Abstract
Type-I interferons (IFN-I) play an important role in the innate immune response to several retroviruses. They seem to be effective in controlling the in vivo infection, though many of the clinical signs of retroviral infection may be due to their continual presence which over-stimulates the immune system and activates apoptosis. IFN-I not only affect the immune system, but also operate directly on virus replication. Most data suggest that the in vitro treatment with IFN-I of retrovirus infected cells inhibits the final stages of virogenesis, avoiding the correct assembly of viral particles and their budding, even though the mechanism is not well understood. However, in some retroviruses IFN-I may also act at a previous stage as some retroviral LTRs posses sequences homologous to the IFN-stimulated response element (ISRE). When stimulated, ISREs control viral transcription. HIV-1 displays several mechanisms for evading IFN-I, such as through Tat and Nef. Besides IFN-α and IFN-β, some other type I IFN, such as IFN-τ and IFN-ω, have potent antiviral activity and are promising treatment drugs.
1. Retroviruses
Retroviruses are enveloped viruses, with single stranded RNA, and with an unusual replication strategy through a double stranded DNA intermediary. This process is accomplished thanks to the enzyme reverse transcriptase, which directs the synthesis of DNA. Once as DNA, the nucleic acid integrates in the genome of the host cell, where it behaves as another gene. Retroviral particles are surrounded by an envelope, which contains two types of viral glycoproteins: SU (surface) and TM (transmembrane); under the envelope the matrix proteins (MA) confer stability to the viral particle. The core or capsid is mostly formed by capsid proteins (CA); it lodges inside the RNA surrounded by the proteins of the nucleocapsid (NC), and the enzymatic proteins: protease (PR), retrotranscriptase or reverse transcriptase (RT) and integrase (IN). Some viruses also have the enzyme dUTPase (DU).
Viral particles have two identical copies of the ARN genome, which have a cap sequence in the 5’ end and a polyadenylated tract or poli(A) in the 3’ end, but do not function as mRNA. Four genes are necessary for infectious viruses: gag, pro, pol and env (Figure 1). Gene gag (group specific antigen) codes for the structural internal proteins of the virus (MA, CA, NC and other proteins specific to certain viruses with undetermined function). Enzymatic proteins necessary for viral replication are coded by genes pro (protease, PR). and pol (polymerase, IN, RT and, in some retroviruses, DU). In some retroviruses, pro is in the same ORF as either gag or pol. Gene env (envelope) codes for the proteins present in the envelope (SU and TM). Deltaretroviruses, such as human T-cell leukemia virus (HTLV) or bovine leukemia virus (BLV) have two additional genes, tax and rex [100], whereas lentiviruses posses, besides the mentioned genes, other genes (such as tat, rev, vif, vpr, and others) which encode for non-structural proteins relevant for the regulation of expression, viral replication, pathogenesis, etc [100].
Depending on the virus, virions attach to a number of different specific cellular receptors via their envelope glycoproteins. In most cases, the virus envelope and the cell membrane fuse, allowing the virion core to enter the cytoplasm; in fewer instances, entry involves receptor-mediated endocytosis. Decapsidation, mediated by membrane proteases or by lytic enzymes in the phagolysosomes, is almost immediate. Thanks to the RT, a dsDNA molecule is synthesized using the RNA as template. During this stage, the RT adds 300-1200 base pairs (bp) in each end of the nucleic acid, which become the structures known as Long Terminal Repeats (LTRs, see below). The new dsDNA molecule crosses the nuclear membrane, and integrates (by ways of the viral integrase) in the chromosomal DNA of the host cell. Initially it was believed that this process happened mostly at random, but nowadays it is recognized that different retroviruses display strong preferences in selecting their target sites in the genomes of their host cells [for a review, see 22]. For instance, murine leukemia virus (MuLV) prefers to integrate at transcription start sites, and human immunodeficiency virus (HIV)-1 displays a strong preference for highly active genes [130]. Many of the biological characteristics of retroviruses, such as the activation of oncogenes, or the possibility that retroviruses become endogenous and be transmitted through generations as Mendelian genes, derive from this stage. When the viral genome is integrated in the genome of the host cell it is termed “provirus”.
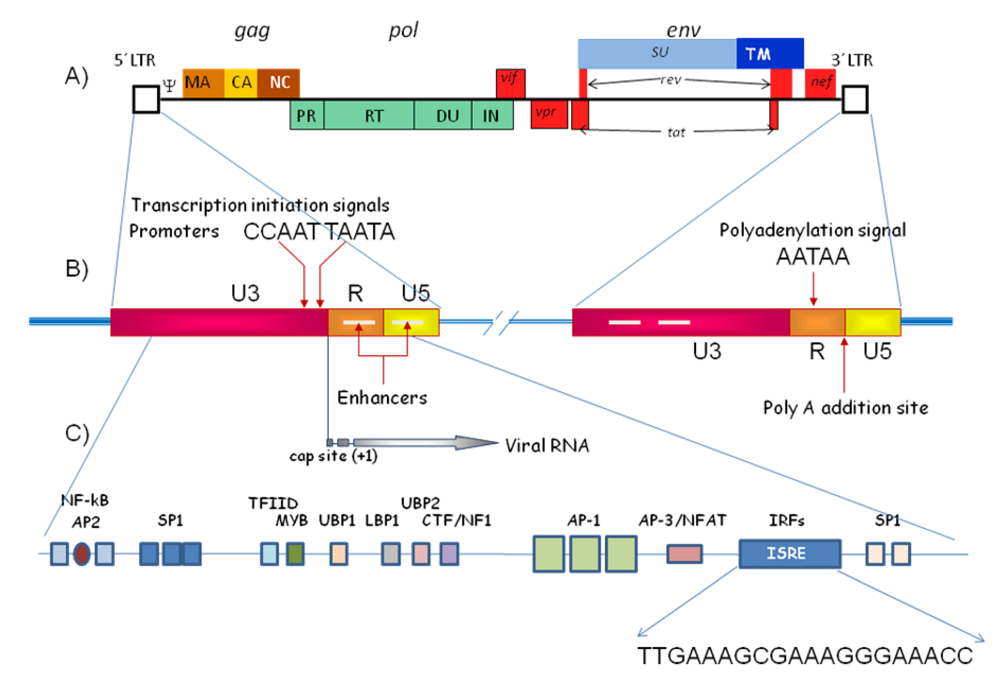
Figure 1.
A) Schematic representation of the genomic structure of HIV-1, B) Blow-up of the two Long Terminal Repeats (LTR) which flank the proviral sequence, C) Some of the regulatory elements in the 5’ LTR, showing the sequence of the Interferon-Stimulated Response Elements (ISRE).
The provirus stays integrated in the host DNA during periods of time of variable length. At some point, mRNA is transcribed from the viral DNA, and once in the cytoplasm it is translated into the specific proteins of the virus. These proteins, along with viral RNA transcribed from the DNA, assemble to form new viral particles that are released from the cell budding from the plasma membrane.
1.1. Long Terminal Repeats (LTR)
The RNA genome of retroviral particles is flanked by two short repeated sequences (R), associated to the regions U5 and U3 in the 5’and 3’ends, respectively. U5 and U3 are duplicated during the reverse transcription of the RNA genome into dsDNA, providing the provirus with a new terminal sequence U3-R-U5 flanking each end, which binds covalently to the host DNA (Figure 1). These new sequences are termed Long Terminal Repeats (LTRs); their size ranges between 300 and 1200 bp, depending on the retrovirus, and they are the control centers for gene expression, containing all of the requisite signals for this function: enhancers, promoters, transcription initiation (capping), transcription terminator and polyadenylation signal.
The transcription starts at the beginning of R, is capped and proceeds through U5 and the rest of the provirus, usually terminating by the addition of a poly A tract just after the R sequence in the 3’ LTR. The LTR contains two functional domains: a promoter region, and an enhancer region (Figure 1), which bind several protein factors (both general and tissue-specific). Enhancers are an important class of transcriptional regulatory elements, downstream from the transcription start site (i.e. cis-acting). Enhancers identified up-to-date show very little homology; they usually have a core domain, about 10-15 bp long, that binds a specific cellular factor (such as hormones, mitogens, cytokines, nutrients or toxins), which regulates their expression [53,111]. Most of these elements have a positive effect on transcription, but some do have a negative effect. The different members of these families vary significantly in their relative abundance, in their functional activity and in their interactions with other additional proteins, generating a complex transcriptional enhancers’ network, which is considered crucial [34,75,134,139]. Thus, the LTR sequences of retroviruses in general, but especially of lentiviruses, offer various possibilities for regulation. This, along with the activity of the accessory regulatory proteins, allows the virus to alter specific biochemical routes of the cell, and enhance viral survival and its spread within the host, permitting them even to invade cellular environments not permissible to other viruses, such as cells which are not dividing.
Some retroviruses contain additional enhancer sites, homologous to sequences detected in genes which are sensitive to the action of interferon (IFN), termed Interferon Stimulated Response Elements or ISREs (see below) (Figure 1). These sequences allow them to respond in a dose dependent manner to the presence of IFN, which thus directs transcription.
The 3’ LTR is not normally functional as a promoter, although it has exactly the same sequence arrangement as the 5’ LTR. The function of the 3’ LTR is to act in transcription termination and polyadenylation.
2. Interferon
Interferons (IFNs) are a multigene family of inducible cytokines, which regulate immunity in infectious diseases and tumours [11,17]. IFNs are commonly grouped into two types [107]. Type I IFNs (IFN-I), which possess antiviral activity and are associated with the innate immunity, are also known as viral IFNs and include amongst others IFN-α (leukocyte IFN), IFN-β (fibroblast IFN), IFN-ω, and IFN-τ. Type II IFN (IFN-II), produced by activated T-cells and natural killer cells, is also known as immune IFN (IFN-γ) [16]. Type I IFNs induce antiproliferative and antiviral responses, and not only play an important role in the innate immune response, but also influence the generation of the adaptive immune responses [7,52,117].
The induction of type I IFNs is regulated by two signal transduction pathways, both of which are activated by viruses [6,56,143]: the classical and the Toll-like receptors (TLR) pathways.
Most cells, including fibroblasts, hepatocytes, and conventional dendritic cells, use the so-called classical pathway. These cells have cytosolic receptors (CR) [6] that are able to recognize viral nucleic acids within the infected cells. CR includes the RIG-I-like RNA helicase receptor (RLH) family: retinoic acid inducible gene-I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) [138]. The presence of viral dsRNA activates the routes of NF-κB and Interferon Regulatory Factor (IRF)-3, the main regulatory factors for IFN transcription. NF-κB and IRF-3 translocate to the nucleus and bind the promoter for the IFN-β gene, forming a complex named “enhanceasome”, which combines with other co-activators and RNA-polymerase, in order to transcribe IFN-β. Thus, cells secrete initially IFN-β. In the following amplification stage, IFN-β triggers the expression of IRF-7. This factor has been proposed as the main regulator of the expression of IFN-I genes [64], and, in collaboration with IRF-3, triggers the synthesis of IFN-α [56,95] (Figure 2).
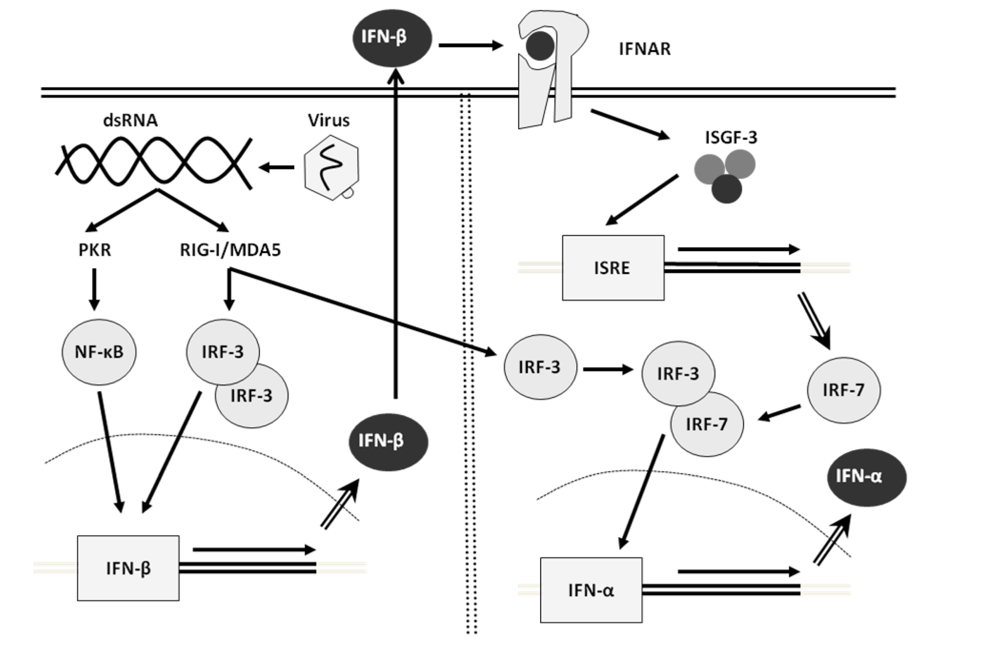
Figure 2.
Simplified mechanism of regulation of the induction of the interferon genes in most cells. PKR, RNA dependent protein kinase; IRF, interferon regulatory factor; ISGF, IFN-stimulated gene factor; IFNAR, interferon alpha receptor; ISRE, interferon stimulated response elements. Ovals represent proteins, squares represent genes.
Plasmacytoid dendritic cells (pDCs), also known as precursors of type 2 dendritic cells or natural IFN-producing cells, use the Toll-like receptors (TLR) pathway, expressed in the cell surface or in the endosomes, sensitive to viral DNA and RNA. The TLR signal activates IRF-7, which is constitutively expressed in these cells [50,56,58,62,65,123], directly originating the secretion of high levels of IFN-α [31,37,49,56] (Figure 3).
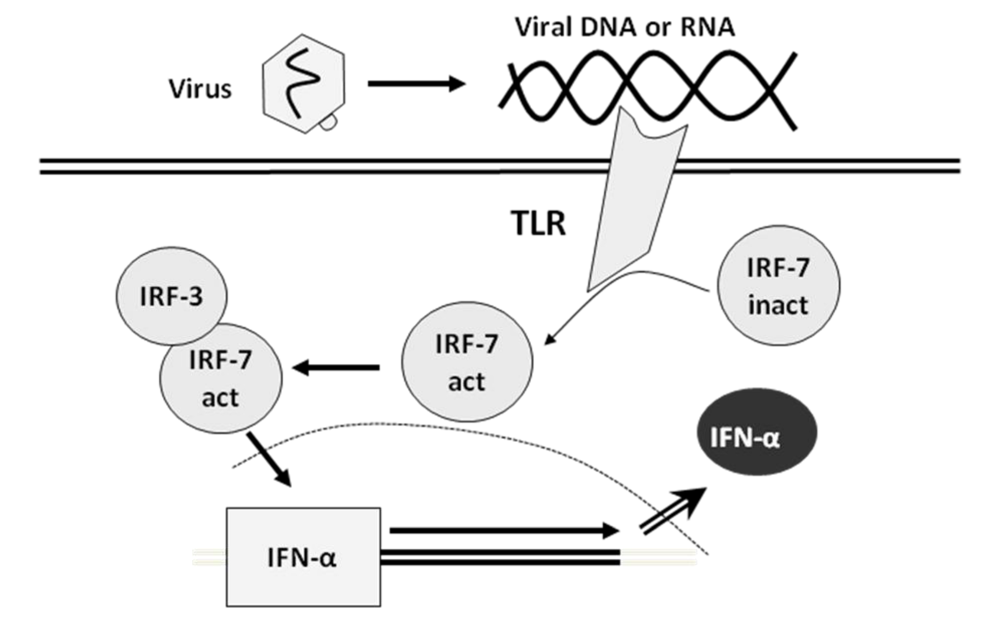
Figure 3.
Simplified mechanism of regulation of the induction of the interferon genes in dendritic cells. IRF, interferon regulatory factor. Ovals represent proteins, squares represent genes.
Once type I IFN is synthesized, it binds to the interferon alpha receptor (IFNAR), the specific receptor for IFN-I on the cell membrane, formed by two subunits: IFNAR-1 and IFNAR-2 (Figure 4). The binding of IFN-I to IFNAR-1 and 2 produces the heterodimerization of both subunits [10,98,135], which activates the tyrosine kinases TYK-2 and JAK-1. This phosphorylates a related transcription factor which is in the cytoplasm, and which is involved in the signal transduction of several molecules, termed STAT (Signal Transduction and Activator of Transcription) [12,85]. Of the several STAT proteins, IFN-α or -β phosphorylate STAT-1 and STAT-2, which bind p48 (IRF-9), forming a trimer (IFN-stimulated gene factor-3, ISGF-3). ISGF-3 translocates to the nucleus and binds the so-called Interferon-Stimulated Response Elements (ISRE) in IFN-inducible genes (ISGs). As mentioned before, ISREs are cis-acting DNA regulatory sequences; they have a consensus sequence a/gNGAAANNGAAACT [142,146], very similar to the IRF elements (IRF-E, whose consensus sequence G(A)AAAg/ct/cGAAAg/ct/c is present within the promoters of IFN-I and of most ISGs) [96] (Figure 4). Antiviral activities associated with IFN-α include, among others, the induction of protein Mx GTPase, activation of protein kinase R (PKR) leading to mRNA translation, and activation of the 2’,5’-OAS/RNase L system, resulting in RNA degradation [54,119].
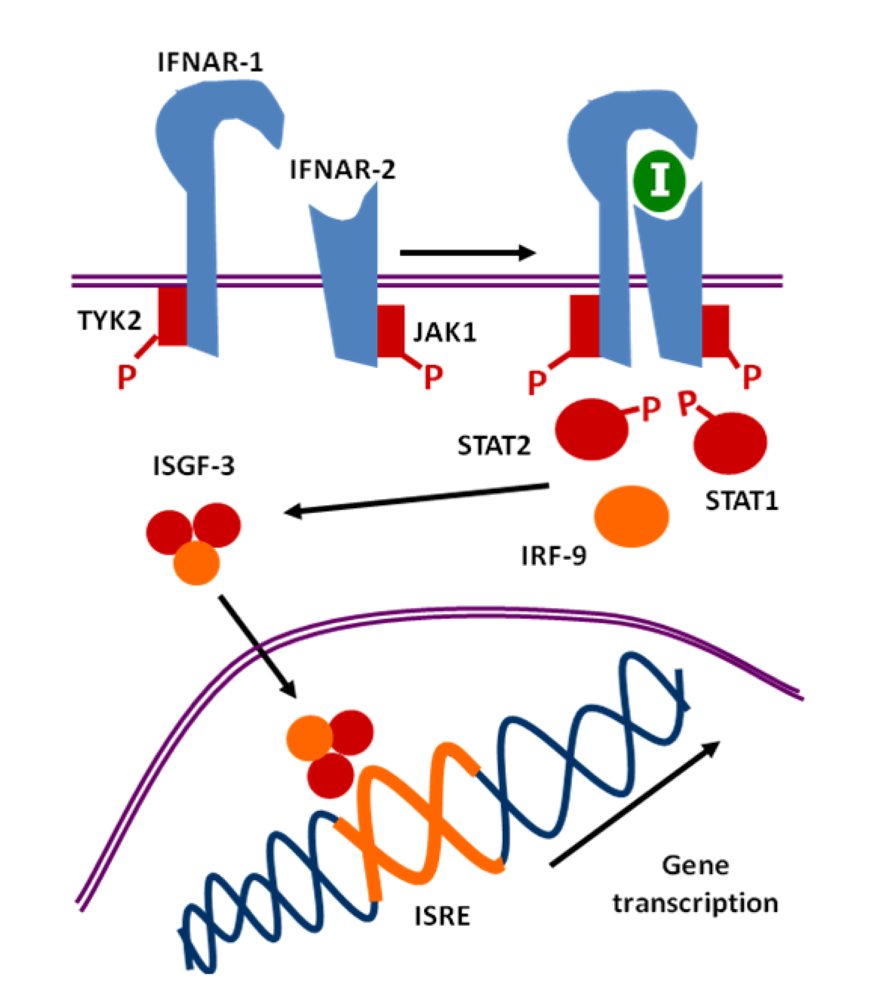
Figure 4.
Induction mechanism of genes by type I interferons. IFNAR, IFN-α receptor; IRF, interferon regulatory factor; ISGF, IFN-stimulated gene factor; ISRE, interferon-stimulated response element; TYK, tyrosine kinase; JAK, Janus kinase; STAT, Signal Transduction and Activator of Transcription.
IFN-I plays an important role in the defense against viral infections, both through IFN-induced proteins and enzymes mentioned above (innate response), as well as through its effect on the adaptive immune response, since the maturation of dendritic cells by IFN is important for the stimulation of T-cells [52,117]. In addition, type I IFNs increase the expression of class I MHC molecules in all cells, contributing to the removal of the infected cells [7]. In HIV infections, results are controversial; some researchers have proposed that the effect of IFN may be beneficial, while other deleterious, since it may activate apoptosis of CD4+ T-cells, both non-infected and HIV-infected, through the expression of TNF-related apoptosis inducing ligand (TRAIL) and death receptor (DR) 5 in their membrane [60].
When IFN-I is administered to HIV-, simian immunodeficiency virus (SIV)-, or feline immunodeficiency virus (FIV)-infected individuals clinical signs are seen to improve [30,33,60,106,117], supporting the hypothesis that type I IFNs play important roles in the response to these retroviruses, as well as to other retroviruses, such as HTLV-I [46,79,117], BLV [78], feline leukemia virus (FeLV) [28,149], or MuLV [2,52].
These effects of IFN-I may be observed both in vivo and in vitro. In the following paragraphs evidence will be discussed on the observations on
- IFN-I levels in natural and experimental retroviral infection, and whether they are protective or deleterious, and association to specific syndromes, such as dementia
- Possible impairment of pDCs as the cause of altered IFN levels
- Effects of IFN on virogenesis
- Effects of IFN at the cellular level: induction of apoptosis in infected and non infected cells
- Effects of IFN at the molecular level: presence of ISREs in retroviral genome (LTRs)
- Mechanisms by which retroviruses may evade the effect of IFN-I
Most data originate from studies performed with IFN-α. However, due to their similarity, in the next sections either IFN-α or IFN-β are referred to as IFN-I. In the last section, some results obtained with other type I IFNs are reviewed.
5. Effects of IFN at the Cellular Level: Apoptosis
In the preceding section, it has been discussed that type I IFNs inhibit the final release of retrovirus particles from the cells, resulting in an accumulation of cell-associated virions, or producing defective noninfectious virus particles, apparently due to the interference with the processing of viral proteins and their assembly into complete virions, without altering significantly retroviral protein synthesis. This prompted researchers to hypothesize that these effects of IFN-I are a result of a cellular interaction with IFN rather than a result of the antiviral activity of IFN [32].
One of the effects of IFN on the cell is the induction of apoptosis. Apoptosis plays a critical role in cellular differentiation, in the elimination of cells that sustain genetic damage or undergo uncontrolled cellular proliferation, and in preventing viral replication by eliminating virus-infected cells [24]. The potential of IFN to induce apoptosis has been known for some time. In general, the prolonged activity of IFN-induced proteins leads to cell death by apoptosis, a response that certainly limits spread of virus from one cell to another. IFN activates the expression of genes which contribute to apoptosis. For example, STAT1, which is critical for signaling for both types of IFN, has been proposed to play a role also in apoptosis. PKR molecules regulate the expression of genes involved in apoptosis, possibly involving signaling through the NF-κB pathway [13]. Though IFNs are positive mediators of cell death, there are instances where both type-I and –II IFNs may actually prevent apoptosis [13].
Strong and sustained induction of TRAIL and/or Fas/FasL in response to IFN has been shown to lead to recruitment and activation of the Fas associated death domain (FADD). FADD plays an important role in IFN mediated apoptosis as transfection experiments, using dominant negative mutants of FADD, conferred IFN-resistance to sensitive cells. FADD activation, in turn, activates caspase-8, initiating activation of the caspase cascade. Activated caspase-8 cleaves Bid, a proapoptotic member of Bcl2 family, resulting in disruption of mitochondrial potential, release of cytochome c from the mitochondria into the cytoplasm, where it acts as a cofactor to stimulate the complexing of Apaf1 with caspase-9. This complex potentiates caspase-3 activation, followed by changes in plasma membrane symmetry, cleavage of PARP, chromatin condensation and DNA fragmentation and cell death [24].
It was long envisioned that the effect of IFN on retroviruses could be a consequence of its interaction with the cell membrane. Thus, apoptosis induction and the existence of lipid rafts were predicted [2]. Using selective stains, type I IFNs have been shown to induce apoptosis in FeLV- and FIV-infected cells, to a degree several orders of magnitude higher than in non-treated cells [28,29]. It was concluded that the intracellular and/or the slight changes in the cell membrane derived from the synergic action of viral infection and IFN-I could lead to a reduced release of viral particles (evaluated indirectly by the RT activity). This would limit the spread of the infection to other cells, and originate the selective death of infective cells [28].
The effects of apoptosis induced by IFN are not limited to retrovirus-infected cells. More notably apoptosis stimulated by IFN-I has been proposed as a mechanism to explain non-infected bystander CD4+ T-cells depletion and disease progression in HIV infection [1,60,61,68,92,96]: in vitro, HIV-1 gp120(SU) in the membrane of infected CD4+ T-cells stimulates the production of IFN in dendritic cells, which induces the expression of TRAIL by infected and non-infected CD4+ T-cells [61]. According to this model, in progressing individuals with high plasma viral loads, HIV binds to CD4 on pDCs, resulting in their activation, IFN-α production and migration from blood to lymphoid tissues. IFN-α binds to its receptor on infected and non infected CD4+ T-cells (resulting in STAT-1/2 regulated expression of membrane TRAIL), and HIV gp120(SU) binds to CD4 on these cells. This latter event is required for the expression of the TRAIL death receptor 5 (DR5). Given that a high percentage of viral particles are not infective, but they still carry gp120(SU), DR5 is expressed on many non-infected CD4+ T-cells. TRAIL binds to DR5, resulting in CD4+ T-cell apoptosis. Contrariwise, in non progressing patients with very low plasma viral loads, pDCs do not produce IFN-α and the expression of TRAIL is not induced, and the previous cascade, resulting in apoptosis, is not initiated [60]. According to another hypothesis, the viral protein Nef, a virulence factor that plays multiple roles in HIV replication, is capable of inducing proapoptotic effects in uninfected bystander cells, and antiapoptotic effects in infected cells. Nef activates the synthesis and secretion of a set of chemokines/cytokines that activate STAT1 and STAT3, as well as IRF-3, leading to the synthesis of IFN-β, which, in turn, induces STAT2 phosphorylation [92].
This cytotoxic mechanism and the stimulation of NK cells [145] may be responsible for the high degree of apoptosis observed in non-infected CD4+ T-cells in HIV-1 infected patients [42]. Other mechanisms involve the upregulation by HIV-1 of the gene expression of p53, associated with HIV-mediated IFN-I synthesis [68], or the induction of IRF-1 by HIV-1, as this factor is clearly involved in apoptosis of activated T-cells and in the context of the machinery that T- cells use to induce apoptosis in their target cells [96].
6. Direct Effects of Type I Interferons at the Molecular Level: ISREs
As mentioned previously, ISREs are DNA sequences which bind with the well-conserved N-terminal region of the interferon regulatory factors (IRFs). By this mechanism, IRFs regulate the expression of genes stimulated by IFN, activating or either repressing gene transcription, depending on the target gene. IRFs are involved in multiple biological processes including regulation of immune responses and host defense, cytokine signaling, cell growth regulation and hematopoietic development. ISREs are present on the promoters of the target genes, i.e. the IFN-α or IFN-β genes and some IFN-stimulated genes (ISG) (for a review of different ISRE sequences, see 142]. Sequences homologous to the ISREs have been identified in several retroviruses. For example, in HIV a sequence (TTGAAAGCGAAAGGGAAACC) homologous to ISRE has been identified in the 5’ HIV-1 LTR downstream of the HIV-1 transcription start site (Figure 1). This sequence is a binding site for several IRFs and its deletion results in impaired LTR promoter activity and altered synthesis of viral RNA and proteins [15]. In vivo it recruits IRF-1 and IRF-3 [144]. Other researchers have reported that it has a binding site for IRF-1 and IRF-2 [15,96], but only IRF-1 is able to stimulate HIV-LTR transcription (and reactivates provirus from latency), interacts with Tat [124], and increases HIV-1 replication [96,125]. HIV is able to induce IRF-1 early upon infection, before the expression of Tat, and it is possible that it plays an important role in the early phases of HIV infection and as a strategy to counteract IFN-mediated host defenses [124,125] On the other hand, IRF-8 represses IRF-1-Tat-mediated transactivation of the LTR by interfering with IRF-1-Tat association, and it inhibits HIV-1 replication in CD4+ cells [96,99].
In FeLV the sequence GGTTTCATTTTCG, matching the consensus NAGTTTCNNTTTCNC/T [146], was found at nucleotide 798, in a region just upstream from that which encodes the Gag-Pol precursor polyprotein gPr80; however, as it is not in the LTR regulatory region, it is difficult to link it with the initiation of transcription of the viral genome [28].
The region situated immediately downstream from the transcription start site (U5) in the BLV LTR is involved in regulation of viral gene expression. It contains a positive regulatory element in its 5’ region (nt +230 to +275) that functions either at the transcriptional or past-transcriptional level. This U5 region contains a sequence (nt +251 to +261) highly similar to an ISRE (TACTTTCTGTTTCTCG), which binds IRF-1 and IRF-2, but not ISGF3, and thus can be considered an IRF binding site, rather than a classical ISRE. Its failure to confer IFN responsiveness is most probably due to its inability to bind ISGF3. This motif is required for optimal basal gene expression from the BLV LTR [78].
Up till now, no ISRE sequences have been identified in HTLV, FIV or other retroviruses, despite the effects of IFN on these viruses, which suggests that the molecular mechanisms involved may be more complex than anticipated and are still to be clarified.
7. Mechanisms by which Retroviruses May Evade the Effects of Type I Inteferons
Viruses have evolved different mechanisms that allow them to evade the antiviral response induced by IFN. This anti-IFN activity may be produced at three distinct time points: during IFN synthesis, during signaling, and through the alteration of the antiviral proteins induced by IFN. Some viral proteins are suppressors of IFN gene expression through their general inhibitory effect on host gene transcription [56]. However, they can also evade specifically the action of IFN. The major strategies include [45,112] (a) competition for binding to IFN receptors; (b) inhibition of IFN production and secretion; (c) blockage of the signal of IFN; and (d) inhibition of the antiviral proteins induced by IFN or of their actions (Figure 5).
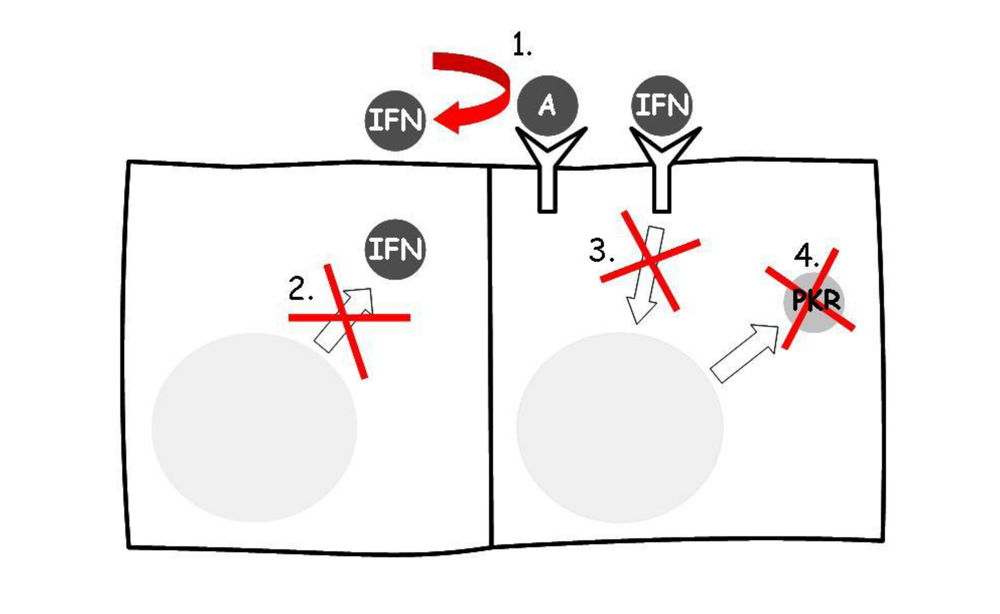
Figure 5.
Mechanisms by which viruses circumvent the effect of IFN. 1. Competition with IFN for receptors (IFNAR); 2. Inhibition of IFN synthesis and secretion; 3. Inhibition of the IFN signaling, which may happen at different levels; 4. Inhibition of the proteins stimulated by IFN, mostly of the PKR system.
The mechanisms by which retroviruses evade IFN control belong to the fourth category. The presence of increasing levels of IFN in the serum of AIDS patients while viral replication continues and the disease progresses indicates that HIV-1 must employ a mechanism to evade the antiviral effects of IFN. However, the complexity of the virus-host interactions and the profound disregulation of the host cytokine network exerted by the virus at different stages during the infection have made these studies extremely difficult. In response to viral infection, IFN induces a number of genes including the dsRNA-dependent protein kinase R (PKR). PKR exerts its anti-viral activity by phosphorylating the alpha subunit of eukaryotic translation initiation factor-2 (eIF-2), which results in the shut-down of protein synthesis in the cell. However, PKR activity is inhibited directly by HIV via the major regulatory protein, Tat [41]. This protein, in association with cellular factors, enhances the efficiency of transcription by cellular RNA polymerase 1000-fold, mainly by preventing premature termination of transcription. It triggers efficient RNA chain elongation by binding to TAR RNA, which forms the initial portion of the HIV-1 transcript. HIV-1 TAR is a highly-conserved stable RNA stem loop that interacts with Tat protein to regulate viral transcription [19]. HIV-1 Tat protein has been shown to act also as a substrate homologue of eIF-2, competing with it and preventing the phosphorylation of this factor, allowing protein synthesis and viral replication to proceed in the cell. Tat protein reduces the activity PKR significantly, while TAR RNA blocks its activation [41].
Nef, a crucial determinant for HIV replication and pathogenesis, is believed also to play a major role in the evasion of the virus from the effect of IFN, by manipulating the phenotypical, morphological and functional characteristics of pDCs, rendering them incapable of activating CD8+ T-cells and down-regulating their proliferation and functional competence [110].
The involvement of IRFs in HIV-1 replication allows to speculate that targeting IRFs (IRF-1, IRF-2 and IRF-8) can be also regarded as a mechanism utilized by HIV-1 to evade the antiviral effect of IFN [96]. Other proteins of HIV degrade PKR and induce an inhibitor of RNaseL [96,128].
HTLV blunts the expression of some genes induced by IFN, probably depressing the MxA, PKR and OAS pathways. Recently it has been described that HTLV-I did not have an effect either on the initiation of the IFN process (as it did not affect the cell surface presentation of IFNAR1 and IFNAR2 or IFN-α binding). However, it reduced the phosphorylation of TYK2 and STAT2, and to a lesser extent, of JAK1 and STAT1, suggesting that the virus inhibits the IFN-induced specific activation of these cellular proteins [45]. Evidence that Gag and Pro may be responsible for this HTLV-I mediated IFN-α inhibition has also been provided [45]. These effects have also been ascribed to the viral protein Tax, a pleiotropic transcription factor that interferes with several of the cellular mechanisms and modulates transcription of a wide range of cellular genes [20]. It has been proposed that Tax protein of HTLV-I acts as an IFN-α antagonist, as it negatively modulates IFN-α induced JAK-STAT pathway by competing with STAT2 for coactivator CBP/p300, thereby inhibiting the transcription activation of STAT2-containing ISGF3 complex [150]. Tax may prevent by this mechanism IFN-α from exerting its antiviral, antiproliferative and proapoptototic effects, contributing to persistent viral infection and HTLV-I associated oncogenesis [150]. Nevertheless, it is possible that different HTLV-I gene products, other than Tax, may act at different levels of the IFN signaling pathways, coexisting more than one mechanism to evade IFN action, as has been shown for other viruses.
8. Other Type I Interferons
8.1. Interferon-tau (IFN-τ)
IFN-τ is a noncytotoxic type I IFN responsible for maternal recognition of pregnancy in ruminants. It has been shown to have potent antiviral and antiproliferative activity. Data suggest that its anti-HIV activity is higher than IFN-α/β on primary PBMC and monocyte-derived macrophages infected in vitro by HIV [35]. Also, it induces IL-6 and IL-10 synthesis [91,115]. The IFN-τ antiretroviral activity is not associated with a decrease in either cell viability or immune reactivity [109], supporting the interest for the IFN-τ as an adjuvant therapy drug in HIV infection [26].
Like other IFNs, it seems to affect several steps of the HIV replication cycle. IFN-τ effectively inhibits the early steps of the biological cycle of HIV replication, particularly in human monocyte-derived macrophages, decreasing intracellular HIV RNA and inhibiting the initiation of the reverse transcription of viral RNA into proviral DNA. Anti-HIV effects of IFN-τ are mediated by several modes of action, either directly by IFN-τ, or via other cytokines [91]. The mechanisms proposed are i) synthesis of cellular antiviral factors such as 2’,5’-OAS/RNase L and MxA protein [26], and ii) increased production of MIP-1α, MIP-1β, RANTES, natural ligands of CCR5, the principal coreceptor of HIV in macrophages [114].
The effects of IFN-τ have also been studied on BLV. BLV titers decreased in BLV-infected cells (FLK-BLV) and in peripheral blood mononuclear cells of BLV-infected cattle treated with recombinant bovine IFN-τ (rBoIFN-τ), demonstrating that this cytokine could directly inhibit BLV propagation rather than acting through its immunomodulatory effects [80].
Significant dose-dependent inhibition of reverse transcriptase activity by IFN-τ was detected by day 6 of culture in FIV-infected feline PBL treated with IFN-τ. In addition, the production of the FIV core protein, p25(CA), was blocked. Both the amino- and carboxyl-terminal regions of IFN-τ, as identified by synthetic peptides, appeared to be involved in its antiretroviral activity. IFN-τ antiretroviral activity was not associated with a decrease in either cell viability or immunologic reactivity [109].
IFN-τ has an effect on the immune response of sheep. The immunomodulatory role of recombinant ovine IFN-τ (rOvIFN-τ) included the increase of the proportions of primary antiviral γδ+and CD8+ immune cells in ovine lentivirus (SRLV)-infected lambs. This may represent a cellular mechanism to explain the antiviral and therapeutic efficacy of this cytokine, in addition to its direct antiviral effect [129]. The effect of rOvIFN-τ on the replication of SRLV in goat synovial membrane cells was studied by Juste et al. [71]. The strongest inhibitory effects were on syncytia formation and release of infectious virions into the cell culture supernatant, though the production of RT was not significantly different in cells treated with IFN-τ and in control cells. These results suggest that, as other type I IFNs, the action of this IFN is mostly on the latter steps of SRLV replication cycle, possibly by blocking virus assembly and/or release, thus limiting the spread of the infection [71]. In vivo results demonstrated a 90% reduction in SRLV titres in lambs infected experimentally with SRLV and that received early rOvIFN-τ treatment [72].
8.2. Interferon-omega (IFN-ω)
IFN-ω is also a potent inhibitor of HIV replication in vitro; both laboratory and primary isolates of HIV-1 are more sensitive to IFN-ω than to IFN-α2, and protein synthesis is inhibited by IFN-ω to a greater degree than by IFN-α2. Data suggest that the expression of ISGs, particularly that of ISG-15, is higher and more sustained on treatment with the former than with the latter, which may confer a higher therapeutic index to IFN-ω in controlling HIV infection [82].
The in vitro effect of commercially available recombinant feline interferon omega (rFeIFN-ω) has been evaluated on the expression and replication of FeLV [28] and FIV [29,136]. Very similarly to IFN-α, rFeIFN-ω induced a marked inhibition of RT activity (and thus of infectivity), whereas it had no effect on protein synthesis.
In summary, type I IFNs seem to play an important role in the innate immune response against retroviruses, but their effect is not well established. In vivo IFN-I levels vary throughout retroviral infections, though findings are contradictory, as for some researchers, high levels seem to be beneficial for the host to fight infection, while for others, they would provide a mechanism for deleting by apoptosis non-infected bystander cells. However, exogenous treatment with IFN-I appear to be generally valuable. In vitro findings support that type I IFNs undoubtedly inhibit replication of retroviruses, though there is not an unanimous agreement about whether it is at the early or late stages of virogenesis, and of whether it is by directly affecting the replication process or by altering cellular viability through apoptosis. Some retroviruses have ISRE-like sequences, which might modulate viral expression, and some are able to evade the effects of IFN, possibly through the regulatory proteins.
References and Notes
- Abbate, I.; Dianzani, F.; Capobianchi, M. R. Activation of signal transduction and apoptosis in healthy lymphomonocytes exposed to bystander HIV-1-infected cells. Clin. Exp. Immunol. 2000, 122, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Aboud, M.; Kimchi, R.; Bakhanashvili, M.; Salzberg, S. Intracellular production of virus particles and viral components in NIH/3T3 cells chronically infected with Moloney murine leukemia virus: effect of interferon. J. Virol. 1981, 40, 830–838. [Google Scholar] [PubMed]
- Aboud, M.; Malik, Z.; Bari, S.; Kimchi, R.; Hassan, Y.; Salzberg, S. Effect of interferon on the formation and release of intracellular virions in NIH/3T3 cells chronically infected with Moloney murine leukemia virus. J. Interferon Res. 1983, 3, 33–44. [Google Scholar] [PubMed]
- Agy, M.B.; Acker, R.L.; Sherbert, C.H.; Katze, M.G. Interferon treatment inhibits virus replication in HIV-1- and SIV-infected CD4+ T-cell lines by distinct mechanisms: evidence for decreased stability and aberrant processing of HIV-1 proteins. Virology 1995, 214, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Cordero, M.; Almeida, J.; Orfao, A. Different subsets of peripheral blood dendritic cells show distinct phenotypic and functional abnormalities in HIV-1 infection. AIDS 2005, 19, 261–271. [Google Scholar] [PubMed]
- Alsharifi, M.; Müllbacher, A.; Regner, M. Interferon type I responses in primary and secondary infections. Immunol. Cell. Biol. 2008, 86, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, M.; Chassin, D.; Desoutter, J.F.; Bouchaert, I.; Baillet, M.; Hanau, D.; Guillet, J.G.; Hosmalin, A. Downregulation of major histocompatibility class I on human dendritic cells by HIV Nef impairs antigen presentation to HIV-specific CD8+ T lymphocytes. AIDS Res. Hum. Retroviruses 2001, 17, 1365–1370. [Google Scholar] [PubMed]
- Babé, L.M.; Unal, A.; Craik, C.S. Obstruction of HIV-1 particle release by interferon-α occurs before viral protease processing and is independent of envelope glycoprotein. J. Interferon Cytokine Res. 1997, 17, 287–93. [Google Scholar] [PubMed]
- Baca-Regen, L.; Heinzinger , N.; Stevenson , M.; Gendelman, HE. A interferon-induced antiretroviral activities: restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus type 1-infected monocytes . J. Virol. 1994, 68, 7559–7565. [Google Scholar] [PubMed]
- Balachandran, S.; Kim, C.N.; Yeh, W.C.; Mak, T.W.; Bhalla, K.; Barber, G.N. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998, 17, 6888–6902. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Powell, T.D.; Sellins, K.S.; Radecki, S.V.; Cohen, J.J.; Milhausen, M.J. The biological effects of five feline IFN-α subtypes. Vet. Immunol. Immunopathol. 2004, 99, 153–167. [Google Scholar] [PubMed]
- Bandyopadhyay, SK.; Leonard Jr., G.T. ; Bandyopadhyay, T.; Stark, G.R.; Sen, G.C. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 1995, 270, 19624–19629. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. The interferons and cell death: guardians of the cell or accomplices of apoptosis? Semin. Cancer Biol.. 2000, 10, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Barron, M.A.; Blyveis., N.; Palmer, B.E.; MaWhinney, S.; Wilson, C.C. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals . J. Infect. Dis. 2003, 187, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Marsili, G.; Sgarbanti, M.; Ensoli, B.; Hiscott, J. IRF regulation of HIV-1 long terminal repeat activity. J. Interferon Cytokine Res. 2002, 22, 27–37. [Google Scholar] [PubMed]
- Bekisz, J.; Schmeisser, H.; Hernandez, J.; Goldman, ND.; Zoon, KC. Human interferons α, beta and omega. Growth Factors 2004, 22, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Belardelli, F.; Ferrantini, M.; Proietti, E.; Kirkwood, J.M. Interferon-α in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Shearer, G.M. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin. Immunol. 2008, 126, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bolinger, C.; Boris-Lawrie, K. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome . Retrovirology 2009, 6:8, 20. [Google Scholar]
- Boxus, M.; Twizere, J.C.; Legros, S.; Dewulf, J.F.; Kettmann, R.; Willems, L. The HTLV-1 Tax interactome. Retrovirology 2008, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell 2003, 115, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072. [Google Scholar] [CrossRef] [PubMed]
- Chawla-Sarkar, M.; Lindner, DJ.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef]
- Chehimi, J.; Campbell, D.E.; Azzoni, L.; Bacheller, D.; Papasavvas, E.; Jerandi, G.; Mounzer, K.; Kostman, J.; Trinchieri, G.; Montaner, L.J. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals . J. Immunol. 2002, 168, 4796–4801. [Google Scholar] [PubMed]
- Clayette, P.; Martin, M.; Dereuddre-Bosquet, N.; Tournay, V.; Gras, G.; Martal, J.; Dormont, D. IFN-tau, a new interferon type I with antiretroviral activity. Pathol. Biol. (Paris) 1999, 47, 553–559. [Google Scholar] [PubMed]
- Coccia, E.M.; Krust, B.; Hovanessian, A.G. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J. Biol. Chem. 1994, 269, 23087–23094. [Google Scholar] [PubMed]
- Collado, V.M.; Gómez-Lucía, E.; Tejerizo, G.; Miró, G.; Escolar, E.; Martín, S.; Doménech, A. Effect of type I interferons on the expression of feline leukaemia virus. Vet. Microbiol. 2007, 123, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Collado, V.M.; Doménech, A.; Ballesteros, N.A.; Ramos, P.; Miró, G.; Gómez-Lucía, E. Efecto in vitro del interferón de tipo I en la expresión del virus de la inmunodeficiencia felina (FIV) . Salamanca, Spain, 2009; p. 274. [Google Scholar]
- Collado, V.M. unpublished results . [CrossRef] [PubMed]
- Colonna, M.; Krug, A.; Cella, M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr. Opin. Immunol. 2002, 14, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Czarniecki, C.W.; Sreevalsan, T.; Friedman, R.M.; Panet, A. Dissociation of interferon effects on murine leukemia virus and encephalomyocarditis virus replication in mouse cells . J. Virol. 1981, 37, 827–831. [Google Scholar] [PubMed]
- de Mari, K.; Maynard, L.; Sanquer, A.; Lebreux, B.; Eun, H.M. Therapeutic effects of recombinant feline interferon-omega on feline leukemia virus (FeLV)-infected and FeLV/feline immunodeficiency virus (FIV)-coinfected symptomatic cats. J. Vet. Intern. Med. 2004, 18, 477–478. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A; Elder, J.H. Demonstration that orf2 encodes the feline immunodeficiency virus transactivating (Tat) protein and characterization of a unique gene product with partial rev activity . J. Virol. 1999, 73, 608–617. [Google Scholar] [PubMed]
- Dereuddre-Bosquet, N.; Clayette, P.; Martin, M.; Mabondzo, A.; Frétier, P.; Gras, G.; Martal, J.; Dormont, D. Anti-HIV potential of a new interferon, interferon-tau (trophoblastin). J. Acquir. ImmuneDefic. Syndr. Hum. Retrovirol. 1996, 11, 241–246. [Google Scholar] [CrossRef]
- Dianzani, F.; Castilletti, C.; Gentile, M.; Gelderblom, H.R.; Frezza, F.; Capobianchi, M.R. Effects of IFN α on late stages of HIV-1 replication cycle. Biochimie 1998, 80, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; Reis e Sousa, C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Diop, O.M.; Ploquin, M.J.; Mortara, L.; Faye, A.; Jacquelin, B.; Kunkel, D.; Lebon, P.; Butor, C.; Hosmalin, A.; Barré-Sinoussi, F.; Müller-Trutwin, M.C. Plasmacytoid dendritic cell dynamics and α interferon production during Simian immunodeficiency virus infection with a nonpathogenic outcome. J. Virol. 2008, 82, 5145–5152. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H.; Pozniak, A.; Gazzard, B.; Qazi, N.; Gilmour, J.; Gotch, F.; Patterson, S. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood 2001, 98, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H.; Gazzard, B.; Gotch, F.; Patterson, S. Dysfunction and infection of freshly isolated blood myeloid and plasmacytoid dendritic cells in patients infected with HIV-1. Blood 2003, 101, 4505–4511. [Google Scholar] [CrossRef] [PubMed]
- Endo-Munoz, L.; Warby, T.; Harrich, D.; McMillan, NA. Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription . Virol. J. 2005, 2:17, 13. [Google Scholar]
- Estaquier, J.; Idziorek, T.; de Bels, F.; Barré-Sinoussi, F.; Hurtrel, B.; Aubertin, A.M.; Venet, A.; Mehtali, M.; Muchmore, E.; Michel, P.; Mouton, Y.; Girard, M.; Ameisen, J.C. Programmed cell death and AIDS: significance of T-cell apoptosis in pathogenic and nonpathogenic primate lentiviral infections. Proc. Natl. Acad. Sci. USA 1994, 91, 9431–9435. [Google Scholar] [CrossRef]
- Eyster, M.E.; Goedert, J.J.; Poon, M.C.; Preble, O.T. Acid-labile α interferon. A possible preclinical marker for the acquired immunodeficiency syndrome in hemophilia. N. Engl. J. Med. 1983, 309, 583–586. [Google Scholar] [PubMed]
- Feldman, S.; Stein, D.; Amrute, S.; Denny, T.; Garcia, Z.; Kloser, P.; Sun, Y.; Megjugorac, N.; Fitzgerald-Bocarsly, P. Decreased interferon-α production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin. Immunol. 2001, 101, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Ratner, L. Human T-cell leukemia virus type 1 blunts signaling by interferon α. Virology 2008, 374, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Heyden, N.V.; Ratner, L. Α interferon inhibits human T-cell leukemia virus type 1 assembly by preventing Gag interaction with rafts. J. Virol. 2003, 77, 13389–13395. [Google Scholar] [CrossRef] [PubMed]
- Finke, J. S.; Shodell, M.; Shah, K.; Siegal, F. P.; Steinman, R.M. Dendritic cell numbers in the blood of HIV-1 infected patients before and after changes in antiretroviral therapy. J. Clin. Immunol. 2004, 24, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, S.; Riboldi, E.; Facchetti, F.; Avolio, M.; Fabbri, M.; Tosti, G.; Becker, PD.; Guzman, CA.; Sozzani, S.; Caruso, A. HIV-1 matrix protein p17 induces human plasmacytoid dendritic cells to acquire a migratory immature cell phenotype. Proc. Natl. Acad. Sci. USA 2008, 105, 3867–3872. [Google Scholar] [CrossRef]
- Fitzgerald-Bocarsly, P.; Feng, D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007, 89, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, L.; Ozato, K. The role of the interferon regulatory factor (IRF) family in dendritic cell development and function. Cytokine Growth Factor Rev. 2007, 18, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Baca, L.M.; Turpin, J.; Kalter, D.C.; Hansen, B.; Orenstein, J.M.; Dieffenbach, C.W.; Friedman, R.M.; Meltzer, M.S. Regulation of HIV replication in infected monocytes by IFN-α. Mechanisms for viral restriction. J. Immunol. 1990, 145, 2669–2676. [Google Scholar] [PubMed]
- Gerlach, N.; Schimmer, S.; Weiss, S.; Kalinke, U.; Dittmer, U. Effects of type I interferons on Friend retrovirus infection. J. Virol. 2006, 80, 3438–3444. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lucia, E.; Tejerizo, G.; Doménech, A. Effect of steroid hormones on retroviruses. In Oncogenic Viruses. Research Trends. 2007; Johannes, L.T.; Nova Biomedical: New York, USA. [Google Scholar]
- Goodbourn, S.; Didcock, L.; Randall, R.E. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 2000, 81, 2341–2364. [Google Scholar] [PubMed]
- Göttlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.D.; Nara, P.L.; Maheshwari, R.K.; Sidhu, G.S.; Bernbaum, J.G.; Hoekzema, D.; Meltzer, M.S.; Gendelman, H.E. Loss of infectivity by progeny virus from α interferon-treated human immunodeficiency virus type 1-infected T cells is associated with defective assembly of envelope gp120. J. Virol. 1992, 66, 7543–7548. [Google Scholar] [PubMed]
- Hardy, A.W.; Graham, D.R.; Shearer, G.M.; Herbeuval, J.P. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-α. Proc. Natl. Acad. Sci. U S A. 2007, 104, 17453–17458. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Donhauser, N.; Chaipan, C.; Schuster, P.; Puffer, B.; Daniels, R.S.; Greenough, T.C.; Kirchhoff, F.; Schmidt, B. CD4 binding affinity determines human immunodeficiency virus type 1-induced α interferon production in plasmacytoid dendritic cells. J. Virol. 2008, 82, 8900–8905. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Shearer, G.M. HIV-1 immunopathogenesis: how good interferon turns bad. Clin Immunol. 2007, 123, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Grivel, J.C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Nilsson, J.; Boasso, A.; Hardy, A.W.; Kruhlak, M.J.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Andersson, J.; Shearer, G.M. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. U S A. 2006, 103, 7000–7005. [Google Scholar] [CrossRef] [PubMed]
- Hishizawa, M.; Imada, K.; Kitawaki, T.; Ueda, M.; Kadowaki, N.; Uchiyama, T. Depletion and impaired interferon-α-producing capacity of blood plasmacytoid dendritic cells in human T-cell leukaemia virus type I-infected individuals. Br. J. Haematol. 2004, 125, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Takaoka, A.; Taniguchi, T. Regulation of the type I IFN induction: a current view. Int. Immunol. 2005, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Bhatnagar ,N.; Ballmaier, M.; Schubert, U.; Henklein, P.; Volgmann, T.; Heiken, H.; Schmidt, R.E.; Meyer-Olson, D. Exogenous HIV-1 Vpr disrupts IFN-α response by plasmacytoid dendritic cells (pDCs) and subsequent pDC/NK interplay . Immunol. Lett. 2009, 125, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Huthoff, H.; Towers, G.J. Restriction of retroviral replication by APOBEC3G/F and TRIM5α. Trends Microbiol. 2008, 16, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Ouellet, M.; Tremblay, M.J. Microarray study reveals that HIV-1 induces rapid type-I interferon-dependent p53 mRNA up-regulation in human primary CD4+ T cells . Retrovirology 2009, 6:5, 14. [Google Scholar]
- Jameson, P.; Essex, M. Inhibition of feline leukemia virus replication by human leukocyte interferon. Antiviral Res. 1983, 3, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Jarrett,O. Strategies of retrovirus survival in the cat . Vet. Microbiol. 1999, 69, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Juste, R.A.; Ott, T.L.; Kwang, J.; Bazer, F.W.; de la Concha-Bermejillo, A. Effects of recombinant interferon-tau on ovine lentivirus replication. J. Interferon Cytokine Res. 1996, 16, 989–994. [Google Scholar] [PubMed]
- Juste, R.A.; Ott, T.L.; Kwang, J.; Bazer, F.W.; de La Concha-Bermejillo, A. Effects of recombinant ovine interferon-tau on ovine lentivirus replication and progression of disease. J. Gen. Virol. 2000, 81, 525–532. [Google Scholar] [PubMed]
- Kamga, I.; Kahi, S.; Develioglu, L.; Lichtner, M.; Marañón, C.; Deveau, C.; Meyer, L.; Goujard, C.; Lebon, P.; Sinet, M.; Hosmalin, A. Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J. Infect. Dis. 2005, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Karpov, A.V. Endogenous and exogenous interferons in HIV-infection. Eur. J. Med. Res. 2001, 6, 507–524. [Google Scholar] [PubMed]
- Kawaguchi, Y; Norimine, J; Miyazawa, T; Kai, C; Mikami, T. Sequences within the feline immunodeficiency virus long terminal repeat that regulate gene expression and respond to activation by feline herpesvirus type 1 . Virology 1992, 190, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Stoddart, C.A.; Linquist-Stepps, V.; Moreno, M.E.; McCune, J.M. IFN-α secretion by type 2 predendritic cells up-regulates MHC class I in the HIV-1-infected thymus. J. Immunol. 2002, 168, 325–331. [Google Scholar] [PubMed]
- Keir, M.E.; Rosenberg, M.G.; Sandberg, J.K.; Jordan, K.A.; Wiznia, A.; Nixon, D.F.; Stoddart, C.A.; McCune, J.M. Generation of CD3+CD8low thymocytes in the HIV type 1-infected thymus. J. Immunol. 2002, 169, 2788–2796. [Google Scholar] [PubMed]
- Kiermer, V.; Van Lint, C.; Briclet, D.; Vanhulle, C.; Kettmann, R.; Verdin, E.; Burny, A.; Droogmans, L. An interferon regulatory factor binding site in the U5 region of the bovine leukemia virus long terminal repeat stimulates Tax-independent gene expression. J. Virol. 1998, 72, 5526–5534. [Google Scholar] [PubMed]
- Kinpara, S.; Hasegawa, A.; Utsunomiya, A.; Nishitsuji, H.; Furukawa, H.; Masuda, T.; Kannagi, M. Stromal cell-mediated suppression of human T-cell leukemia virus type 1 expression in vitro and in vivo by type I interferon. J. Virol. 2009, 83, 5101–5108. [Google Scholar] [CrossRef] [PubMed]
- Kohara, J.; Yokomizo, Y. In vitro and in vivo effects of recombinant bovine interferon-tau on bovine leukemia virus. J. Vet. Med. Sci. 2007, 69, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Kornbluth, R.S.; Oh, P.S.; Munis, J.R.; Cleveland, P.H.; Richman, D.D. Interferons and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J. Exp. Med. 1989, 169, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Künzi, M.S.; Pitha, P.M. Role of interferon-stimulated gene ISG-15 in the interferon-omega-mediated inhibition of human immunodeficiency virus replication. J. Interferon Cytokine Res. 1996, 16, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Künzi, M.S.; Farzadegan, H.; Margolick, J.B.; Vlahov, D.; Pitha, P.M. Identification of human immunodeficiency virus primary isolates resistant to interferon-α and correlation of prevalence to disease progression. J. Infect. Dis. 1995, 171, 822–828. [Google Scholar] [PubMed]
- Lehner, T.; Wang, Y.; Pido-Lopez, J.; Whittall, T.; Bergmeier, L.A.; Babaahmady, K. The emerging role of innate immunity in protection against HIV-1 infection. Vaccine 2008, 26, 2997–3001. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E. Physiological significance of STAT proteins: investigations through gene disruption in vivo. Cell. Mol. Life Sci. 1999, 55, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.A.; Scott, I.; Mackewicz, C. Protection from HIV/AIDS: the importance of innate immunity. Clin. Immunol. 2003, 108, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Fitzgerald, P.A.; Siegal, F.P. Severe acquired immune deficiency syndrome in male homosexuals: diminished capacity to make interferon-α in vitro associated with severe opportunistic infections. J. Infect. Dis. 1983, 148, 962–966. [Google Scholar] [PubMed]
- Luo, C.; Wang, K.; Liu de, Q.; Li, Y.; Zhao, Q.S. The functional roles of lipid rafts in T cell activation; immune diseases and HIV infection and prevention. Cell. Mol. Immunol. 2008, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Macchi, B.; Faraoni, I.; Mastino, A.; D'Onofrio, C.; Romeo, G.; Bonmassar, E. Protective effect of interferon beta on human T cell leukaemia virus type I infection of CD4+ T cells isolated from human cord blood. Cancer Immunol. Immunother. 1993, 37, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.N.; Barry, A.P.; Vanderford, T.H.; Kozyr, N.; Chavan, R.; Klucking, S.; Barrat, F.J.; Coffman, R.L.; Staprans, S.I.; Feinberg, M.B. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Maneglier, B.; Rogez-Kreuz, C.; Dereuddre-Bosquet, N.; Martal, J.; Devillier, P.; Dormont, D.; Clayette, P. Anti-HIV effects of IFN-tau in human macrophages: role of cellular antiviral factors and interleukin-6. Pathol. Biol. (Paris) 2008, 56, 492–503. [Google Scholar] [PubMed]
- Mangino, G.; Percario, Z.A.; Fiorucci, G.; Vaccari, G.; Manrique, S.; Romeo, G.; Federico, M.; Geyer, M.; Affabris, E. In vitro treatment of human monocytes/macrophages with myristoylated recombinant Nef of human immunodeficiency virus type 1 leads to the activation of mitogen-activated protein kinases, IkappaB kinases, and interferon regulatory factor 3 and to the release of beta interferon. J. Virol. 2007, 81, 2777–2791. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Monier, M.N.; Fradagrada, A.; Mitchell, K.; Baychelier, F.; Eid, P.; Johannes, L.; Lamaze, C. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol. Biol. Cell. 2006, 17, 2896–2909. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, M.C.; Flynn, C.; Huitron-Rezendiz, S.; Watry, D.D.; Zandonatti, M.; Fox, H.S. Early antiretroviral treatment prevents the development of central nervous system abnormalities in simian immunodeficiency virus-infected rhesus monkeys. AIDS 2009, 23, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Marié, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-α genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Marsili, G.; Borsetti, A.; Sgarbanti, M.; Remoli, A.L.; Ridolfi, B.; Stellacci, E.; Ensoli, B.; Battistini, A. On the role of interferon regulatory factors in HIV-1 replication. Ann. N Y Acad. Sci. 2003, 1010, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Meylan, P.R.; Guatelli, J.C.; Munis, J.R.; Richman, D.D.; Kornbluth, R.S. Mechanisms for the inhibition of HIV replication by interferons-α, -beta, and -gamma in primary human macrophages. Virology 1993, 193, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, K.E.; Lewerenz, M.; Reboul, J.; Lutfalla, G.; Uzé, G. The type I interferon receptor: structure, function, and evolution of a family business. J. Interferon Cytokine Res. 1999, 19, 1069–1098. [Google Scholar] [PubMed]
- Munier, S.; Delcroix-Genête, D.; Carthagéna, L.; Gumez, A.; Hazan, U. Characterization of two candidate genes, NCoA3 and IRF8, potentially involved in the control of HIV-1 latency. Retrovirology 2005, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.A.; Gibbs, E.P.J.; Horzinek, M.C.; Studdert, M.J. Retroviridae, 3 edAcademic Press: San Diego, USA, 1999; pp. 363–389. [Google Scholar]
- Niermann, G.L.; Buehring, G.C. Hormone regulation of bovine leukemia virus via the long terminal repeat. Virology 1997, 239, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Ohtsuki, Y.; Sonobe, H.; Furihata, M.; Miyoshi, I. Suppressive effects of interferons on the production and release of human T-lymphotropic virus type-I (HTLV-I). Arch. Virol. 1990, 115, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U S A. 2006, 103, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Pacanowski, J.; Kahi, S.; Baillet, M.; Lebon, P.; Deveau, C.; Goujard, C.; Meyer, L.; Oksenhendler, E.; Sinet, M.; Hosmalin, A. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection . Blood 2001, 98, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Pacanowski, J.; Develioglu, L.; Kamga, I.; Sinet, M.; Desvarieux, M.; Girard, P. M.; Hosmalin, A. Early plasmacytoid dendritic cell changes predict plasma HIV load rebound during primary infection. J. Infect. Dis. 2004, 190, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, E.; Passeri, B.; Amadori, M.; Isola, P.; Di Pede, P.; Telera, A.; Vescovini, R.; Quintavalla, F.; Pistello, M. Low-dose interferon-α treatment for feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 2006, 109, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Poli, G.; Orenstein, J.M.; Kinte,r A.; Folks, T.M.; Fauci, A.S. Interferon-α but not AZT suppresses HIV expression in chronically infected cell lines . Science 1989, 244, 575–577. [Google Scholar] [PubMed]
- Pontzer, C.H.; Yamamoto, J.K.; Bazer, F.W.; Ott, T.L.; Johnson, H.M. Potent anti-feline immunodeficiency virus and anti-human immunodeficiency virus effect of IFN-tau. J. Immunol. 1997, 158, 4351–4357. [Google Scholar] [PubMed]
- Quaranta, M.G.; Mattioli, B.; Giordani, L.; Viora, M. HIV-1 Nef equips dendritic cells to reduce survival and function of CD8+ T cells: a mechanism of immune evasion. FASEB J. 2004, 18, 1459–1461. [Google Scholar] [PubMed]
- Ramji, D.P; Foka, P. CCAAT/enhancer-binding proteins: structure, function and regulation . Biochem. J. 2002, 365, 561–575. [Google Scholar] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Rho, M.B.; Wesselingh, S.; Glass, J.D.; McArthur, J.C.; Choi, S.; Griffin, J.; Tyor, W.R. A potential role for interferon-α in the pathogenesis of HIV-associated dementia. Brain Behav. Immun. 1995, 9, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Rogez, C.; Martin, M.; Dereuddre-Bosquet, N.; Martal, J.; Dormont, D.; Clayette, P. Anti-human immunodeficiency virus activity of tau interferon in human macrophages: involvement of cellular factors and beta-chemokines. J. Virol. 2003, 77, 12914–12920. [Google Scholar] [CrossRef] [PubMed]
- Rogez-Kreuz, C.; Manéglier, B.; Martin, M.; Dereuddre-Bosquet, N.; Martal, J.; Dormont, D.; Clayette, P. Involvement of IL-6 in the anti-human immunodeficiency virus activity of IFN-tau in human macrophages. Int. Immunol. 2005, 17, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Rossol, S.; Voth, R.; Laubenstein, H.P.; Müller, W.E.; Schröder, H.C.; Meyer zum Büschenfelde, K.H.; Hess, G. Interferon production in patients infected with HIV-1. J. Infect. Dis. 1989, 159, 815–821. [Google Scholar] [PubMed]
- Saito, M.; Nakagawa, M.; Kaseda, S.; Matsuzaki, T.; Jonosono, M.; Eiraku, N.; Kubota, R.; Takenouchi, N.; Nagai, M.; Furukawa, Y.; Usuku, K.; Izumo, S.; Osame, M. Decreased human T lymphotropic virus type I (HTLV-I) provirus load and alteration in T cell phenotype after interferon-α therapy for HTLV-I-associated myelopathy/tropical spastic paraparesis. J. Infect. Dis. 2004, 189, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Mael, A.A.; Ikeda, Y. Α interferon enhances TRIM5α-mediated antiviral activities in human and rhesus monkey cells. J. Virol. 2007, 81, 10201–10206. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Sas, A.R.; Bimonte-Nelson, H.A.; Tyor, W.R. Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of Interferon-α in the brain. AIDS 2007, 21, 2151–2159. [Google Scholar] [CrossRef] [PubMed]
- Sas, A.R.; Bimonte-Nelson, H.; Smothers, C.T.; Woodward, J.; Tyor, W.R. Interferon-α causes neuronal dysfunction in encephalitis. J. Neurosci. 2009, 29, 3948–3955. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Ashlock, B. M.; Foster, H.; Fujimura, S. H.; Levy, J. A. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology 2005, 343, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Severa, M.; Fitzgerald, K.A. TLR-mediated activation of type I IFN during antiviral immune responses: fighting the battle to win the war. Curr. Top. Microbiol. Immunol. 2007, 316, 167–192. [Google Scholar] [PubMed]
- Sgarbanti, M.; Borsetti, A.; Moscufo, N.; Bellocchi, MC.; Ridolfi, B.; Nappi, F.; Marsili, G.; Marziali, G.; Coccia, EM.; Ensoli, B.; Battistini, A. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J. Exp. Med. 2002, 195, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, M.; Marsili, G.; Remoli, A.L.; Ridolfi, B.; Stellacci, E.; Borsetti, A.; Ensoli, B.; Battistini, A. Analysis of the signal transduction pathway leading to human immunodeficiency virus-1-induced interferon regulatory factor-1 upregulation. Ann. N Y Acad. Sci. 2004, 1030, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, Y.; Pitha, P.M. Α interferon inhibits early stages of the human immunodeficiency virus type 1 replication cycle. J. Virol. 1992, 66, 1321–1328. [Google Scholar] [PubMed]
- Shirazi, Y.; Pitha, P.M. Interferon α-mediated inhibition of human immunodeficiency virus type 1 provirus synthesis in T-cells. Virology 1993, 193, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Short, J.A.L. Viral evasion of interferon stimulated genes. Bioscience Horizons 2009, 2, 212–224. [Google Scholar] [CrossRef]
- Singh, B.; Ott, T.L.; Bazer, F.W.; de la Concha-Bermejillo, A. Phenotypic and ultrastructural characteristics of bronchoalveolar lavage cells of lentivirus-infected lambs treated with recombinant ovine IFN-tau. J. Interferon Cytokine Res. 2001, 21, 677–686. [Google Scholar] [PubMed]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response . Cell Death Differ. 2005, Suppl 1, 971–978. [Google Scholar] [CrossRef]
- Smith, M.S.; Thresher, R.J.; Pagano, J.S. Inhibition of human immunodeficiency virus type 1 morphogenesis in T cells by α interferon. Antimicrob. Agents Chemother. 1991, 35, 62–67. [Google Scholar] [PubMed]
- Soumelis, V.; Scott, I.; Gheyas, F.; Bouhour, D.; Cozon, G.; Cotte, L.; Huang, L.; Levy, J.A.; Liu, Y.J. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients . Blood 2001, 98, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Scott, I.; Liu, Y.J.; Levy, J. Natural type 1 interferon producing cells in HIV infection. Hum. Immunol. 2002, 63, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Sparger, E.E.; Shacklett, B.L.; Renshaw-Gegg, L.; Barry, P.A.; Pedersen, N.C.; Elder, J.H.; Luciw, P.A. Regulation of gene expression directed by the long terminal repeat of the feline immunodeficiency virus. Virology 1992, 187, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Yamamoto, J.K. Feline immunodeficiency virus lacks sensitivity to the antiviral activity of feline IFN-gamma. J. Interferon Cytokine Res. 2001, 21, 1039–46. [Google Scholar] [PubMed]
- Taylor, M.D.; Korth, M.J.; Katze, M.G. Interferon treatment inhibits the replication of simian immunodeficiency virus at an early stage: evidence for a block between attachment and reverse transcription. Virology 1998, 241, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.V.; Locarnini, S.A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell. Biol. 2007, 85, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.J.; Elder, J.; Neil, J.C. Cis- and trans-regulation of feline immunodeficiency virus: identification of functional binding sites in the long terminal repeat. J. Gen. Virol. 1994, 75, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Tilton, J.C.; Johnson, A.J.; Luskin, M.R.; Manion, M.M.; Yang, J.; Adelsberger, J.W.; Lempicki, R.A.; Hallahan, C.W.; McLaughlin, M.; Mican, J.M.; Metcalf, J.A.; Iyasere, C.; Connors, M. Diminished production of monocyte proinflammatory cytokines during human immunodeficiency virus viremia is mediated by type I interferons. J. Virol. 2006, 80, 11486–11497. [Google Scholar] [CrossRef] [PubMed]
- Tilton, J.C.; Manion, M.M.; Luskin, M.R.; Johnson, A.J.; Patamawenu, A.A.; Hallahan, C.W.; Cogliano-Shutta, N.A.; Mican, J.M.; Davey Jr., R.T.; Kottilil, S.; Lifson, J.D.; Metcalf, J.A.; Lempicki, R.A.; Connors, M. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished α interferon production in vitro. J. Virol. 2008, 82, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Kim, S.; Taylor, M.W. REFINEMENT: a search framework for the identification of interferon-responsive elements in DNA sequences--a case study with ISRE and GAS. Comput. Biol. Chem. 2006, 30, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and type I interferons. J. Biol. Chem. 2007, 282, 15319–15324. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C.; Amella, C.A.; Emiliani, S.; John, M.; Jie, T.; Verdin, E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 1997, 71, 6113–6127. [Google Scholar] [PubMed]
- Vieillard, V.; Costagliola, D.; Simon, A.; Debré, P; French Asymptomatiques à Long Terme (ALT) Study Group. Specific adaptive humoral response against a gp41 motif inhibits CD4 T-cell sensitivity to NK lysis during HIV-1 infection . AIDS 2006, 20, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Transcriptional regulation of interferon-stimulated genes. Eur. J. Biochem. 1991, 200, 1–11. [Google Scholar] [CrossRef]
- Yamamoto, J.K.; Ho, E.; Pedersen, N.C. A feline retrovirus induced T-lymphoblastoid cell-line that produces an atypical α type of interferon. Vet. Immunol. Immunopathol. 1986, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Morita, R.; Takaori-Kondo, A.; Kadowaki, N.; Kitawaki, T.; Hori, T.; Uchiyama, T. Natural α interferon-producing cells respond to human immunodeficiency virus type 1 with α interferon production and maturation into dendritic cells. J. Virol. 2003, 77, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, N.S.; Myles, M.H.; Mathiason-DuBard, C.K.; Dreitz, M.J.; Mullins, J.I.; Hoover, E.A. Α interferon (2b) in combination with zidovudine for the treatment of presymptomatic feline leukemia virus-induced immunodeficiency syndrome. Antimicrob. Agents Chemother. 1990, 34, 1749–1756. [Google Scholar] [PubMed]
- Zhang, J.; Yamada, O.; Kawagishi, K.; Araki, H.; Yamaoka, S.; Hattori, T.; Shimotohno, K. Human T-cell leukemia virus type 1 Tax modulates interferon-α signal transduction through competitive usage of the coactivator CBP/p300. Virology 2008, 379, 306–313. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.