
High-Density Genetic Map and QTL Analysis in Cunninghamia lanceolate: Insights into Growth and Wood-Color Traits

Abstract
1. Introduction
2. Materials and Methods
2.1. Mapping Population
2.2. Measurement of Heartwood Traits
2.3. SLAF Marker Generation
2.4. SLAF-Seq Data Analysis and Genotyping
2.5. Map Construction
2.6. QTL Analysis
3. Results
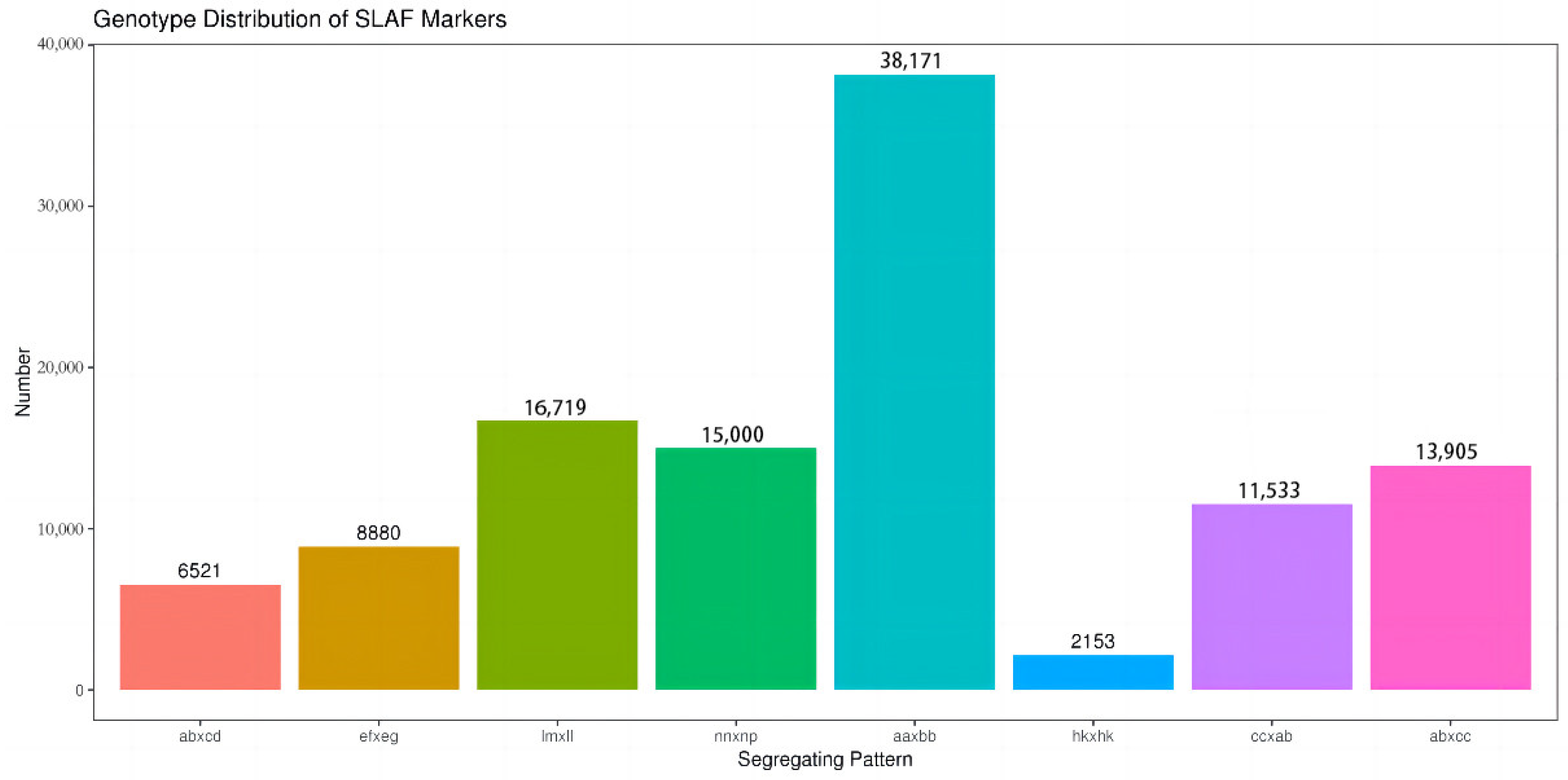
3.1. Analysis of SLAF-Seq Data and SLAF Markers
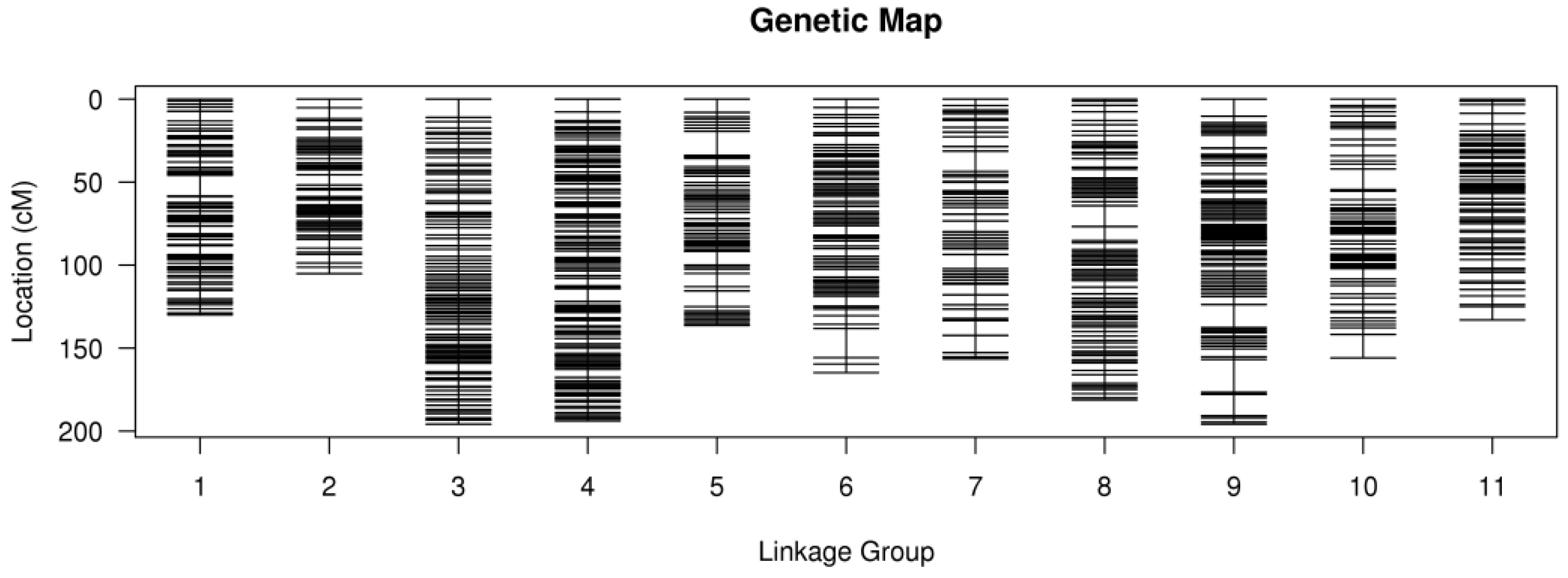
3.2. The C. lanceolata Genetic Maps
3.3. Evaluation of the Genetic Map
3.4. Analysis of Variation in Quantitative Traits
3.5. QTL Description
4. Discussion
4.1. Mapping Population
4.2. The Segregation of SLAF Markers
4.3. Linkage Mapping
4.4. QTL Mapping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bian, L.; Shi, J.; Zheng, R.; Chen, J.; Wu, H. Genetic parameters and genotype–environment interactions of Chinese fir (Cunninghamia lanceolata) in Fujian Province. Can. J. For. Res. 2014, 44, 582–592. [Google Scholar] [CrossRef]
- Duan, H.; Hu, D.; Li, Y.; Zheng, H. Characterization of a collection of Chinese fir elite genotypes using sequence-related amplified polymorphism markers. J. For. Res.-Jpn. 2016, 27, 1105–1110. [Google Scholar] [CrossRef]
- Shi, J.; Zhen, Y.; Zheng, R. Proteome profiling of early seed development in Cunninghamia lanceolata (Lamb.) Hook. J. Exp. Bot. 2010, 61, 2367–2381. [Google Scholar] [CrossRef]
- Zheng, R.; Shi, J.; Xiao, H.; Su, S.; Xu, L.; Ouyang, L.; Zhang, Z.; Ye, D.; Fang, Y. Genetic variation and early selection of growth traits in 8-year-old open-pollinated progenies of the 3rd germplasm of Chinese fir. J. Nanjing For. Univ. Nat. Sci. Ed. 2014, 6, 38–42. [Google Scholar]
- Wen, H.; Deng, X.; Zhang, Y.; Wei, X.; Wang, G.; Zhou, B.; Xiang, W.; Zhu, N. Cunninghamia lanceolata variant with red-heart wood: A mini-review. Dendrobiology 2018, 79, 156–167. [Google Scholar] [CrossRef]
- Searle, S.D.; Owen, J.V. Variation in basic wood density and percentage heartwood in temperate Australian Acacia species. Aust. For. 2005, 68, 11. [Google Scholar] [CrossRef]
- Zeng, Z.; Yang, X.; Xiao, F.; Ye, J.; Yu, J.; Wang, X. Study on variability of timber characters and gene resources utilization for Chenshan Red-heart Chinese fir. Jiangxi For. Sci. Technol. 2001, 3, 1–6. [Google Scholar]
- Chen, X.; Xiao, F.; Yu, L.; Lou, Y.; Xu, H. Estimation of genetic parameters of Cunninghamia lanceolata growth traits based on mixed linear model. J. For. Environ. 2018, 38, 419–424. [Google Scholar]
- Ericsson, T.; Fries, A. High heritability for heartwood in north Swedish Scots pine. Theor. Appl. Genet. 1999, 98, 732–735. [Google Scholar] [CrossRef]
- Yanjie, L.; Apiolaza, L.A.; Clemens, A. Genetic variation in heartwood properties and growth traits of Eucalyptus bosistoana. Eur. J. For. Res. 2018, 137, 565–572. [Google Scholar]
- Pâques, L.E. Genetic Control of Heartwood Content in Larch. Silvae Genet. 2001, 50, 69–75. [Google Scholar]
- Schenk, L.; Ellert, U.; Neuhauser, H. Inheritance Pattern of Growth and Wood Traits in Teak (Tectona grandis L.f.). Silvae Genet. 2009, 58, 97–101. [Google Scholar]
- Róger, M.; Berrocal, A. Wood colour variation in sapwood and heartwood of young trees of Tectona grandis and its relationship with plantation characteristics, site, and decay resistance. Ann. For. Sci. 2010, 67, 109. [Google Scholar]
- Montes, C.S.; Hernández, R.E.; Beaulieu, J.; Weber, J.C. Genetic variation in wood color and its correlations with tree growth and wood density of Calycophyllum spruceanum at an early age in the Peruvian Amazon. New For. 2008, 35, 57–73. [Google Scholar] [CrossRef]
- Moya, R.; Marín, J.D.; Murillo, O.; Leandro, L. Wood physical properties, color, decay resistance and stiffness in Tectona grandis clones with evidence of genetic control. Silvae Genet. 2013, 62, 142–152. [Google Scholar] [CrossRef]
- Mosedale, J.; Charrier, B.; Janin, G. Genetic control of wood colour, density and heartwood ellagitannin concentration in European oak (Quercus petraea and Q. robur). Forestry 1996, 69, 111–124. [Google Scholar] [CrossRef]
- Hannrup, B.; Cahalan, C.; Chantre, G.; Grabner, M.; Karlsson, B.; Bayon, I.L.; Jones, G.L.; Müller, U.; Pereira, H.; Rodrigues, J.C.; et al. Genetic parameters of growth and wood quality traits in Picea abies. Scand. J. For. Res. 2004, 19, 14–29. [Google Scholar] [CrossRef]
- Shao, S.; Yu, M.; Qian, H.; Jin, Z.; De, Y.I. Study on genetic diversity of an artificial Chinese fir forest under interference and natural conditions. J. Zhejiang For. Sci. Technol. 2004, 24, 12–14. [Google Scholar]
- Zheng, H.; Hu, D.; Wang, R.; Wu, S. Genetic divergence of the Chinese Fir fast-growing genotypes revealed by SRAP markers. J. Southwest For. Univ. 2017, 37, 14–20. [Google Scholar]
- Li, M.; Chen, X.; Huang, M.; Wu, P.; Ma, X. Genetic diversity and relationships of ancient Chinese fir (Cunninghamia lanceolata) genotypes revealed by sequence-related amplified polymorphism markers. Genet. Resour. Crop. Evol. 2017, 64, 1087–1099. [Google Scholar] [CrossRef]
- You, Y.; Hong, J. Application of RAPD marker to genetic variation of Chinese fir provenances. Sci. Silvae Sin. 1998, 34, 32–38. [Google Scholar]
- Li, M.; Shi, J.; He, Z.; Yi, N. Study on molecular genetic variation of superior trees in Chinese fir (Cunninghamia lanceolata) (Lamb.) Hook. Sci. Silvae Sin. 2001, 37, 137–141. [Google Scholar]
- Li, M.; Shi, J.; Li, F.; Gan, S. Molecular characterization of elite genotypes with in a second-generation Chinese fir (Cunninghamia lanceolata) breeding population using RAPD markers. Sci. Silvae Sin. 2007, 43, 50–55. [Google Scholar]
- Ouyang, L.; Chen, J.; Zheng, R.; Xu, Y.; Lin, Y.; Huang, J.; Ye, D.; Fang, Y.; Shi, J. Genetic diversity among the germplasm collections of the Chinese fir in 1st breeding population upon SSR markers. J. Nanjing For. Univ. Nat. Sci. Ed. 2014, 38, 21–26. [Google Scholar]
- Xu, Y.; Chen, J.; Zhao, Y.; Wang, Y.; Wang, X.; Liu, W.; Shi, J.; Zheng, R.; Ouyang, L.; Zhang, Z.; et al. Variation of EST-SSR molecular markers among provenances of Chinese fir. J. Nanjing For. Univ. Nat. Sci. Ed. 2014, 38, 1–8. [Google Scholar]
- Duan, H.; Hu, R.; Wu, B.; Chen, D.; Huang, K.; Dai, J.; Chen, Q.; Wei, Z.; Cao, S.; Sun, Y.; et al. Genetic characterization of red-colored heartwood genotypes of Chinese fir using simple sequence repeat (SSR) markers. Genet. Mol. Res. 2015, 14, 18552–18561. [Google Scholar] [CrossRef]
- Li, M.; Huang, M.; Su, L.; Chen, X.; Ma, X. Genetic diversity of germplasm resources of the king of Chinese fir in Fujian provenances. J. For. Environ. 2016, 36, 312–318. [Google Scholar]
- Qi, M. Genetic diversity of wide cross population of Cunninghamia lanceolata and Platycladu orientalis. Bull. Bot. Res. 2008, 28, 299–303. [Google Scholar]
- Qi, M.; Wang, H.; Peng, J.; Zhou, J.; Weng, C.; He, G. Genetic diversity and heterotic grouping of Cunninghamia lanceolata breeding garden. South China For. Sci. 2018, 46, 21–25. [Google Scholar]
- Muchero, W.; Guo, J.J.; DiFazio, S.P.; Chen, J.G.; Ranjan, P.; Slavov, G.T.; Gunter, L.E.; Jawdy, S.; Bryan, A.C.; Sykes, R.; et al. High-resolution genetic mapping of allelic variants associated with cell wall chemistry in Populus. BMC Genom. 2015, 16, 24. [Google Scholar] [CrossRef]
- Bdeir, R.; Muchero, W.; Yordanov, Y.; Tuskan, G.A.; Busov, V.; Gailing, O. Quantitative trait locus mapping of Populus bark features and stem diameter. BMC Plant Biol. 2017, 17, 224. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Shi, J.; Wang, M.; Yu, R.; Chen, X. The construction of molecular linkage frame map in Chinese fir. J. Nanjing For. Univ. Nat. Sci. Ed. 2000, 24, 22–26. [Google Scholar]
- Tong, C.; Shi, J. Constructing Genetic Linkage Map Chinese Fir s in Chinese Fir Using F1 Progeny. Acta Genet. Sin. 2004, 31, 1149–1156. [Google Scholar] [PubMed]
- Bindler, G.; Plieske, J.; Bakaher, N.; Gunduz, I.; Ivanov, N.; Hoeven, R.V.; Ganal, M.; Donini, P. A high density genetic map of tobacco (Nicotiana tabacum L.) obtained from large scale microsatellite marker development. Theor. Appl. Genet. 2011, 123, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Fang, L.; Xin, H.; Wang, L.; Li, S. Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Biol. 2012, 12, 148. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Yu, S.; Su, T.; Zhi, S.; Zhang, F.; Wang, W.; Zhang, D.; Zhao, X.; Yu, Y. Construction of a sequence-based bin map and mapping of QTLs for downy mildew resistance at four developmental stages in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol. Breed. 2016, 36, 44. [Google Scholar] [CrossRef]
- Tao, A.; Huang, L.; Wu, G.; Afshar, R.K.; Qi, J.; Xu, J.; Fang, P.; Lin, L.; Zhang, L.; Lin, P. High-density genetic map construction and QTLs identification for plant height in white jute (Corchorus capsularis L.) using specific locus amplified fragment (SLAF) sequencing. BMC Genom. 2017, 18, 355. [Google Scholar] [CrossRef]
- Fang, L.; Liu, H.; Wei, S.; Wei, S.; Keefover-Ring, K.; Yin, T. High-density genetic map of Populus deltoides constructed by using specific length amplified fragment sequencing. Tree Genet. Genomes 2018, 14, 10. [Google Scholar] [CrossRef]
- Chen, X.B.; Xu, H.N.; Xiao, F.M.; Sun, S.W.; Lou, Y.F.; Zou, Y.X.; Xu, X.Q. Genetic diversity and paternity analyses in a 1.5th generation seed orchard of Chenshan red-heart Chinese fir. J. Nanjing For. Univ. Nat. Sci. Ed. 2021, 45, 87–92. [Google Scholar]
- Liu, D.; Ma, C.; Hong, W.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.; Deng, D.; Xin, H.; Song, J.; et al. Construction and Analysis of High-Density Linkage Map Using High-Throughput Sequencing Data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef]
- West Mal Van, L.; Leeuwen, H.V.; Kozik, A.; Daniel, J.K.; Doerge, R.W.; Clair, D.A.S.; Michelmore, R.W. High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Res. 2006, 16, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W. MapQTL®5: Software for the Mapping of Quantitative Trait Loci in Experimental Populations; Kyazma BV: Wageningen, The Netherlands, 2004. [Google Scholar]
- Grattapaglia, D.; Sederoff, R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudotestcross: Mapping strategy and RAPD markers. Genetics 1994, 137, 1121–1137. [Google Scholar] [CrossRef]
- Hall, M.C.; Willis, J.H. Transmission ratio distortion in intraspecific hybrids of Mimulus guttatus: Implications for genomic divergence. Genetics 2005, 170, 375–386. [Google Scholar] [CrossRef] [PubMed]
- MaKnox, M.R.; Ellis, T.H.N. Excess heterozygosity contributes to genetic map expansion in pea recombinant inbred populations. Genetics 2002, 162, 861–873. [Google Scholar]
- Jin, J.; Li, C.; Wang, R.; Zheng, H.; Yao, M.; Chen, L. Large-scale SNP discovery and genotyping for constructing a high-density genetic map of tea plant using specific-locus amplified fragment sequencing (SLAF-seq). PLoS ONE 2015, 10, e0128798. [Google Scholar]
- Zhang, Q.; Liu, W.; Liu, N.; Zhang, Y.; Liu, S.; Wei, X.; Liu, Y. Segregation type of heterozygous loci in F1 and construction of molecular markers linkage map in Apricot. Acta Hortic. Sin. 2011, 38, 1983–1990. [Google Scholar]
- Luo, L.; Zhang, Y.M.; Xu, S. A quantitative genetics model for viability selection. Heredity 2005, 94, 347–355. [Google Scholar] [CrossRef]
- Zhang, L.; Deng, Q.; Li, P.; Wang, S.; Zheng, A.; Li, Z.; Li, H.; Li, S.; Wang, J. Effects of missing marker and segregation distortion on QTL mapping in F2 populations. Theor. Appl. Genet. 2010, 121, 1071–1082. [Google Scholar] [CrossRef]
- Xu, J.; Shi, J. C banding and fluorescent banding pattern of the chromosomes of Cunninghamia lanceolate. Mol. Plant Breed. 2007, 5, 515–520. [Google Scholar]
- Li, H.; Hearne, S.; Bänziger, M.; Li, Z.; Wang, J. Statistical properties of QTL linkage mapping in biparental genetic populations. Heredity 2010, 105, 257–267. [Google Scholar] [CrossRef]
- Doerge, R.W. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 2002, 3, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, Y.; Xu, J.; Mei, Z.; Shi, Y.; Liu, P.; He, J.; Wang, X.; Meng, Y.; Feng, S.; et al. High-density genetic map construction and stem total polysaccharide content-related QTL exploration for Chinese endemic dendrobium (Orchidaceae). Front. Plant Sci. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, C.; Zhang, Y. Modeling segregation distortion for viability selection. I. Reconstruction of linkage maps with distorted markers. Theor. Appl. Genet. 2007, 114, 295–305. [Google Scholar] [CrossRef]
- Lowe, K.M.; Riaz, S.; Walker, M.A. Variation in recombination rates across Vitis species. Tree Genet. Genomes 2009, 5, 71–80. [Google Scholar] [CrossRef]
- Cao, S.; Deng, H.; Zhao, Y.; Zhang, Z.; Tian, Y.; Sun, Y.; Li, Y.; Zheng, H. Metabolite profiling and transcriptome analysis unveil the mechanisms of red-heart Chinese fir (Cunninghamia lanceolata (lamb.) hook) heartwood coloration. Front. Plant Sci. 2022, 13, 594. [Google Scholar] [CrossRef]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Storey, J.D. Relaxed significance criteria for linkage analysis. Genetics 2006, 173, 2371–2381. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lander, E.S.; Botstein, D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Kochevenko, A.; Jiang, Y.; Seiler, C.; Surdonja, K.; Kollers, S.; Reif, J.C.; Korzun, V.; Graner, A. Identification of QTL hot spots for malting quality in two elite breeding lines with distinct tolerance to abiotic stress. BMC Plant Biol. 2018, 18, 106. [Google Scholar] [CrossRef]
- Guan, P.; Shen, X.; Mu, Q.; Wang, Y.; Peng, H. Dissection and validation of a QTL cluster linked to Rht-B1 locus controlling grain weight in common wheat (Triticum aestivum L.) using near-isogenic lines. Theor. Appl. Genet. 2020, 133, 2639–2653. [Google Scholar] [CrossRef] [PubMed]
- Daniela, T.M.; Nadia, V.; Ezio, P.; Alberto, A.; Chiara, B.; Shawn, A.M.; Todd, C.M.; Erik, R.R.; Roberto, B. High density SNP mapping and QTL analysis for time of leaf budburst in Corylus avellana L. PLoS ONE 2018, 13, e0195408. [Google Scholar]



{kind=link}
{kind=link}
| Reads number (M) | 120,007 |
| Q30 percentage (%) | 93.78 |
| Guanine cytosine (GC) percentage (%) | 37.75 |
| SLAF amount | 532,450 |
| The parents’ mean sequencing depth of SLAFs | 39.89× |
| The progeny’ mean sequencing depth of SLAFs | 10.72× |
| Markers for Mapping | 2574 |
| Number of Markers | Size (cM) | Average Interval between Markers (cM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Female Map | Male Map | Integrated Map | Female Map | Male Map | Integrated Map | Female Map | Male Map | Integrated Map | |
| LG1 | 106 | 86 | 177 | 155.63 | 100.13 | 130.06 | 1.48 | 1.18 | 0.74 |
| LG2 | 105 | 5 | 108 | 113.72 | 30.70 | 105.10 | 1.09 | 7.67 | 0.98 |
| LG3 | 354 | 41 | 387 | 182.20 | 145.13 | 195.82 | 0.52 | 3.63 | 0.51 |
| LG4 | 181 | 279 | 444 | 184.10 | 183.25 | 193.82 | 1.02 | 0.66 | 0.44 |
| LG5 | 128 | 83 | 193 | 130.33 | 89.61 | 136.31 | 1.03 | 1.09 | 0.71 |
| LG6 | 226 | 52 | 269 | 149.69 | 139.87 | 164.80 | 0.67 | 2.74 | 0.61 |
| LG7 | 74 | 180 | 243 | 157.64 | 132.28 | 156.31 | 2.16 | 0.74 | 0.65 |
| LG8 | 269 | 23 | 288 | 175.74 | 160.64 | 181.24 | 0.66 | 7.30 | 0.63 |
| LG9 | 80 | 192 | 266 | 188.19 | 175.11 | 195.79 | 2.38 | 0.92 | 0.74 |
| LG10 | 15 | 88 | 98 | 81.33 | 192.01 | 155.93 | 5.81 | 2.21 | 1.61 |
| LG11 | 94 | 9 | 101 | 147.19 | 87.66 | 133.02 | 1.58 | 10.96 | 1.33 |
| Total | 1632 | 1038 | 2574 | 1665.76 | 1436.39 | 1748.40 | 1.03 | 1.40 | 0.68 |
| LG ID | Percentage of Double Crossovers (%) | Percentage of Missing Data (%) |
|---|---|---|
| LG1 | 16.17 | 0.00 |
| LG2 | 5.34 | 0.00 |
| LG3 | 6.88 | 0.00 |
| LG4 | 10.09 | 0.00 |
| LG5 | 16.14 | 0.00 |
| LG6 | 9.28 | 0.00 |
| LG7 | 11.23 | 0.00 |
| LG8 | 6.89 | 0.00 |
| LG9 | 6.92 | 0.00 |
| LG10 | 10.09 | 0.00 |
| LG11 | 5.96 | 0.00 |
| Traits | Mean | VC | Min | Max | N |
|---|---|---|---|---|---|
| Diameter at breast height (cm) | 14.38 | 0.299 | 5.4 | 25.14 | 70 |
| heartwood diameter (cm) | 8.12 | 0.391 | 2.62 | 16.87 | 70 |
| heartwood proportion (%) | 55.43 | 0.158 | 37 | 77 | 70 |
| heartwood a* | 8.99 | 0.086 | 7.34 | 10.73 | 70 |
| heartwood b* | 17.49 | 0.062 | 15.45 | 21.20 | 70 |
| heartwood L* | 72.34 | 0.036 | 65.95 | 78.20 | 70 |
| sapwood a* | 5.42 | 0.133 | 3.75 | 6.72 | 70 |
| sapwood b* | 15.39 | 0.062 | 13.05 | 17.57 | 70 |
| sapwood L* | 82.32 | 0.019 | 79.40 | 86.40 | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Xiong, C.; Lou, Y.; Xu, H.; Cheng, Q.; Sun, S.; Xiao, F. High-Density Genetic Map and QTL Analysis in Cunninghamia lanceolate: Insights into Growth and Wood-Color Traits. Forests 2023, 14, 1591. https://doi.org/10.3390/f14081591
Chen X, Xiong C, Lou Y, Xu H, Cheng Q, Sun S, Xiao F. High-Density Genetic Map and QTL Analysis in Cunninghamia lanceolate: Insights into Growth and Wood-Color Traits. Forests. 2023; 14(8):1591. https://doi.org/10.3390/f14081591
Chicago/Turabian StyleChen, Xingbin, Caiyun Xiong, Yongfeng Lou, Haining Xu, Qiangqiang Cheng, Shiwu Sun, and Fuming Xiao. 2023. "High-Density Genetic Map and QTL Analysis in Cunninghamia lanceolate: Insights into Growth and Wood-Color Traits" Forests 14, no. 8: 1591. https://doi.org/10.3390/f14081591
APA StyleChen, X., Xiong, C., Lou, Y., Xu, H., Cheng, Q., Sun, S., & Xiao, F. (2023). High-Density Genetic Map and QTL Analysis in Cunninghamia lanceolate: Insights into Growth and Wood-Color Traits. Forests, 14(8), 1591. https://doi.org/10.3390/f14081591