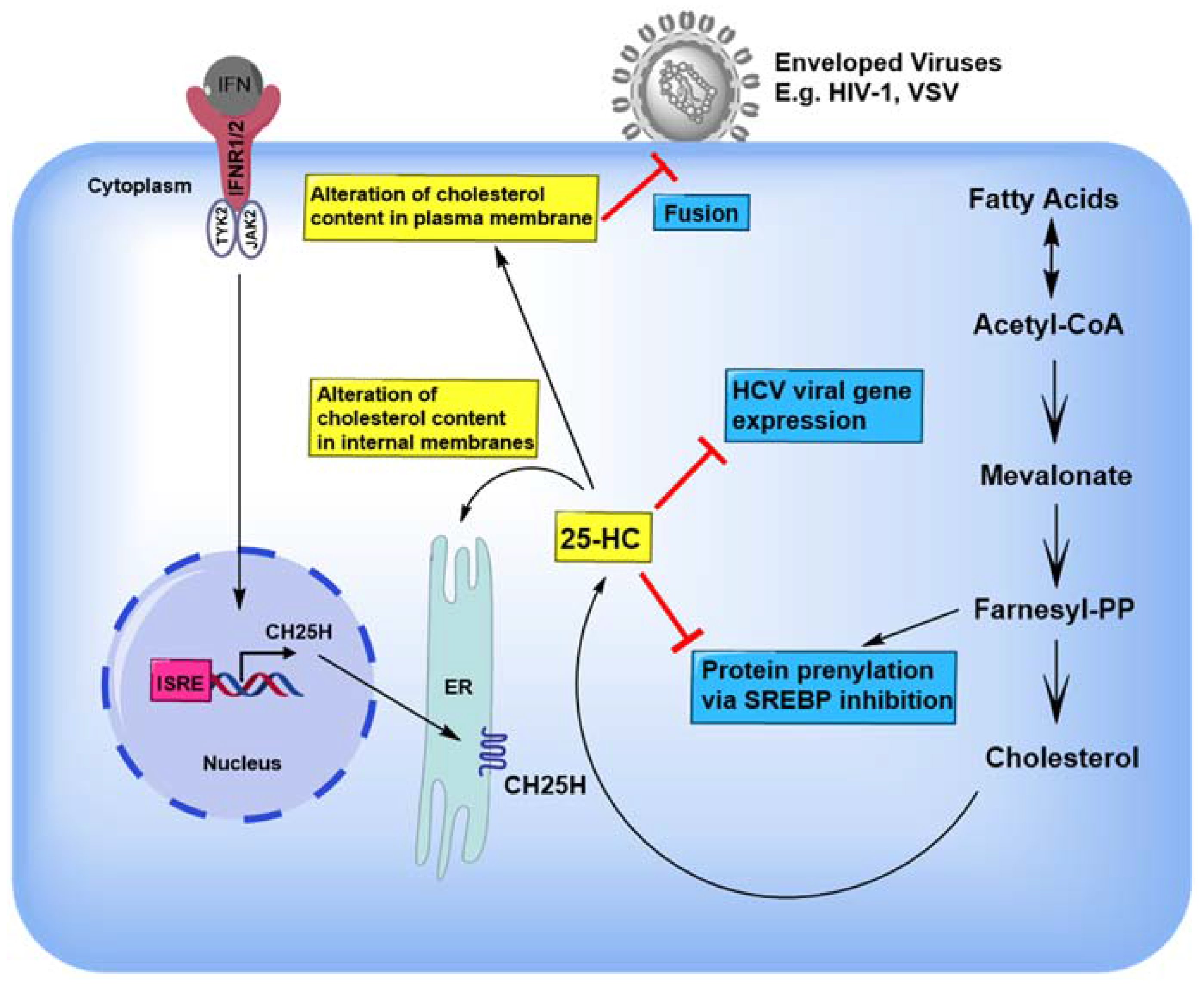
4. 25-HC Inhibits a Variety of Viruses by Modulating Sterol Synthesis
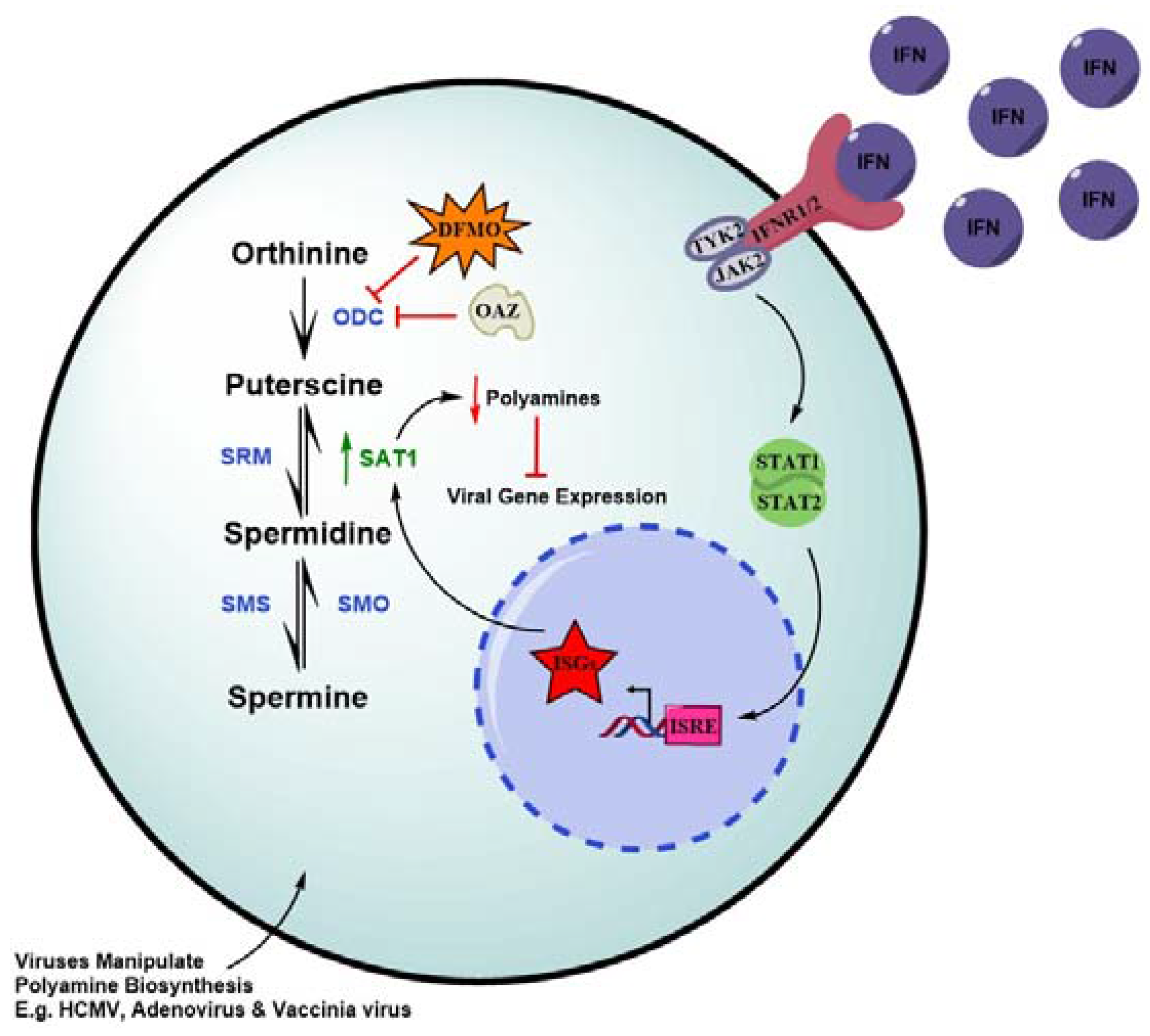
5. IFNs Deplete Polyamines as a Strategy to Check Viral Infection
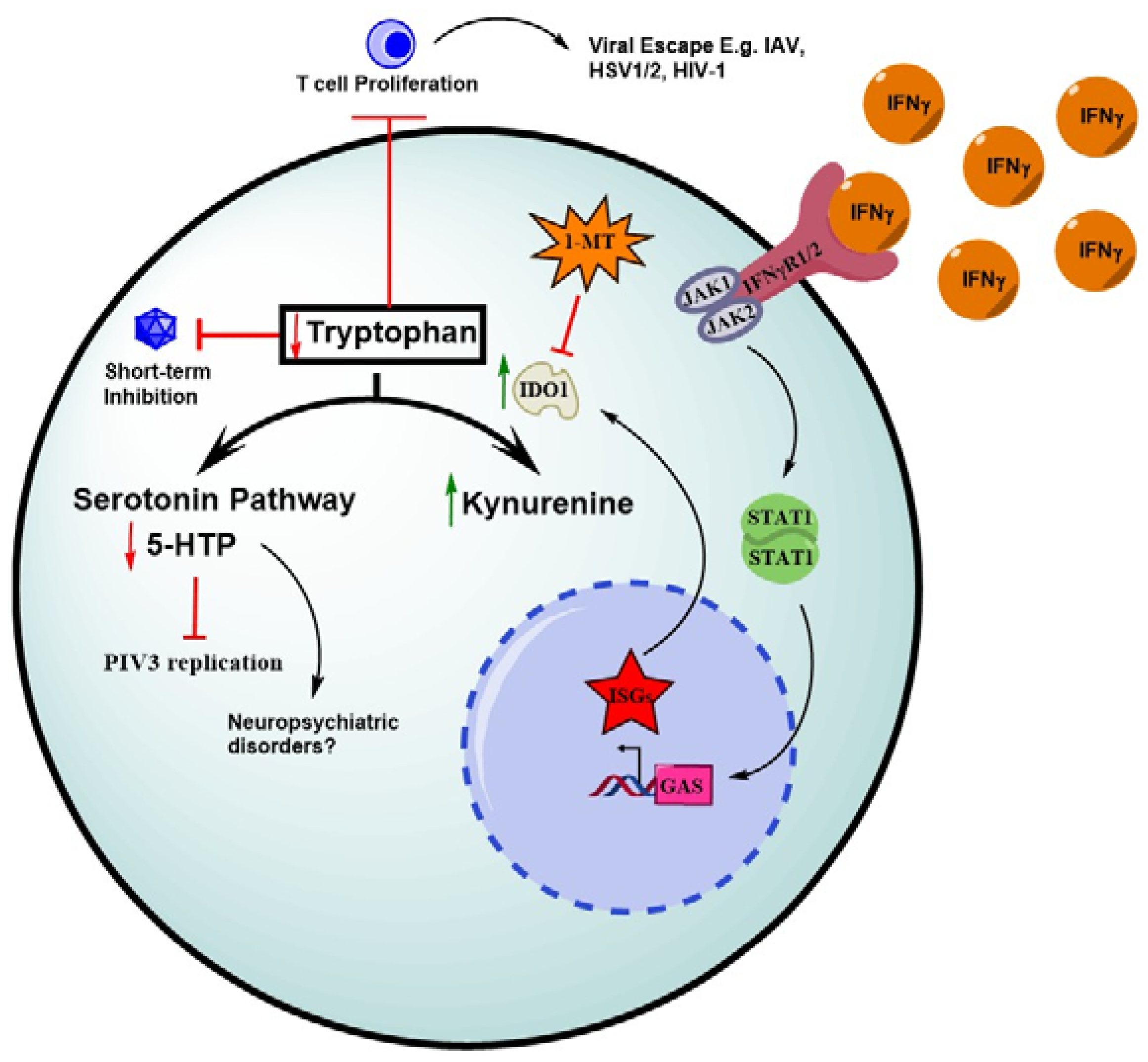
6. IFN-Mediated Depletion of Tryptophan through IDO1 Induction
7. SAMHD1 Restricts Viral DNA Synthesis by Depleting Cellular dNTP Pool
8. Targeting Metabolic Pathways for Viral Therapy: A Novel Therapeutic Approach
9. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Slama, L.; Le Camus, C.; Serfaty, L.; Pialoux, G.; Capeau, J.; Gharakhanian, S. Metabolic disorders and chronic viral disease: The case of HIV and HCV. Diabetes Metab. 2009, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bantug, G.R.; Galluzzi, L.; Kroemer, G.; Hess, C. The spectrum of T cell metabolism in health and disease. Nat. Rev. Immunol. 2018, 18, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Goldrath, A.W.; Glass, C.K. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity 2017, 46, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Buitendijk, M.; Eszterhas, S.K.; Howell, A.L. Toll-like receptor agonists are potent inhibitors of human immunodeficiency virus-type 1 replication in peripheral blood mononuclear cells. AIDS Res. Hum. Retrovir. 2014, 30, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Rathmell, J.C. Metabolic pathways in T cell fate and function. Trends Immunol. 2012, 33, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H.; Habel, K. The nutritional requirements for the propagation of poliomyelitis virus by the hela cell. J. Exp. Med. 1956, 104, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Munger, J. Dynamics of the Cellular Metabolome during Human Cytomegalovirus Infection. PLoS Pathog. 2006. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Lagos, D.; Enver, T.; Boshoff, C. Kaposi’s sarcoma herpesvirus microRNAs induce metabolic transformation of infected cells. PLoS Pathog. 2014, 10, e1004400. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.L. Temporal Proteome and Lipidome Profiles Reveal Hepatitis C Virus-Associated Reprogramming of Hepatocellular Metabolism and Bioenergetics. PLoS Pathog. 2010. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Camarda, R.; Lagunoff, M. Vaccinia Virus Requires Glutamine but Not Glucose for Efficient Replication. Journal of Virology 2014, 88, 4366–4374. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, A.; Kavanagh Williamson, M.; Huthoff, H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Ritter, J.B.; Wahl, A.S.; Freund, S.; Genzel, Y.; Reichl, U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Syst. Biol. 2010, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the Keys to the Kitchen: Viral Manipulation of the Host Cell Metabolic Network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, S.D.; Weichhart, T. Effects of Interferons and Viruses on Metabolism. Front. Immunol. 2016, 7, 630. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Pulliam, T.H.; Dimaio, T.A.; Thalhofer, A.B.; Delgado, T.; Lagunoff, M. Glycolysis, Glutaminolysis, and Fatty Acid Synthesis Are Required for Distinct Stages of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2017, 91, e02237-16. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. By A. Isaacs and J. Lindenmann, 1957. J. Interferon Res. 1987, 7, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; MacDonald, M.R.; Rice, C.M. To translate, or not to translate: Viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol. 2015, 25, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Sanchez, D.J.; Aliyari, R.; Lu, S.; Cheng, G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 2012, 109, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. RIG-I family RNA helicases: Cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007, 18, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Ahlers, L.R.H.; Goodman, A.G. Nucleic acid sensing and innate immunity: Signaling pathways controlling viral pathogenesis and autoimmunity. Curr. Clin. Microbiol. Rep. 2016, 3, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Jackel-Cram, C.; Qiao, L.; Xiang, Z.; Brownlie, R.; Zhou, Y.; Babiuk, L.; Liu, Q. Hepatitis C virus genotype-3a core protein enhances sterol regulatory element-binding protein-1 activity through the phosphoinositide 3-kinase-Akt-2 pathway. J. Gen. Virol. 2010, 91, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Watterson, S.; Shui, G.; Lacaze, P.; Khondoker, M.; Dickinson, P.; Sing, G.; Rodríguez-Martín, S.; et al. Host Defense against Viral Infection Involves Interferon Mediated Down-Regulation of Sterol Biosynthesis. PLoS Biol. 2011, 9, e1000598. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Singaravelu, R.; O’Hara, S.; Jones, D.M.; Chen, R.; Taylor, N.G.; Srinivasan, P.; Quan, C.; Roy, D.G.; Steenbergen, R.H.; Kumar, A.; et al. MicroRNAs regulate the immunometabolic response to viral infection in the liver. Nat. Chem. Biol. 2015, 11, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Moog, C.; Aubertin, A.M.; Kirn, A.; Luu, B. Oxysterols, but not cholesterol, inhibit human immunodeficiency virus replication in vitro. Antivir. Chem. Chemother. 1998, 9, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Anggakusuma; Romero-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.; Xu, S.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Shimojima, M.; Fukushi, S.; Yoshikawa, T.; Fukuma, A.; Taniguchi, S.; Morikawa, S.; Saijo, M. Characterization of Glycoprotein-Mediated Entry of Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2016, 90, 5292–5301. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, Y.Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.Y.; Zhang, N.N.; Watanabe, M.; Dong, H.L.; Liu, P.; et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zeng, L.; Zhang, L.; Guo, Z.Z.; Lu, S.F.; Ming, S.L.; Li, G.L.; Wan, B.; Tian, K.G.; Yang, G.Y.; et al. Cholesterol 25-hydroxylase acts as a host restriction factor on pseudorabies virus replication. J. Gen. Virol. 2017, 98, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Lembo, D.; Cagno, V.; Civra, A.; Poli, G. Oxysterols: An emerging class of broad spectrum antiviral effectors. Mol. Aspects Med. 2016, 49, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Civra, A.; Cagno, V.; Donalisio, M.; Biasi, F.; Leonarduzzi, G.; Poli, G.; Lembo, D. Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol. Sci. Rep. 2014, 4, 7487. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, Q.; Liu, X.; Bai, J.; Zhao, Y.; Wang, X.; Jiang, P. Cholesterol 25-hydroxylase is an interferon-inducible factor that protects against porcine reproductive and respiratory syndrome virus infection. Vet. Microbiol. 2017, 210, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Pereiro, P.; Forn-Cuni, G.; Dios, S.; Coll, J.; Figueras, A.; Novoa, B. Interferon-independent antiviral activity of 25-hydroxycholesterol in a teleost fish. Antivir. Res. 2017, 145, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava-Ranjan, P.; Bergeron, E.; Chakrabarti, A.K.; Albarino, C.G.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. 25-Hydroxycholesterol Inhibition of Lassa Virus Infection through Aberrant GP1 Glycosylation. mBio 2016, 7, e01808-16. [Google Scholar] [CrossRef] [PubMed]
- Burri, D.J.; da Palma, J.R.; Kunz, S.; Pasquato, A. Envelope glycoprotein of arenaviruses. Viruses 2012, 4, 2162–2181. [Google Scholar] [CrossRef] [PubMed]
- Cagno, V.; Civra, A.; Rossin, D.; Calfapietra, S.; Caccia, C.; Leoni, V.; Dorma, N.; Biasi, F.; Poli, G.; Lembo, D. Inhibition of herpes simplex-1 virus replication by 25-hydroxycholesterol and 27-hydroxycholesterol. Redox Biol. 2017, 12, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J. Virol. 2013, 87, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Li, S. Oxysterol-binding proteins: Sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog. Lipid Res. 2013, 52, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Arita, M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Roulin, P.S.; Lotzerich, M.; Torta, F.; Tanner, L.B.; van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.L.; Feingold, K.R.; Moser, A.H.; Grunfeld, C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 1993, 98, 67–82. [Google Scholar] [CrossRef]
- Huang, Y.L.; Morales-Rosado, J.; Ray, J.; Myers, T.G.; Kho, T.; Lu, M.; Munford, R.S. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J. Biol. Chem. 2014, 289, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Dushkin, M.I.; Kovshik, G.G. Effect of toll-like receptor agonists on the formation of macrophage/foam cells upon acute peritonitis in mice. Bull. Exp. Biol. Med. 2013, 156, 49–52. [Google Scholar] [CrossRef] [PubMed]
- York Autumn, G.; Williams Kevin, J.; Argus Joseph, P.; Zhou Quan, D.; Brar, G.; Vergnes, L.; Gray Elizabeth, E.; Zhen, A.; Wu Nicholas, C.; Yamada Douglas, H.; et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; MacDonald, M.R. Polyamines: Small Molecules with a Big Role in Promoting Virus Infection. Cell Host Microbe 2016, 20, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Mounce, B.C.; Olsen, M.E.; Vignuzzi, M.; Connor, J.H. Polyamines and Their Role in Virus Infection. Microbiol. Mol. Biol. Rev. 2017, 81, e00029-17. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Mammalian polyamine metabolism and function. IUBMB Life 2009, 61, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Schipper, R.G.; Penning, L.C.; Verhofstad, A.A. Involvement of polyamines in apoptosis. Facts and controversies: Effectors or protectors? Semin. Cancer Biol. 2000, 10, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Sirisoma, N.S.; Kuppusamy, P.; Zweier, J.L.; Woster, P.M.; Casero, R.A., Jr. The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. USA 1998, 95, 11140–11145. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.C.; Mehta, D.J.; Gerner, E.W. Polyamine-dependent gene expression. Cell. Mol. Life Sci. 2003, 60, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Mounce, B.C.; Poirier, E.Z.; Passoni, G.; Simon-Loriere, E.; Cesaro, T.; Prot, M.; Stapleford, K.A.; Moratorio, G.; Sakuntabhai, A.; Levraud, J.P.; et al. Interferon-Induced Spermidine-Spermine Acetyltransferase and Polyamine Depletion Restrict Zika and Chikungunya Viruses. Cell Host Microbe 2016, 20, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013, 41, D1040–D1046. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Hall, J. Endogenous polyamine function—The RNA perspective. Nucleic Acids Res. 2014, 42, 11275–11290. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. Modulation of protein synthesis by polyamines. IUBMB Life 2015, 67, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.; Roizman, B. Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc. Natl. Acad. Sci. USA 1971, 68, 2818–2821. [Google Scholar] [CrossRef] [PubMed]
- Tyms, A.S.; Williamson, J.D. Inhibitors of polyamine biosynthesis block human cytomegalovirus replication. Nature 1982, 297, 690. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, W.; Holowczak, J.A. Polyamines in vaccinia virions and polypeptides released from viral cores by acid extraction. J. Virol. 1975, 16, 1254–1264. [Google Scholar] [PubMed]
- Park, M.H.; Cooper, H.L.; Folk, J.E. Identification of hypusine, an unusual amino acid, in a protein from human lymphocytes and of spermidine as its biosynthetic precursor. Proc. Natl. Acad. Sci. USA 1981, 78, 2869–2873. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A). J. Biochem. 2006, 139, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.E.; Filone, C.M.; Rozelle, D.; Mire, C.E.; Agans, K.N.; Hensley, L.; Connor, J.H. Polyamines and Hypusination Are Required for Ebolavirus Gene Expression and Replication. mBio 2016, 7, e00882-16. [Google Scholar] [CrossRef] [PubMed]
- Baumann, S.; Sander, A.; Gurnon, J.R.; Yanai-Balser, G.; VanEtten, J.L.; Piotrowski, M. Chlorella viruses contain genes encoding a complete polyamine biosynthetic pathway. Virology 2007, 360, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Keinanen, T.A.; Ivanova, O.N.; Hyvonen, M.T.; Khomutov, A.R.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Hepatitis C virus alters metabolism of biogenic polyamines by affecting expression of key enzymes of their metabolism. Biochem. Biophys. Res. Commun. 2017, 483, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Evageliou, N.F.; Haber, M.; Vu, A.; Laetsch, T.W.; Murray, J.; Gamble, L.D.; Cheng, N.C.; Liu, K.; Reese, M.; Corrigan, K.A.; et al. Polyamine Antagonist Therapies Inhibit Neuroblastoma Initiation and Progression. Clin. Cancer Res. 2016, 22, 4391–4404. [Google Scholar] [CrossRef] [PubMed]
- Bassiri, H.; Benavides, A.; Haber, M.; Gilmour, S.K.; Norris, M.D.; Hogarty, M.D. Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl. Pediatr. 2015, 4, 226–238. [Google Scholar] [PubMed]
- Mounce, B.C.; Cesaro, T.; Moratorio, G.; Hooikaas, P.J.; Yakovleva, A.; Werneke, S.W.; Smith, E.C.; Poirier, E.Z.; Simon-Loriere, E.; Prot, M.; et al. Inhibition of Polyamine Biosynthesis Is a Broad-Spectrum Strategy against RNA Viruses. J. Virol. 2016, 90, 9683–9692. [Google Scholar] [CrossRef] [PubMed]
- Creaven, P.J.; Pendyala, L.; Petrelli, N.J. Evaluation of alpha-difluoromethylornithine as a potential chemopreventive agent: Tolerance to daily oral administration in humans. Cancer Epidemiol. Biomark. Prev. 1993, 2, 243–247. [Google Scholar]
- Sainio, E.L.; Pulkki, K.; Young, S.N. L-Tryptophan: Biochemical, nutritional and pharmacological aspects. Amino Acids 1996, 10, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Leklem, J.E. Quantitative aspects of tryptophan metabolism in humans and other species: A review. Am. J. Clin. Nutr. 1971, 24, 659–672. [Google Scholar] [PubMed]
- King, N.J.; Thomas, S.R. Molecules in focus: Indoleamine 2,3-dioxygenase. Int. J. Biochem. Cell Biol. 2007, 39, 2167–2172. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, M.; Yamamoto, Y.; Kawasoe, M.; Arioka, Y.; Murakami, Y.; Hoshi, M.; Saito, K. Studies on tissue and cellular distribution of indoleamine 2,3-dioxygenase 2: The absence of IDO1 upregulates IDO2 expression in the epididymis. J. Histochem. Cytochem. 2012, 60, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Shirey, K.A.; Carlin, J.M. Synergistic transcriptional activation of indoleamine dioxygenase by IFN-gamma and tumor necrosis factor-alpha. J. Interferon Cytokine Res. 2003, 23, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Manlapat, A.K.; Kahler, D.J.; Chandler, P.R.; Munn, D.H.; Mellor, A.L. Cell-autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19+ dendritic cells. Eur. J. Immunol. 2007, 37, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Crabtree, J.M.; Sage, L.K.; Tompkins, S.M.; Tripp, R.A. Interferon Lambda Upregulates IDO1 Expression in Respiratory Epithelial Cells After Influenza Virus Infection. J. Interferon Cytokine Res. 2015, 35, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Adams, O.; Besken, K.; Oberdorfer, C.; MacKenzie, C.R.; Russing, D.; Daubener, W. Inhibition of human herpes simplex virus type 2 by interferon gamma and tumor necrosis factor alpha is mediated by indoleamine 2,3-dioxygenase. Microbes Infect. 2004, 6, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Ito, H.; Ando, K.; Ishikawa, T.; Hara, A.; Taguchi, A.; Saito, K.; Takemura, M.; Imawari, M.; Moriwaki, H.; et al. Upregulation of indoleamine 2,3-dioxygenase in hepatocyte during acute hepatitis caused by hepatitis B virus-specific cytotoxic T lymphocytes in vivo. Liver Int. 2009, 29, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Riezu-Boj, J.I.; Gil-Guerrero, L.; Casares, N.; Aldabe, R.; Sarobe, P.; Civeira, M.P.; Heeney, J.L.; Rollier, C.; Verstrepen, B.; et al. Upregulation of indoleamine 2,3-dioxygenase in hepatitis C virus infection. J. Virol. 2007, 81, 3662–3666. [Google Scholar] [CrossRef] [PubMed]
- Terajima, M.; Leporati, A.M. Role of Indoleamine 2,3-Dioxygenase in Antiviral Activity of Interferon-gamma Against Vaccinia Virus. Viral Immunol. 2005, 18, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Ribaudo, M.; Guo, J.T.; Barik, S. Identification of Interferon-Stimulated Gene Proteins That Inhibit Human Parainfluenza Virus Type 3. J. Virol. 2016, 90, 11145–11156. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Schultze, J.L. New Insights into IDO Biology in Bacterial and Viral Infections. Front. Immunol. 2014, 5, 384. [Google Scholar] [CrossRef] [PubMed]
- Gaelings, L.; Soderholm, S.; Bugai, A.; Fu, Y.; Nandania, J.; Schepens, B.; Lorey, M.B.; Tynell, J.; Vande Ginste, L.; Le Goffic, R.; et al. Regulation of kynurenine biosynthesis during influenza virus infection. FEBS J. 2017, 284, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Klonowski, K.D.; Tompkins, S.M.; Tripp, R.A.; Mellor, A.L. Induction and role of indoleamine 2,3 dioxygenase in mouse models of influenza a virus infection. PLoS ONE 2013, 8, e66546. [Google Scholar] [CrossRef] [PubMed]
- Panasiuk, A.; Prokopowicz, D.; Zak, J.; Matowicka-Karna, J.; Osada, J.; Wysocka, J. Activation of blood platelets in chronic hepatitis and liver cirrhosis P-selectin expression on blood platelets and secretory activity of beta-thromboglobulin and platelet factor-4. Hepatogastroenterology 2001, 48, 818–822. [Google Scholar] [PubMed]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Whitmire, J.K.; Marchese, P.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc. Natl. Acad. Sci. USA 2008, 105, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Contaldo, C.; Georgiev, P.; El-Badry, A.M.; Recher, M.; Kurrer, M.; Cervantes-Barragan, L.; Ludewig, B.; Calzascia, T.; Bolinger, B.; et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat. Med. 2008, 14, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Herbeuval, J.P.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Fuchs, D.; Shearer, G.M. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 2007, 109, 3351–3359. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Shearer, G.M. HIV-induced type I interferon and tryptophan catabolism drive T cell dysfunction despite phenotypic activation. PLoS ONE 2008, 3, e2961. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; Croteau, J.D.; Shirk, E.N.; Engle, E.L.; Zink, M.C.; Graham, D.R. Distinct Patterns of Tryptophan Maintenance in Tissues during Kynurenine Pathway Activation in Simian Immunodeficiency Virus-Infected Macaques. Front. Immunol. 2016, 7, 605. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.-S.; Firek, A.F.; Langridge, W.H.R. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines 2015, 3, 703–729. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Urade, Y.; Tokuda, M.; Hayaishi, O. Induction of indoleamine 2,3-dioxygenase in mouse lung during virus infection. Proc. Natl. Acad. Sci. USA 1979, 76, 4084–4086. [Google Scholar] [CrossRef] [PubMed]
- Guillonneau, C.; Mintern, J.D.; Hubert, F.X.; Hurt, A.C.; Besra, G.S.; Porcelli, S.; Barr, I.G.; Doherty, P.C.; Godfrey, D.I.; Turner, S.J. Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Fallarini, S.; Paoletti, T.; Panza, L.; Lombardi, G. Alpha-galactosylceramide modulates the induction of indoleamine 2,3-dioxygenase in antigen presenting cells. Biochem. Pharmacol. 2008, 76, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Sage, L.K.; Huang, L.; Barber, J.; Klonowski, K.D.; Mellor, A.L.; Tompkins, S.M.; Tripp, R.A. Inhibition of indoleamine 2,3-dioxygenase enhances the T-cell response to influenza virus infection. J. Gen. Virol. 2013, 94, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Sage, L.K.; Poore, S.; Johnson, S.; Tompkins, S.M.; Tripp, R.A. Drug analog inhibition of indoleamine 2,3-dioxygenase (IDO) activity modifies pattern recognition receptor expression and proinflammatory cytokine responses early during influenza virus infection. J. Leukoc. Biol. 2014, 96, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Müllers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of diverse retroviruses by SAMHD1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.T.; White, T.E.; Brandariz-Nunez, A.; Diaz-Griffero, F.; Weitzman, M.D. SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J. Virol. 2013, 87, 12949–12956. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhu, M.; Pan, X.; Zhu, Y.; Yan, H.; Jiang, T.; Shen, Y.; Dong, X.; Zheng, N.; Lu, J.; et al. Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun. 2014, 450, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Goutieres, F.; Aicardi, J.; Barth, P.G.; Lebon, P. Aicardi-Goutieres syndrome: An update and results of interferon-alpha studies. Ann. Neurol. 1998, 44, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, W.; Cao, X. Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol. Lett. 2000, 74, 221–224. [Google Scholar] [CrossRef]
- Pauls, E.; Jimenez, E.; Ruiz, A.; Permanyer, M.; Ballana, E.; Costa, H.; Nascimiento, R.; Parkhouse, R.M.; Pena, R.; Riveiro-Munoz, E.; et al. Restriction of HIV-1 replication in primary macrophages by IL-12 and IL-18 through the upregulation of SAMHD1. J. Immunol. 2013, 190, 4736–4741. [Google Scholar] [CrossRef] [PubMed]
- Riess, M.; Fuchs, N.V.; Idica, A.; Hamdorf, M.; Flory, E.; Pedersen, I.M.; Konig, R. Interferons Induce Expression of SAMHD1 in Monocytes through Down-regulation of miR-181a and miR-30a. J. Biol. Chem. 2017, 292, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Peng, X.; Liu, F.; Cheng, L.; Xie, T.; Lu, X.; Wu, H.; Wu, N. Interferon-induced sterile alpha motif and histidine/aspartic acid domain-containing protein 1 expression in astrocytes and microglia is mediated by microRNA-181a. Aids 2016, 30, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- St Gelais, C.; de Silva, S.; Amie, S.M.; Coleman, C.M.; Hoy, H.; Hollenbaugh, J.A.; Kim, B.; Wu, L. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4(+) T-lymphocytes cannot be upregulated by interferons. Retrovirology 2012, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Schaller, T.; Galao, R.P.; Amie, S.M.; Kim, B.; Olivieri, K.; Neil, S.J.; Malim, M.H. Evidence for IFNalpha-induced, SAMHD1-independent inhibitors of early HIV-1 infection. Retrovirology 2013, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nunez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ryoo, J.; Oh, C.; Hwang, S.; Ahn, K. SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology 2015, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Hwang, S.-Y.; Choi, J.; Oh, C.; Ahn, K. Reply to SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1074. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; St Gelais, C.; de Silva, S.; Yount, J.S.; Tang, C.; Ji, X.; Shepard, C.; Xiong, Y.; Kim, B.; Wu, L. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Wu, L. SAMHD1 knockout mice: Modeling retrovirus restriction in vivo. Retrovirology 2013, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; Schumann, T.; Gerbaulet, A.; Nguyen, L.A.; Schubert, N.; Alexopoulou, D.; Berka, U.; Lienenklaus, S.; Peschke, K.; Gibbert, K. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep. 2013, 4, 689. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hao, C.; DeLucia, M.; Swanson, S.; Florens, L.; Washburn, M.P.; Ahn, J.; Skowronski, J. CyclinA2-Cyclin-dependent Kinase Regulates SAMHD1 Protein Phosphohydrolase Domain. J. Biol. Chem. 2015, 290, 13279–13292. [Google Scholar] [CrossRef] [PubMed]
- White, T.E.; Brandariz-Nunez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 2013, 13, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Suarez, H.; Alvarez, S.; Lopez-Martin, S.; Lenzi, G.M.; Vences-Catalan, F.; Levy, S.; Kim, B.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; et al. CD81 association with SAMHD1 enhances HIV-1 reverse transcription by increasing dNTP levels. Nat. Microbiol. 2017, 2, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Baldauf, H.M.; Keppler, O.T.; Fackler, O.T. Restrictions to HIV-1 replication in resting CD4(+) T lymphocytes. Cell Res. 2013, 23, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics 2017: Monitoring Health for the SDGs, Sustainable Development Goals; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Li, T.C.; Chan, M.C.; Lee, N. Clinical Implications of Antiviral Resistance in Influenza. Viruses 2015, 7, 4929–4944. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Fry, A.M.; Gubareva, L.V. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir. Ther. 2012, 17, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Thorlund, K.; Awad, T.; Boivin, G.; Thabane, L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect. Dis. 2011, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Reece, P.A. Neuraminidase inhibitor resistance in influenza viruses. J. Med. Virol. 2007, 79, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; et al. Targeting Metabolic Reprogramming by Influenza Infection for Therapeutic Intervention. Cell Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Kurkjian, C.; Infante, J.R.; Bauer, T.M.; Burris, H.A., 3rd; Greco, F.A.; Shih, K.C.; Thompson, D.S.; Lane, C.M.; Finney, L.H.; et al. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.; Habib, N.; Guzzardi, M.A.; Kitsberg, D.; Bomze, D.; Ezra, E.; Uygun, B.E.; Uygun, K.; Trippler, M.; Schlaak, J.F.; et al. Nuclear receptors control pro-viral and antiviral metabolic responses to hepatitis C virus infection. Nat. Chem. Biol. 2016, 12, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Bonzo, J.A.; Ferry, C.H.; Matsubara, T.; Kim, J.H.; Gonzalez, F.J. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J. Biol. Chem. 2012, 287, 7345–7356. [Google Scholar] [CrossRef] [PubMed]
- Holloway, M.G.; Miles, G.D.; Dombkowski, A.A.; Waxman, D.J. Liver-specific hepatocyte nuclear factor-4alpha deficiency: Greater impact on gene expression in male than in female mouse liver. Mol. Endocrinol. 2008, 22, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.S.; Schieffer, D.; Cherry, S. AMP-Activated Kinase Restricts Rift Valley Fever Virus Infection by Inhibiting Fatty Acid Synthesis. PLoS Pathog. 2012, 8, e1002661. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; He, M.; Jung, J.U.; Lu, C.; Gao, S.J. Suppression of Kaposi’s Sarcoma-Associated Herpesvirus Infection and Replication by 5′-AMP-Activated Protein Kinase. J. Virol. 2016, 90, 6515–6525. [Google Scholar] [CrossRef] [PubMed]
- Pold, R.; Jensen, L.S.; Jessen, N.; Buhl, E.S.; Schmitz, O.; Flyvbjerg, A.; Fujii, N.; Goodyear, L.J.; Gotfredsen, C.F.; Brand, C.L.; et al. Long-term AICAR administration and exercise prevents diabetes in ZDF rats. Diabetes 2005, 54, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Idrovo, J.P.; Yang, W.L.; Jacob, A.; Aziz, M.; Nicastro, J.; Coppa, G.F.; Wang, P. AICAR attenuates organ injury and inflammatory response after intestinal ischemia and reperfusion. Mol. Med. 2015, 20, 676–683. [Google Scholar] [PubMed]
- Mounce, B.C.; Cesaro, T.; Vlajnić, L.; Vidiņa, A.; Vallet, T.; Weger-Lucarelli, J.; Passoni, G.; Stapleford, K.A.; Levraud, J.P.; Vignuzzi, M. Chikungunya Virus Overcomes Polyamine Depletion by Mutation of nsP1 and the Opal Stop Codon To Confer Enhanced Replication and Fitness. J. Virol. 2017, 91, e00344-17. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raniga, K.; Liang, C. Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses 2018, 10, 36. https://doi.org/10.3390/v10010036
Raniga K, Liang C. Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses. 2018; 10(1):36. https://doi.org/10.3390/v10010036
Chicago/Turabian StyleRaniga, Kavita, and Chen Liang. 2018. "Interferons: Reprogramming the Metabolic Network against Viral Infection" Viruses 10, no. 1: 36. https://doi.org/10.3390/v10010036
APA StyleRaniga, K., & Liang, C. (2018). Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses, 10(1), 36. https://doi.org/10.3390/v10010036