
Spatial Variation of Bacterial Diversity in Shiro-Associated and Non-Mycorrhizal Microhabitats of Tuber sinense–Quercus aliena Symbiosis

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site and Soil Sample Collection
2.2. DNA Extraction, PCR Amplification, and High-Throughput Sequencing
2.3. Microbial Diversity and Community Structure Analysis
2.4. Identification of Differential and Indicator Taxa
3. Results
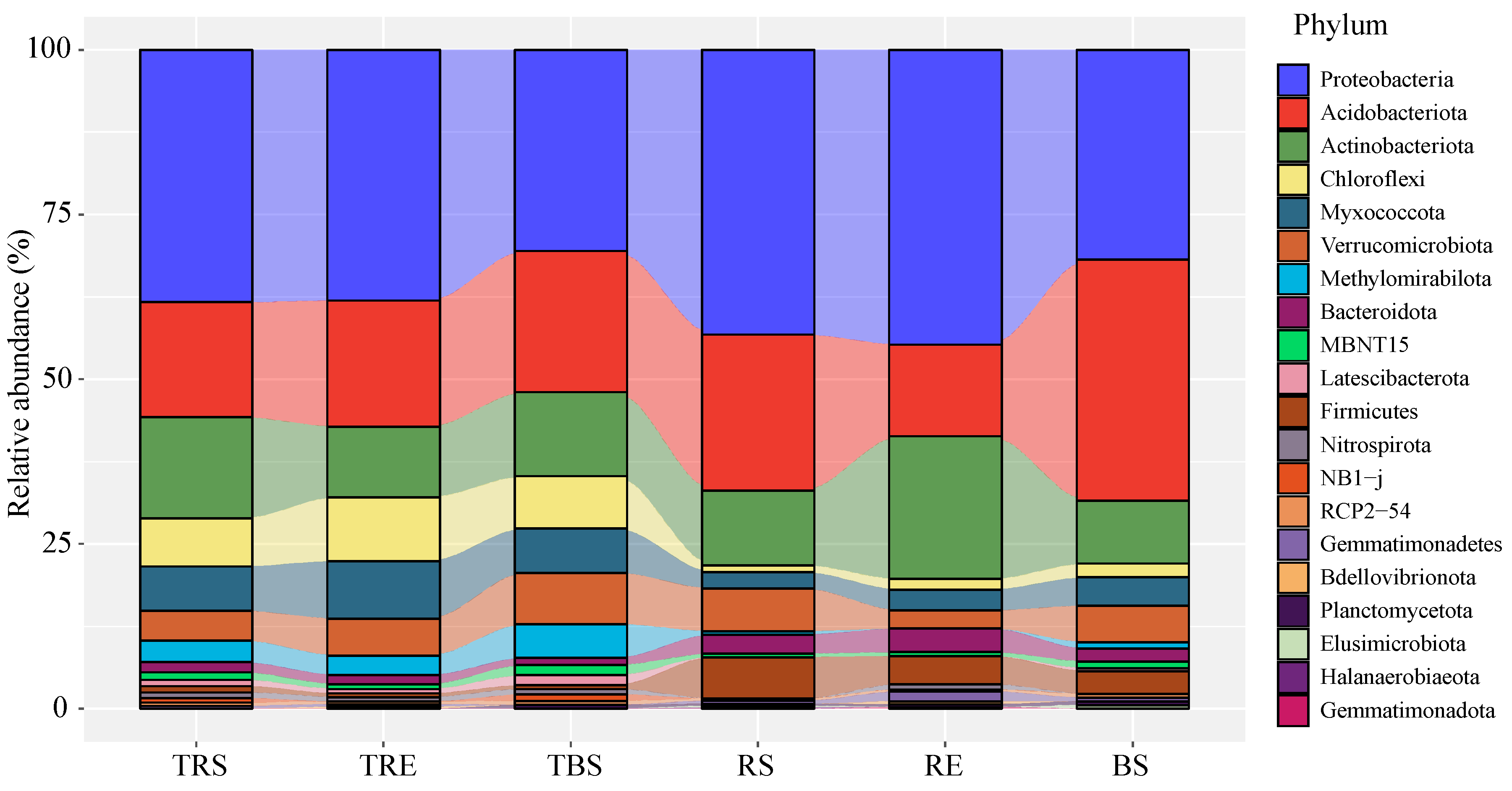
3.1. Comparison of Bacterial Community Composition Between Tuber sinense-Producing and Control Areas
3.2. Alpha Diversity of Bacterial Communities in Producing and Control Areas
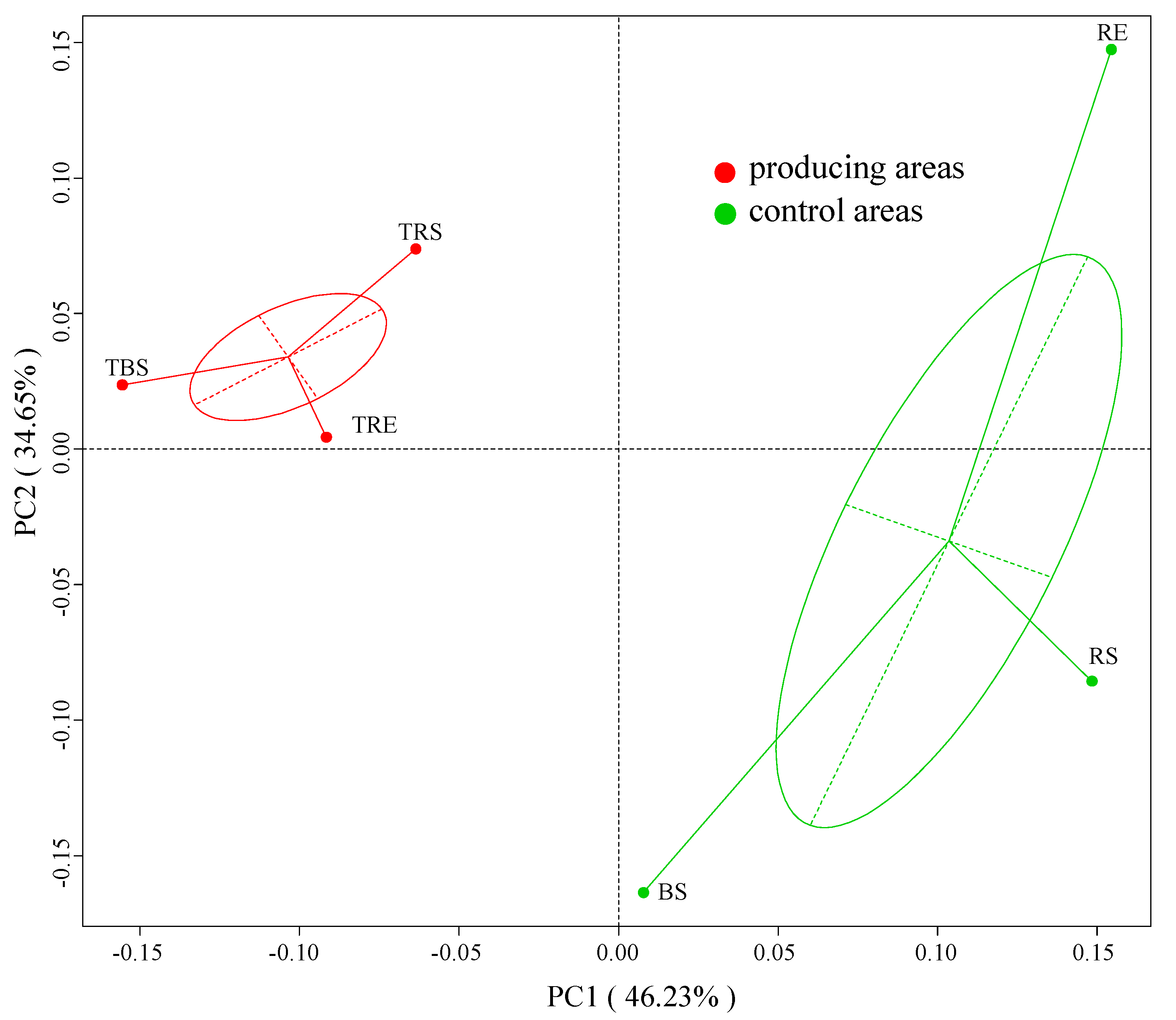
3.3. Beta Diversity of Microbial Communities in Producing and Control Areas
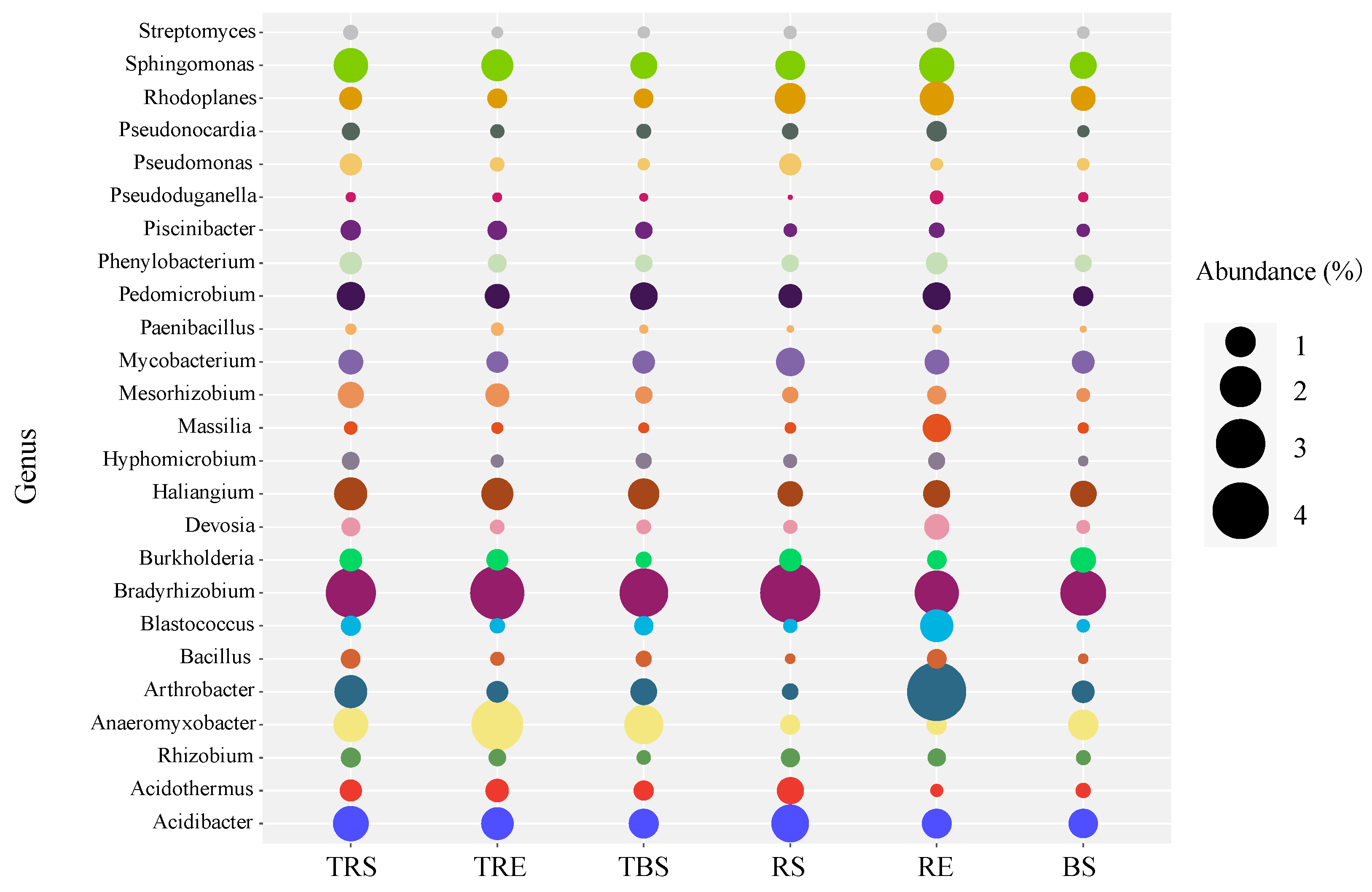
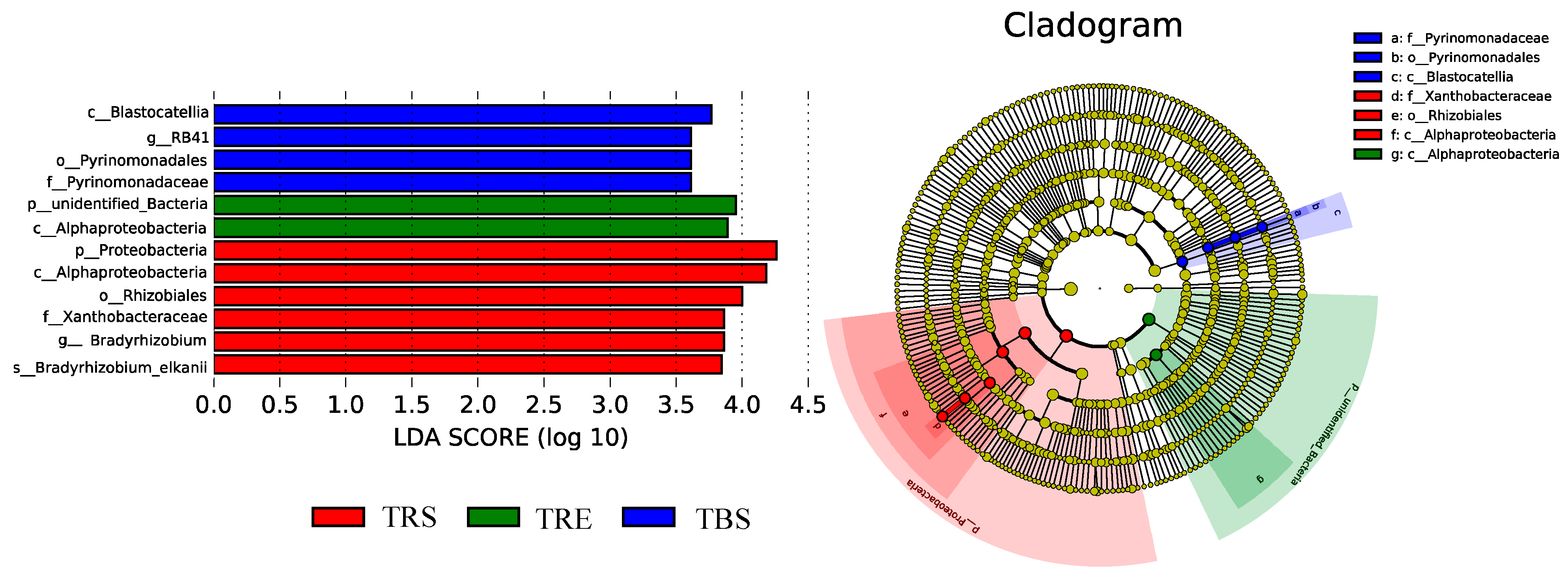
3.4. Spatial Variation in Bacterial Taxa Within Tuber sinense-Producing Regions
3.5. Identification of Potential Keystone Taxa in Different Producing Regions
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kües, U.; Martin, F. On the road to understanding truffles in the underground. Fungal Genet. Biol. 2011, 48, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Zhang, J.L.; Li, T.; Sun, H.J.; Xiong, W.P.; Li, Y. Chinese black truffles: Tuber yigongense sp. nov., taxonomic reassessment of T. indicum s.l., and re-examination of the T. sinense isotype. Mycotaxon 2018, 133, 183–196. [Google Scholar] [CrossRef]
- García-Montero, L.G.; Díaz, P.; Di Massimo, G.; García-Abril, A. A review of research on Chinese Tuber species. Mycol. Prog. 2010, 9, 315–335. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, Z.M.; Zhang, D.C.; Murat, C.; Jeandroz, S.; Le Tacon, F. Phylogenetic and populational study of the Tuber indicum complex. Mycol. Res. 2006, 110, 1034–1045. [Google Scholar] [CrossRef]
- Fan, L.; Li, T.; Xu, Y.; Yan, X. Species diversity, phylogeny, endemism and geography of the truffle genus Tuber in China based on morphological and molecular data. Pers.-Mol. Phylogeny Evol. Fungi 2022, 48, 175–202. [Google Scholar] [CrossRef]
- Ogawa, M. Microbial ecology of ‘Shiro’ in Tricholoma matsutake (S. Ito & Imai) Sing. and its allied species. V. Tricholoma matsutake in Tsuga sieboldii forests. Trans. Mycol. Soc. Jpn. 1977, 18, 34–46. [Google Scholar]
- Stamets, P. Mycelium Running: How Mushrooms Can Help Save the World; Penguin Random House: New York, NY, USA, 2005; p. 339. [Google Scholar]
- Pacioni, G. Effects of Tuber metabolites on the rhizospheric environment. Mycol. Res. 1991, 95, 1355–1358. [Google Scholar] [CrossRef]
- Streiblová, E.; Gryndlerová, H.; Gryndler, M. Truffle brûlé: An efficient fungal life strategy. FEMS Microbiol. Ecol. 2012, 80, 1–8. [Google Scholar] [CrossRef]
- Taschen, E.; Sauve, M.; Vincent, B.; Parladé, J.; van Tuinen, D.; Aumeeruddy-Thomas, Y.; Assenat, B.; Selosse, M.A.; Richard, F. Insight into the truffle brûlé: Tripartite interactions between the black truffle (Tuber melanosporum), holm oak (Quercus ilex) and arbuscular mycorrhizal plants. Plant Soil 2020, 446, 577–594. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.M.; Tang, Y.J.; Xing, Y.M.; Qiao, P.; Li, Y.; Liu, P.G.; Guo, S.X. Chinese black truffle-associated bacterial communities of Tuber indicum from different geographical regions with nitrogen fixing bioactivity. Front. Microbiol. 2019, 10, 2515. [Google Scholar] [CrossRef]
- Siebyła, M.; Szyp-Borowska, I.; Młodzińska, A. Bacterial communities inhabiting the ascomata of the ectomycorrhizal summer truffle (Tuber aestivum). Appl. Soil Ecol. 2024, 199, 105428. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, J.; Xiong, C.; Li, X.; Chen, Z.; Li, P.; Huang, W. Tuber indicum shapes the microbial communities of ectomycorhizosphere soil and ectomycorrhizae of an indigenous tree (Pinus armandii). PLoS ONE 2017, 12, e0175720. [Google Scholar] [CrossRef] [PubMed]
- Rondolini, M.; Zotti, M.; Bragato, G.; Baciarelli Falini, L.; Reale, L.; Donnini, D. The expanding truffle environment: A study of the microbial dynamics in the old productive site and the new Tuber magnatum picco habitat. J. Fungi 2024, 10, 800. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Ceccaroli, P.; Agostini, D.; Zeppa, S.D.; Gioacchini, A.M.; Stocchi, V. Truffle-associated bacteria: Extrapolation from diversity to function. In True Truffle (Tuber spp.) in the World; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; Volume 47, p. 436. [Google Scholar]
- Piñuela, Y.; Alday, J.G.; Oliach, D.; Bolaño, F.; Colinas, C.; Bonet, J.A. Use of inoculator bacteria to promote Tuber melanosporum root colonization and growth on Quercus faginea saplings. Forests 2020, 11, 792. [Google Scholar] [CrossRef]
- Giorgi, V.; Amicucci, A.; Landi, L.; Castelli, I.; Romanazzi, G.; Peroni, C.; Ranocchi, B.; Zambonelli, A.; Neri, D. Effect of bacteria inoculation on colonization of roots by Tuber melanosporum and growth of Quercus ilex seedlings. Plants 2024, 13, 224. [Google Scholar] [CrossRef]
- Frey-Klett, P.; Garbaye, J.; Tarkka, M. The mycorrhiza helper bacteria revisited. New Phytol. 2007, 176, 22–36. [Google Scholar] [CrossRef]
- Seethalakshmi, P.; Kumaresan, T.; Vishnu Prasad Nair, R.; Prathiviraj, R.; Seghal Kiran, G.; Selvin, J. Comparative analysis of commercially available kits for optimal DNA extraction from bovine fecal samples. Arch. Microbiol. 2024, 206, 314. [Google Scholar] [CrossRef]
- Soliman, H.; Ismaeil, M.; Soussa, H.; El-Sayed, W.S. Unveiling organohalide respiration potential in River Nile sediments via 16S rRNA gene amplicon sequencing of endogenous bacterial communities. BMC Microbiol. 2025, 25, 186. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Wu, F.; Ma, S.; Zhou, J.; Han, C.; Hu, R.; Yang, X.; Nie, G.; Zhang, X. Genetic diversity and population structure analysis in a large collection of white clover (Trifolium repens L.) germplasm worldwide. PeerJ 2021, 9, e11325. [Google Scholar] [CrossRef] [PubMed]
- Khleborodova, A.; Gamboa-Tuz, S.D.; Ramos, M.; Segata, N.; Waldron, L.; Oh, S. lefser: Implementation of metagenomic biomarker discovery tool, LEfSe, in R. Bioinformatics 2024, 40, btae707. [Google Scholar] [CrossRef]
- Diaz-Uriarte, R. GeneSrF and varSelRF: A web-based tool and R package for gene selection and classification using random forest. BMC Bioinform. 2007, 8, 328. [Google Scholar] [CrossRef]
- Giorgio, M.; Niccolò, B.G.M.; Benedetta, T.; Luisa, M.; Leonardo, B.F.; Gregory, B.; Pietro, B.; Alberto, A.; Domizia, D.; Emidio, A. Fungal and bacterial diversity in the Tuber magnatum ecosystem and microbiome. Microb. Ecol. 2023, 85, 508–521. [Google Scholar] [CrossRef]
- Sillo, F.; Vergine, M.; Luvisi, A.; Calvo, A.; Petruzzelli, G.; Balestrini, R.; Mancuso, S.; De Bellis, L.; Vita, F. Bacterial communities in the fruiting bodies and background soils of the white truffle Tuber magnatum. Front. Microbiol. 2022, 13, 864434. [Google Scholar] [CrossRef]
- Antony-Babu, S.; Deveau, A.; Van Nostrand, J.D.; Zhou, J.; Le Tacon, F.; Robin, C.; Frey-Klett, P.; Uroz, S. Black truffle-associated bacterial communities during the development and maturation of Tuber melanosporum ascocarps and putative functional roles. Environ. Microbiol. 2014, 16, 2831–2847. [Google Scholar] [CrossRef] [PubMed]
- Alhuthali, S.; Bello, S.K.; Bageel, A.M.; Shori, A.B.; Bataweel, N.M.; Al-Hejin, A.M.; Al-Qarawi, A.A.; Thomas, P.W. Soil physicochemical and metagenomic analyses of bacteria and fungi: Toward desert truffle cultivation in Saudi Arabia. Agronomy 2024, 14, 3021. [Google Scholar] [CrossRef]
- Splivallo, R.; Vahdatzadeh, M.; Maciá-Vicente, J.G.; Molinier, V.; Peter, M.; Egli, S.; Uroz, S.; Paolocci, F.; Deveau, A. Orchard conditions and fruiting body characteristics drive the microbiome of the black truffle Tuber aestivum. Front. Microbiol. 2019, 10, 1437. [Google Scholar] [CrossRef]
- Mello, A.; Ding, G.C.; Piceno, Y.M.; Napoli, C.; Tom, L.M.; DeSantis, T.Z.; Andersen, G.L.; Smalla, K.; Bonfante, P. Truffle brûlés have an impact on the diversity of soil bacterial communities. PLoS ONE 2013, 8, e61945. [Google Scholar] [CrossRef]
- Liu, D.; Pérez-Moreno, J.; He, X.; Garibay-Orijel, R.; Yu, F. Truffle microbiome is driven by fruit body compartmentalization rather than soils conditioned by different host trees. Msphere 2021, 6, e0003921. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.Y.; Peng, M.H.; Wang, J.J.; Ye, W.Y.; Li, Y.L.; Zhang, T.; Wang, A.R.; Zhang, D.M.; Wang, Z.H.; Lu, G.D.; et al. Microbial community associated with ectomycorrhizal Russula symbiosis and dominated nature areas in southern China. FEMS Microbiol. Lett. 2021, 368, fnab028. [Google Scholar] [CrossRef]
- Monaco, P.; Naclerio, G.; Mello, A.; Bucci, A. Role and potentialities of bacteria associated with Tuber magnatum: A mini-review. Front. Microbiol. 2022, 13, 1017089. [Google Scholar] [CrossRef]
- Graziosi, S.; Puliga, F.; Iotti, M.; Amicucci, A.; Zambonelli, A. In vitro interactions between Bradyrhizobium spp. and Tuber magnatum mycelium. Environ. Microbiol. Rep. 2024, 16, e13271. [Google Scholar] [CrossRef]
- Satish, L.; Barak, H.; Keren, G.; Yehezkel, G.; Kushmaro, A.; Ben-Dov, E.; Kagan-Zur, V.; Barak, Z.; Sitrit, Y. The microbiome structure of the symbiosis between the desert truffle Terfezia boudieri and its host plant Helianthemum sessiliflorum. J. Fungi 2022, 8, 1062. [Google Scholar] [CrossRef]
- Liu, D.; Chater, C.C.; Yu, F.; Perez-Moreno, J. Tuber pseudohimalayense ascomata-compartments strongly select their associated bacterial microbiome from nearby pine forest soils independently of their maturation stage. Pedobiologia 2021, 87–88, 150743. [Google Scholar] [CrossRef]
- Deveau, A.; Antony-Babu, S.; Le Tacon, F.; Robin, C.; Frey-Klett, P.; Uroz, S. Temporal changes of bacterial communities in the Tuber melanosporum ectomycorrhizosphere during ascocarp development. Mycorrhiza 2016, 26, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, Q.; Guo, W.; Wu, F.; Chen, J.; Liu, P.; Tian, W.; Qiao, P. Microbial communities of ascocarps and soils in a natural habitat of Tuber indicum. Arch. Microbiol. 2022, 204, 189. [Google Scholar] [CrossRef] [PubMed]
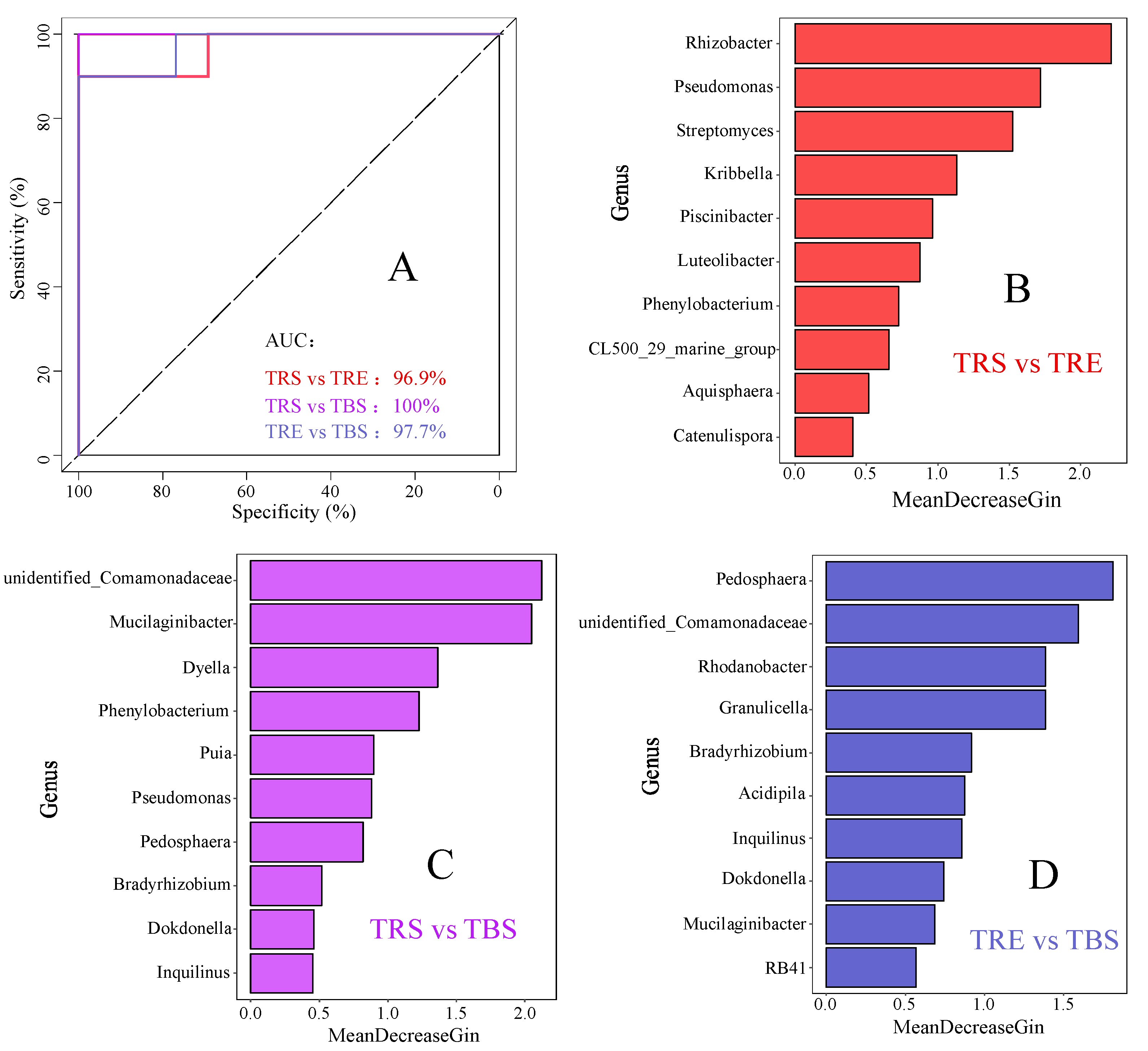

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | OTUs | Goods Coverage | Shannon | Simpson | Chao1 | ACE | PD Whole Tree |
|---|---|---|---|---|---|---|---|
| TRS | 4274 ± 213.7 a | 0.954 ± 0.003 c | 10.26 ± 0.51 a | 0.9973 ± 0.002 a | 20854.6 ± 1042.7 a | 6531.5 ± 326.6 a | 277.24 ± 13.86 b |
| TRE | 3885 ± 194.3 b | 0.960 ± 0.003 b | 9.83 ± 0.49 ab | 0.9959 ± 0.002 b | 8639.2 ± 432.0 b | 5689.7 ± 284.5 b | 254.78 ± 12.74 c |
| TBS | 3857 ± 192.9 b | 0.962 ± 0.003 b | 9.97 ± 0.50 ab | 0.9967 ± 0.002 ab | 8539.9 ± 427.0 b | 5465.5 ± 273.3 b | 262.23 ± 13.11 c |
| RS | 3070 ± 153.5 c | 0.973 ± 0.003 a | 9.39 ± 0.47 b | 0.9945 ± 0.002 c | 5113.4 ± 255.7 c | 4151.2 ± 207.6 c | 302.53 ± 15.13 a |
| RE | 3725 ± 186.3 bc | 0.970 ± 0.003 a | 9.91 ± 0.50 ab | 0.9955 ± 0.002 b | 5804.8 ± 290.2 c | 4730.7 ± 236.5 c | 294.90 ± 14.75 a |
| BS | 3193 ± 159.7 c | 0.971 ± 0.003 a | 9.48 ± 0.47 b | 0.9959 ± 0.002 b | 6364.3 ± 318.2 c | 4387.2 ± 219.4 c | 222.37 ± 11.12 d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, T.; Liu, H.; Xu, R.; Chen, Y.; Liu, J.; Piao, C.; Xue, H.; Liu, R.; Li, Y. Spatial Variation of Bacterial Diversity in Shiro-Associated and Non-Mycorrhizal Microhabitats of Tuber sinense–Quercus aliena Symbiosis. Forests 2025, 16, 982. https://doi.org/10.3390/f16060982
Ma T, Liu H, Xu R, Chen Y, Liu J, Piao C, Xue H, Liu R, Li Y. Spatial Variation of Bacterial Diversity in Shiro-Associated and Non-Mycorrhizal Microhabitats of Tuber sinense–Quercus aliena Symbiosis. Forests. 2025; 16(6):982. https://doi.org/10.3390/f16060982
Chicago/Turabian StyleMa, Tengfei, Haijiao Liu, Risheng Xu, Yafei Chen, Juan Liu, Chungen Piao, Han Xue, Renlu Liu, and Yong Li. 2025. "Spatial Variation of Bacterial Diversity in Shiro-Associated and Non-Mycorrhizal Microhabitats of Tuber sinense–Quercus aliena Symbiosis" Forests 16, no. 6: 982. https://doi.org/10.3390/f16060982
APA StyleMa, T., Liu, H., Xu, R., Chen, Y., Liu, J., Piao, C., Xue, H., Liu, R., & Li, Y. (2025). Spatial Variation of Bacterial Diversity in Shiro-Associated and Non-Mycorrhizal Microhabitats of Tuber sinense–Quercus aliena Symbiosis. Forests, 16(6), 982. https://doi.org/10.3390/f16060982