Abstract
Early detection protocols for the oak wilt fungal pathogen (Bretziella fagacearum) are crucial for forest health practitioners on the boundaries of the growing disease front. Established protocols use oak wilt-specific primers and gel electrophoresis to amplify and detect oak wilt among DNA extracted from nitidulid vectors. However, these protocols are prone to inconclusive results due to the presence of off-target amplification products similar in size to positive control bands. Using sequence-adapted oak wilt primers, we employed a metabarcoding amplicon sequencing approach that resolved inconclusive results and validated true oak wilt positives. We found that these off-target amplification products are co-occurring taxa from natural forest and beetle microbiomes, further necessitating a sequencing approach for early surveillance of oak wilt.
1. Introduction
Oak wilt is a disease caused by the fungal pathogen Bretziella fagacearum (Bretz) Z.W. deBeer, Marinc., T.A. Duong & M.J. Wingf., comb. nov. (Microascales: Ceratocystidaceae), which affects all Quercus spp. but is especially severe in red oaks [1,2]. Fungal mycelium clogs tree vascular systems, leading to leaf wilting and eventual tree death. Below ground, oak wilt is spread through root grafts, transmitting outward to create patches of infected trees [2]. Above ground, new infection centers are caused by nitidulid beetle vectors (Coleoptera: Nitidulidae) [3]. When an infected oak dies, the fungus produces sporulating mats that rupture the bark and expose the infectious spores [2]. Nitidulid beetles, attracted to these fungal mats, pick up and potentially vector these spores to new hosts, with long-distance transmission possible by human transport of infected firewood [2].
As of October 2025, the closest oak wilt-positive tree and nitidulid detection to New Hampshire was in Schenectady County, New York [3]. Due to the ecological and economic threats of oak wilt to the state [4], early monitoring systems have been established, trapping nitidulid beetles and using a targeted nested PCR assay to identify positive samples [3]. This nested PCR approach first amplifies fungal DNA from the sample using universal primers for the internal transcribed spacer region (ITS) [5,6], followed by an internal PCR with primers designed specifically within the ITS region of B. fagacearum [7].
Although prior sequence confirmation of gel-positive PCR products has shown a strong association with B. fagacearum [3], assigning species based exclusively on successful PCR amplification of taxon-specific primers introduces risks of false positives and inconclusive results that could be detrimental to the response of forest health specialists [8]. By employing a metabarcoding approach, sequencing all nested PCR products to identify all taxa amplified by the B. fagacearum-specific primers, we are able to resolve both size and taxonomic identity of the DNA from gel bands, clarifying inconclusive results and identifying co-occurring fungi contributing to potential false positives. Adding sequencing to oak wilt detection protocols improves pathogen detection and contributes to the much-needed modernization of early detection protocols for forest pathogens [9].
In this report, samples of nitidulid beetles trapped from state parks across New Hampshire were subjected to standard oak wilt molecular diagnostics through specific nested PCR and gel electrophoresis [3,10]. Nested primers were modified with sequence adapters to allow for direct amplicon sequencing of the PCR products to clarify inconclusive gel results. Sequencing revealed a variety of off-target amplicons with predicted sizes to within 10 bp of the B. fagacearum control from taxa common to the nitidulid and forest microenvironment. These findings indicate that in an oak wilt surveillance context, i.e., without an infection morphology, sequencing is necessary to identify true oak wilt positives. This updated protocol and pipeline can be used by other forest health practitioners to improve early detection protocols and effectively surveil for oak wilt in a metabarcoding fashion.
2. Materials and Methods
2.1. Field Protocol for Nitidulid Collection
Nitidulid traps were set up and collected at multiple sites in various state parks across New Hampshire in 2023 and 2024, including Greenfield (GF), Mount Sunapee (SN), Bear Brook (BB), White Lake (WL), and Monadnock (MN) state parks (Figure S1). Traps were placed at highly trafficked state campgrounds as well as near oak-importing mills. Trap baiting and collection were based on McLaughlin et al. [3]; briefly, Lindgren funnel traps baited with Colopterus truncatus (Randall) and Caplothorax sayi (Parsons) pheromone lures were collected every two weeks between April and August, and trapped nitidulid beetles (Caplothorax sayi, Colopterus truncatus, Glischrochilus sanguinolentus [Olivier], and Glischrochilus fasciatus [Olivier]) were manually separated from bycatch. Nitidulid beetles from individual collection sites were pooled (3–4 beetle species per tube, up to 12 total beetles), frozen, and sent to the Hubbard Center for Genome Studies (Durham, NH, USA) for processing.
2.2. DNA Extraction
Frozen beetle pools were crushed with pestles and inert beads until homogenized. Lysis buffer was added, and samples were vortexed for 10 min, at which point Proteinase K was added. Remaining extraction steps proceeded on the KingFisher following the MagMAX Ultra nucleic acid isolation protocol (ThermoFisher, Waltham, MA, USA). DNA extractions were quantified by Qubit (ThermoFisher, Waltham, MA, USA).
2.3. Nested PCR, Gel Electrophoresis, and Amplicon Sequencing
Nested PCR for oak wilt detection was performed with standard ITS1 and ITS4 primers as previously described in McLaughlin et al. [3], and with modification of the oak wilt-specific primers to contain Illumina TruSeq adapters (CF01-TS: 5′–ACACTCTTTCCCTACACGACGCTCTTCCGATCTGGCAGGGACTTCTTTCTT–3′ and CF02-TS: 5′–GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAAGGCTTGAGTGGTGAAA–3′). Products from each PCR step were run on a 2% agarose gel and imaged on an E-Gel Electrophoresis System (ThermoFisher, Waltham, MA, USA). B. fagacearum DNA was subjected to the same PCR steps and run on the gels as a positive control. Samples were indexed with dual 8-mers, sequenced on a NovaSeq 6000 SP flow cell using 250 base pair (bp) paired-end reads, and demultiplexed using bcl-convert v4.4.0 (Illumina, San Diego, CA, USA) to generate raw reads.
2.4. Bioinformatic Analysis of Amplicon Sequencing Data
Reads were trimmed using fastp v0.23.4 [11] with parameters “-l 150 -g -Q” to remove poly-G tails and adapters. Trimmed reads were imported into the QIIME2 program q2cli v2024.5.0 [12] and analyzed to produce Amplicon Sequence Variants (ASVs) using the dada2 denoise-paired plugin [13] with trim lengths of 18 to remove primer sequences and truncation lengths of 240. Resulting ASVs were taxonomically identified using MegaBLAST (blastn v2.13.0+) against the NCBI nucleotide database (downloaded 31 August 2024), with parameters specified for use with taxonomy_assignment_BLAST_V2.py [14]. Species, family, and phyla cutoffs were set to 80% with otherwise default settings. ASVs with undetermined species-level taxa were manually annotated using the most resolved taxa among the top BLAST hits. Common co-occurring fungi were identified as non-B. fagacearum ASVs present in at least two samples. ASV counts were manually aggregated by taxonomy.
3. Results and Discussion
3.1. Gel Electrophoresis of ITS1–ITS4 and Nested CF01–CF02-TS PCR Products
Amplification products from both steps of the oak wilt nested PCR were imaged on gel electrophoresis (Figure 1). New, smaller bands were noted in the nested PCR of several surveillance samples (Figure 1B), indicating nested amplification of ITS1–ITS4 products by CF01–CF02-TS. Although positive control bands were easily identifiable from the nested gel (Figure 1B), difficulty arose in determining whether bands near the expected target size (347 bp) in surveillance samples were true oak wilt positives or off-target products amplified by the oak wilt primer. This gel-based size determination may be effective in testing oak wilt when phenotypically evident but leaves inconclusive results in samples with complex fungal communities, especially in an early detection program.
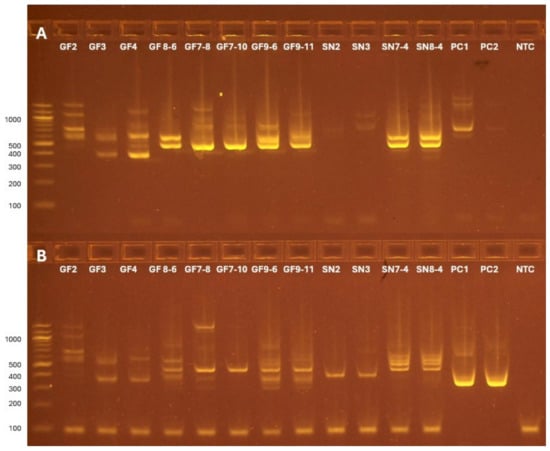
Figure 1.
Gel electrophoresis results for the products of the ITS1–ITS4 (A) and CF01–CF02-TS (B) PCR amplifications on a subset of the samples. (A) ITS1–ITS4 amplification shows a range of typical product sizes [15], with the most abundant products in the 400–700 base pair (bp) range. (B) CF01–CF02-TS amplification of the positive control is indicated by a bright band in the PC1 and PC2 lanes at the expected size of 347 bp (280 bp + 67 bp for TruSeq adapters) [3]. The presence of new bands in the 300–400 bp range in several surveillance samples indicates new nested products amplified by CF01–CF02-TS. (A,B) PC1 is a 1:10 dilution of B. fagacearum DNA, while PC2 is a 1:100 dilution. A no-template control is shown as NTC.
3.2. Sequencing of B. fagacearum Amplification Products
Analysis of the positive control sequencing reads revealed multiple ASVs aligning to the CF01–CF02 locus of B. fagacearum with an expected size of 280 bp, excluding TruSeq adapters [3]. These ASVs were only found in the positive controls, with no other taxa identified. Other bands in the positive control sample first PCR gel electrophoresis results (Figure 1A) suggest potential off-target sites for ITS1–ITS4 amplification in the B. fagacearum genome. However, these products would lack sequencing adapters and would not appear in sequencing results.
3.3. Co-Occuring Fungi Identified in Samples
After filtering, 79 non-B. fagacearum ASVs were identified in sequencing results (Table S1). These ASVs taxonomically resolved to 14 bins, of which the six most prevalent are found in Table 1. GenBank entries of the matched reference sequences show some of these fungi may have originated from forest habitats, as suggested by studies of decaying or damaged pine needles (Dothideomyetes sp.: KX908391, P. pontiformis: KT000144) and detritus in submerged stream habitats (Cladosporium sp.: MW764179). Others may have been fungi vectored on or found inside the beetles, with S. polyspora (OK576253) vectored from beetles in regions of declining pine, R. atrovirens (MH971246) from fungivorous millipedes, and K. quercitrusa (MN497061) from the gut contents of termites, but also found from nitidulid species (CBS 17297). The standard on diagnostics for oak wilt notes that Cladosporium sp. have previously been isolated and amplified using the nested PCR from the bark of oak wilt-suspected trees [10], but other co-occurring taxa are not mentioned. A previous study of fungi from nitidulids in New Hampshire in 2018 also identified Cladosporium sp. using similar trapping methods and did not detect B. fagacearum [16].

Table 1.
The most prevalent co-occurring taxa identified by the nested CF01–CF02-TS metabarcoding protocol. Taxa are sorted in descending number of reads per sample in the present samples. A total of 93% of non-B. fagacearum reads come from Dothideomycetes sp., while the next five most prevalent taxa make up another 6% of the reads.
3.4. Economic Feasibility of Oak Wilt Metabarcoding for Forest Health Practitioners
An economic assessment of oak wilt in 2011 estimated infected tree removal to cost an average of USD 360 per tree [17]. Since oak wilt disease typically progresses as expanding infection centers, 70% of which contain at least three infected trees [17], reducing the formation of new infection centers often necessitates the removal of both diseased and apparently healthy trees within a 15.2 m radius of symptomatic trees [2]. Severing root grafts around infection centers adds further expense, estimated at three dollars per foot (~USD 10/m) [18]. Consequently, the cost of managing a single infection center can reach hundreds to thousands of dollars. These economic considerations highlight the importance of early and accurate detection, as interventions applied before the fungus can reproduce can completely stop local spread [3].
Standard molecular diagnostics for oak wilt already require DNA extraction, nested PCR, and gel electrophoresis at approximately USD 23 per sample [19], without considering the cost of sample collection. Sequencing-based detection adds only modestly to per-sample costs (~USD 13 for indexing, sequencing, and analysis [19], totaling USD 36 per sample), yet may improve reliability and reduce the frequency of inconclusive diagnoses that can delay management response or lead to unnecessary treatments. In this context, the higher upfront investment in direct sequencing may be offset by the long-term benefits of more accurate early detection. Furthermore, sequencing could expand surveillance efforts to detect oak wilt before the pathogen is established in the environment, such as in major invasion pathways like tree product imports [9].
4. Conclusions
This report presents a crucial improvement for early detection and surveillance of oak wilt. Using a metabarcoding protocol, we demonstrate that oak wilt-specific primers amplify off-target taxa from trap-collected nitidulid beetle DNA, leading to inconclusive results with standard gel-based size determination. For forest health practitioners, investing in direct sequencing enables accurate early oak wilt detection and helps prevent costly infection center establishment. The sequence-adapted primer pair and pipeline described here can be used in further oak wilt studies and serve as a framework for adapting surveillance of other forest pathogens to sequencing.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f16111628/s1, Figure S1: New Hampshire state park nitidulid trapping locations, Table S1: ASV table with sequence, amplicon length, full taxonomy, taxa bin, ASV, and amplicons per sample for all non-B. fagacearum ASVs passing filters.
Author Contributions
Conceptualization, L.M.G., R.S.C., M.K., J.L.S., and W.K.T.; methodology, L.M.G., R.S.C., M.K., J.L.S., and W.K.T.; formal analysis, L.M.G. and M.K.; investigation, R.S.C., M.K., J.A.H., A.S.N., and S.D.S.; resources, R.S.C. and W.K.T.; data curation, L.M.G., R.S.C., and J.L.S.; writing—original draft preparation, L.M.G.; writing—review and editing, all; visualization, L.M.G. and M.K.; supervision, W.K.T.; project administration, K.M.; funding acquisition, R.S.C. and W.K.T. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the USDA Forest Service through Grant No. 23-DG-11094200-312 and by the NH-INBRE program and the Center for Integrated Biomedical and Bioengineering Research (CIBBR) through grants from the National Institute of General Medical Sciences of the National Institutes of Health under Award Numbers P20GM103506 and P20GM113131, respectively.
Data Availability Statement
All data supporting the results of the manuscript can be found within the figures and attached Supplementary Materials.
Acknowledgments
We would like to thank USFS collaborators Isabel Munck and Marc DiGirolomo for their help with this project. We would also like to thank Kelsey McLaughlin and Jessica Cancelliere (NY DEC) and Karen Snover-Clift (Cornell University) for their help with field protocols. Additionally, we would like to thank Brett Arenz from the University of Minnesota Plant Disease Clinic for providing the B. fagacerum control DNA.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| ASV | Amplicon Sequence Variant |
| ITS | Internal Transcribed Spacer |
| bp | base pair(s) |
References
- de Beer, Z.W.; Marincowitz, S.; Duong, T.A.; Wingfield, M.J. Bretziella, a New Genus to Accommodate the Oak Wilt Fungus, Ceratocystis fagacearum (Microascales, Ascomycota). MycoKeys 2017, 27, 1–19. [Google Scholar] [CrossRef]
- Koch, K.A.; Quiram, G.L.; Venette, R.C. A Review of Oak Wilt Management: A Summary of Treatment Options and Their Efficacy. Urban For. Urban Green. 2010, 9, 1–8. [Google Scholar] [CrossRef]
- McLaughlin, K.; Snover-Clift, K.; Somers, L.; Cancelliere, J.; Cole, R. Early Detection of the Oak Wilt Fungus (Bretziella fagacearum) Using Trapped Nitidulid Beetle Vectors. For. Pathol. 2022, 52, e12767. [Google Scholar] [CrossRef]
- Cooperative Extension. New Hampshire Statewide Oak Wilt Response Plan; New Hampshire Department of Natural and Cultural Resources, Division of Forests and Lands: Concord, NH, USA, 2022; Available online: https://scholars.unh.edu/extension/1109/ (accessed on 21 August 2025).
- Creer, S.; Deiner, K.; Frey, S.; Porazinska, D.; Taberlet, P.; Thomas, W.K.; Potter, C.; Bik, H.M. The Ecologist’s Field Guide to Sequence-based Identification of Biodiversity. Methods Ecol. Evol. 2016, 7, 1008–1018. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Hajibabaei, M.; Rieseberg, L.H. Environmental DNA. Mol. Ecol. 2012, 21, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Chen, G.Y.; Li, B.; Su, H.; An, Y.L.; Zhen, S.Z.; Ye, J.R. Rapid and Accurate Detection of Ceratocystis fagacearum from Stained Wood and Soil by Nested and Real-time PCR. For. Pathol. 2011, 41, 15–21. [Google Scholar] [CrossRef]
- MacDonald, A.J.; Sarre, S.D. A Framework for Developing and Validating Taxon-specific Primers for Specimen Identification from Environmental DNA. Mol. Ecol. Resour. 2017, 17, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Munck, I.A.; Bonello, P. Modern Approaches for Early Detection of Forest Pathogens Are Sorely Needed in the United States. For. Pathol. 2018, 48, e12445. [Google Scholar] [CrossRef]
- PM 7/1 (2) Bretziella fagacearum (Formerly Ceratocystis fagacearum). EPPO Bull. 2023, 53, 505–517. [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.L. Assign-Taxonomy-with-BLAST 2025. Available online: https://github.com/Joseph7e/Assign-Taxonomy-with-BLAST (accessed on 21 August 2025).
- Fujita, S.-I.; Senda, Y.; Nakaguchi, S.; Hashimoto, T. Multiplex PCR Using Internal Transcribed Spacer 1 and 2 Regions for Rapid Detection and Identification of Yeast Strains. J. Clin. Microbiol. 2001, 39, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- DiGirolomo, M.F.; Munck, I.A.; Dodds, K.J.; Cancelliere, J. Sap Beetles (Coleoptera: Nitidulidae) in Oak Forests of Two Northeastern States: A Comparison of Trapping Methods and Monitoring for Phoretic Fungi. J. Econ. Entomol. 2020, 113, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Haight, R.G.; Homans, F.R.; Horie, T.; Mehta, S.V.; Smith, D.J.; Venette, R.C. Assessing the Cost of an Invasive Forest Pathogen: A Case Study with Oak Wilt. Environ. Manag. 2011, 47, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Cook, B. Oak Wilt Treatment. Available online: https://www.canr.msu.edu/news/oak_wilt_treatment (accessed on 21 August 2025).
- Services and Pricing. Available online: https://hcgs.unh.edu/services-pricing (accessed on 21 August 2025).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).