

Insight into the Complex Genetic Relationship of Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) Advanced Parent Trees Based on SSR and SNP Datasets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Genomic DNA Extraction
2.3. SSR Assay
2.4. SLAF-Seq and SNP Development
2.5. Statistical Analysis
3. Results
3.1. Polymorphism Analysis of Markers and Genetic Diversity
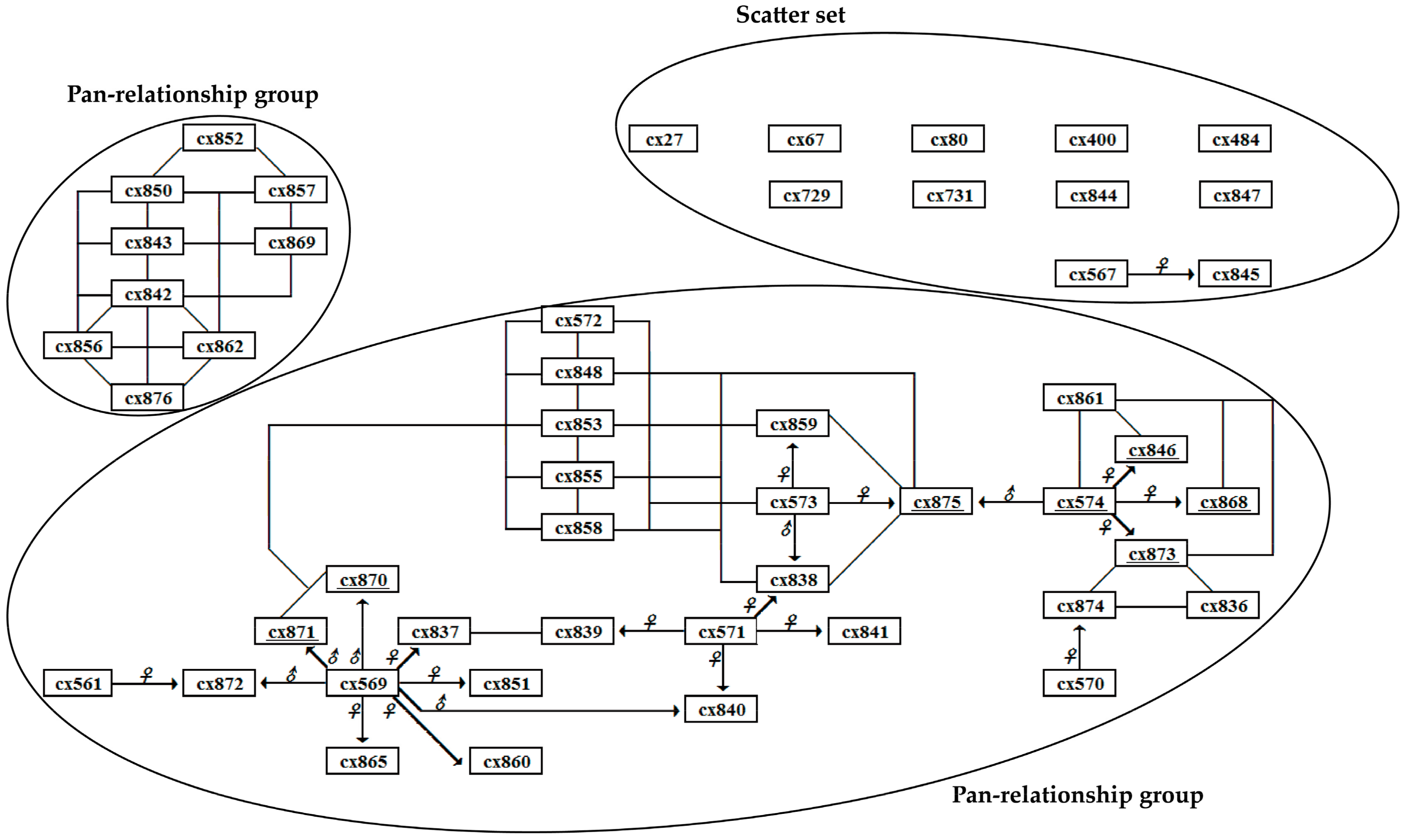
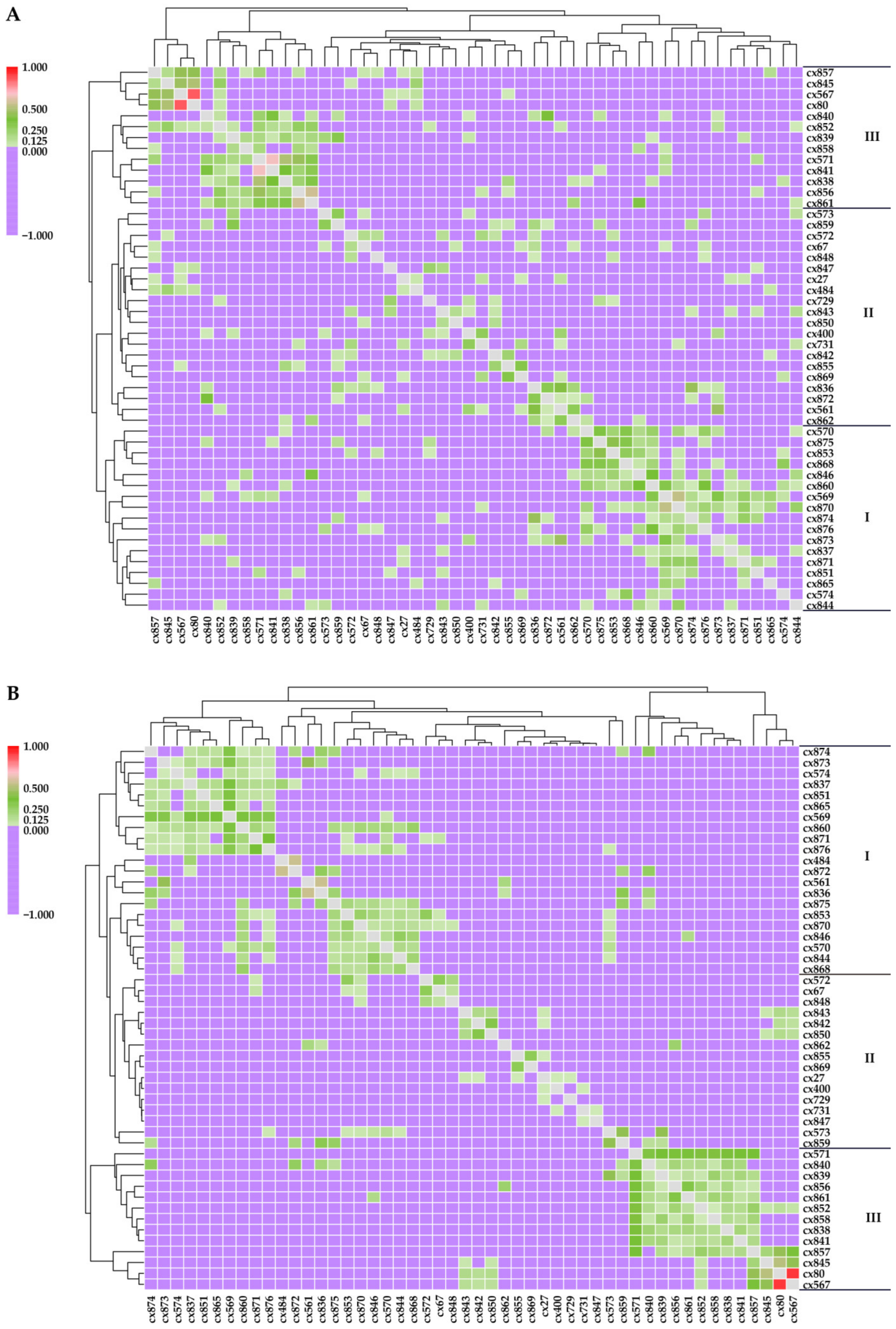
3.2. Genetic Relationships of Parent Trees
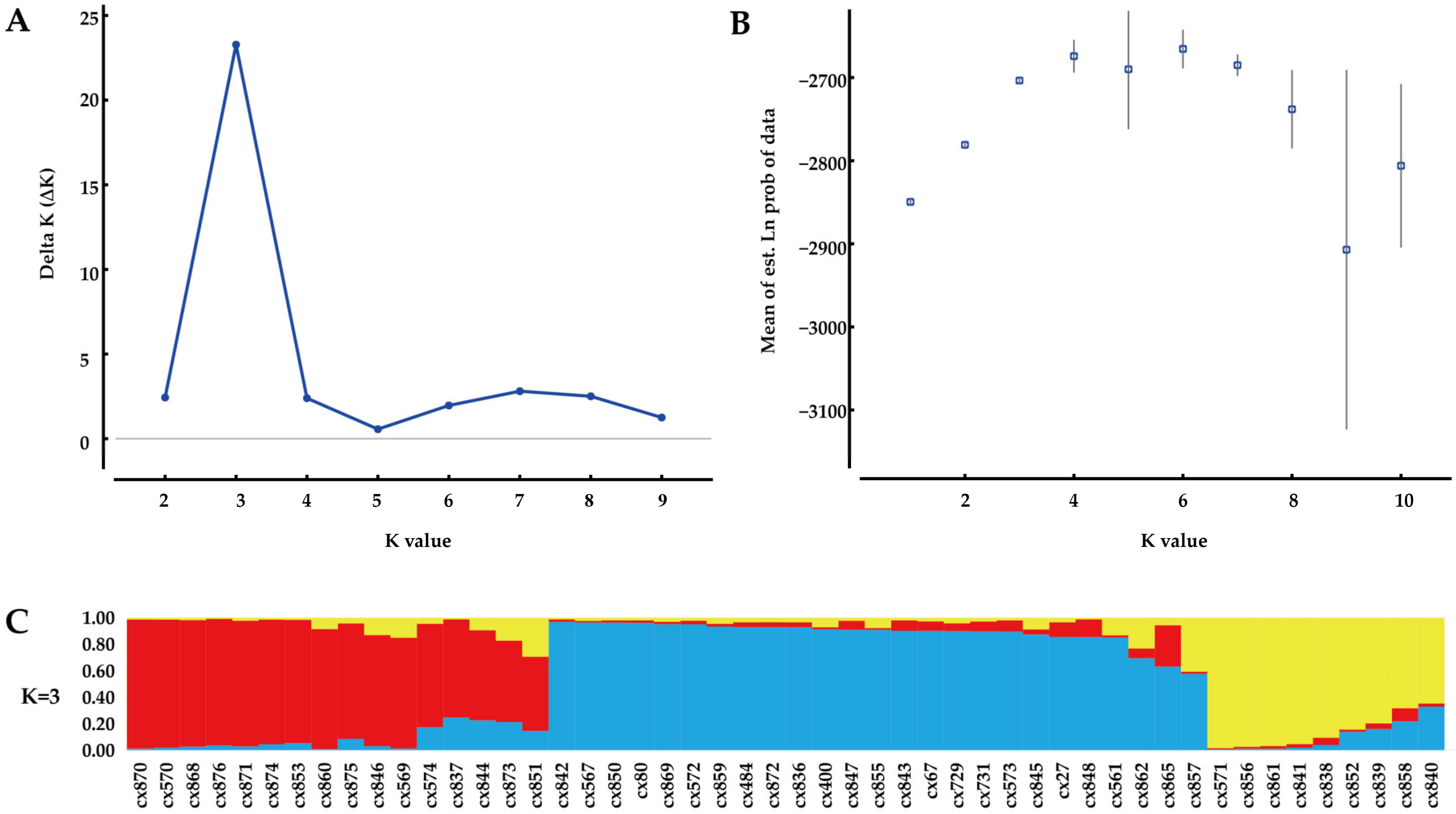
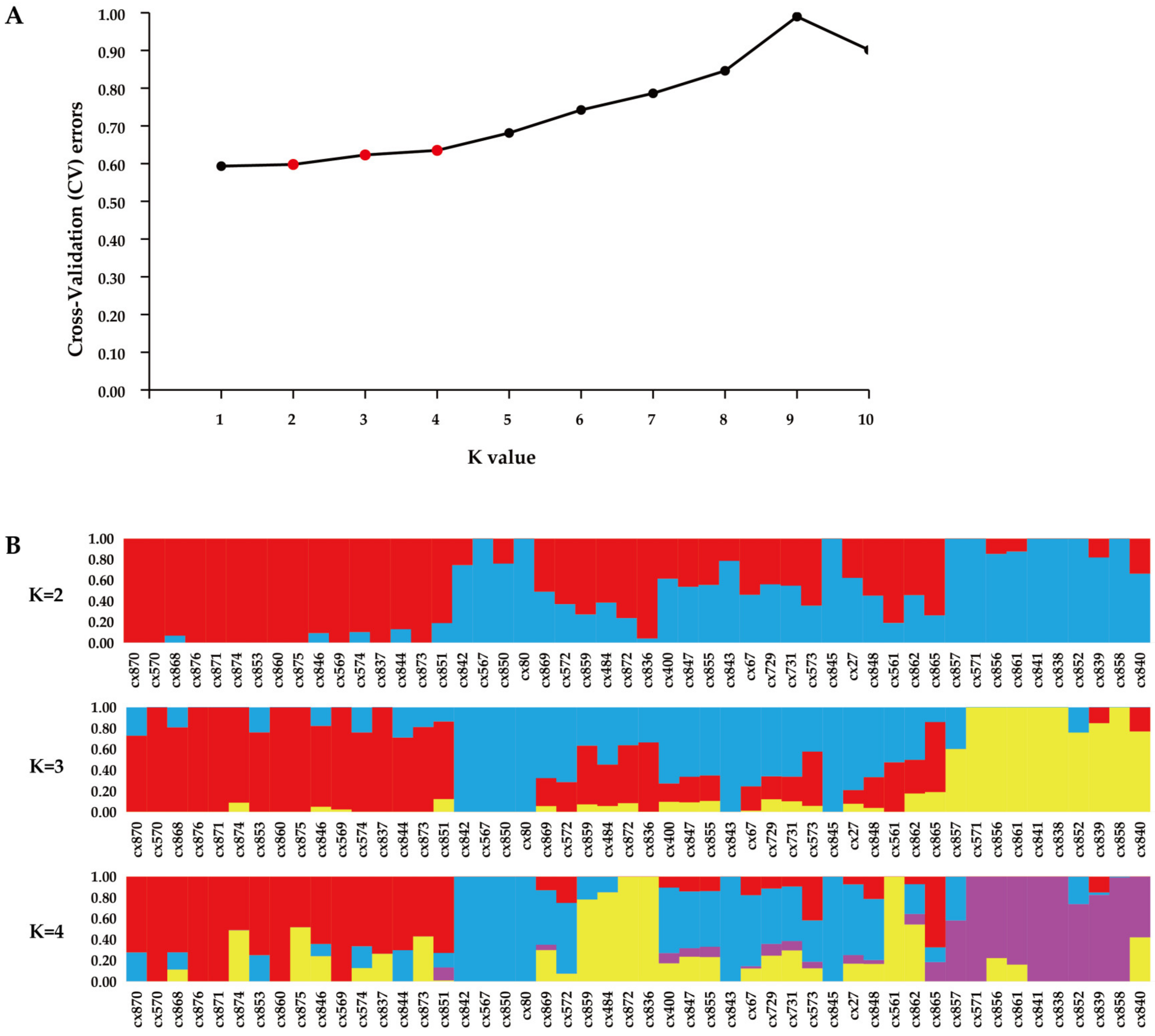
3.3. Genetic Structure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sethuraman, A. Estimating genetic relatedness in admixed populations. G3-Genes Genomes Genet. 2018, 8, 3203–3220. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Zhao, W.; Wennstrom, U.; Gull, B.A.; Wang, X.R. Parentage and relatedness reconstruction in Pinus sylvestris using genotyping-by-sequencing. Heredity 2020, 124, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Visscher, P.M.; Goddard, M.E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009, 91, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Wang, J.L. Effects of sampling close relatives on some elementary population genetics analyses. Mol. Ecol. Resour. 2018, 18, 41–54. [Google Scholar] [CrossRef]
- Yang, B.N.; Sun, H.H.; Qi, J.D.; Niu, S.H.; El-Kassaby, Y.A.; Li, W. Improved genetic distance-based spatial deployment can effectively minimize inbreeding in seed orchard. For. Ecosyst. 2020, 7, 11. [Google Scholar] [CrossRef]
- Howard, N.P.; Peace, C.; Silverstein, K.A.T.; Poets, A.; Luby, J.J.; Vanderzande, S.; Durel, C.E.; Muranty, H.; Denance, C.; van de Weg, E. The use of shared haplotype length information for pedigree reconstruction in asexually propagated outbreeding crops, demonstrated for apple and sweet cherry. Hortic. Res. 2021, 8, 202. [Google Scholar] [CrossRef]
- Yang, B.N.; Niu, S.H.; El-Kassaby, Y.A.; Li, W. Monitoring genetic diversity across Pinus tabuliformis seed orchard generations using SSR markers. Can. J. Forest. Res. 2021, 51, 1534–1540. [Google Scholar] [CrossRef]
- Liesebach, H.; Liepe, K.; Baucker, C. Towards new seed orchard designs in Germany—A review. Silvae Genet. 2021, 70, 84–98. [Google Scholar] [CrossRef]
- Keller, L.F.; Waller, D.M. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- White, T.L.; Adams, W.T.; Neale, D.B. Forest Genetics; CABI Publishing: Wallingford, UK, 2007. [Google Scholar]
- Doerksen, T.K.; Bousquet, J.; Beaulieu, J. Inbreeding depression in intra-provenance crosses driven by founder relatedness in white spruce. Tree Genet. Genomes 2014, 10, 203–212. [Google Scholar] [CrossRef]
- Ford, G.A.; McKeand, S.E.; Jett, J.B.; Isik, F. Effects of inbreeding on growth and quality traits in loblolly pine. For. Sci. 2015, 61, 579–585. [Google Scholar] [CrossRef]
- Van Becelaere, G.; Lubbers, E.L.; Paterson, A.H.; Chee, P.W. Pedigree- vs. DNA marker-based genetic similarity estimates in cotton. Crop Sci. 2005, 45, 2281–2287. [Google Scholar] [CrossRef]
- Yang, J.J.; Zhang, J.; Du, H.S.; Zhao, H.; Mao, A.J.; Zhang, X.F.; Jiang, L.; Zhang, H.Y.; Wen, C.L.; Xu, Y. Genetic relationship and pedigree of Chinese watermelon varieties based on diversity of perfect SNPs. Hortic. Plant J. 2022, 8, 489–498. [Google Scholar] [CrossRef]
- Gao, J.M.; Zhang, S.G.; Qi, L.W.; Zhang, Y.; Wang, C.G.; Song, W.Q. ISSR and AFLP identification and genetic relationships of Chinese elite accessions from the genus Populus. Ann. For. Sci. 2006, 63, 499–506. [Google Scholar] [CrossRef]
- Li, X.Y.; Xu, H.X.; Chen, J.W. Genetic diversity and relationships among 47 loquat varieties revealed by EST-SSR markers. Sci. Hortic. 2013, 160, 375–382. [Google Scholar] [CrossRef]
- Amom, T.; Tikendra, L.; Apana, N.; Goutam, M.; Sonia, P.; Koijam, A.S.; Potshangbam, A.M.; Rahaman, H.; Nongdam, P. Efficiency of RAPD, ISSR, iPBS, SCoT and phytochemical markers in the genetic relationship study of five native and economical important bamboos of North-East India. Phytochemistry 2020, 174, 112330. [Google Scholar] [CrossRef]
- Jiang, K.B.; Xie, H.; Liu, T.Y.; Liu, C.X.; Huang, S.W. Genetic diversity and population structure in Castanopsis fissa revealed by analyses of sequence-related amplified polymorphism (SRAP) markers. Tree Genet. Genomes 2020, 16, 52. [Google Scholar] [CrossRef]
- Han, Z.Q.; Gao, P.; Geng, X.N.; Du, K.; Kang, X.Y. Identification of the male parent of superior half-sib Populus tomentosa individuals based on SSR markers. Mol. Breed. 2017, 37, 155. [Google Scholar] [CrossRef]
- Tsykun, T.; Rellstab, C.; Dutech, C.; Sipos, G.; Prospero, S. Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistipes. Heredity 2017, 119, 371–380. [Google Scholar] [CrossRef]
- Ramesh, P.; Mallikarjuna, G.; Sameena, S.; Kumar, A.; Gurulakshmi, K.; Reddy, B.V.; Reddy, P.C.O.; Sekhar, A.C. Advancements in molecular marker technologies and their applications in diversity studies. J. Biosci. 2020, 45, 123. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Shrivastava, N.; Padh, H. Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 2008, 27, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.Q.; Hu, D.H.; Wei, R.P.; Yan, S.; Wang, R.H. Chinese fir breeding in the high-throughput sequencing era: Insights from SNPs. Forests 2019, 10, 681. [Google Scholar] [CrossRef]
- Howe, G.T.; Jayawickrama, K.; Kolpak, S.E.; Kling, J.; Trappe, M.; Hipkins, V.; Ye, T.; Guida, S.; Cronn, R.; Cushman, S.A.; et al. An Axiom SNP genotyping array for Douglas-fir. BMC Genom. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Capo-Chichi, L.J.A.; Elakhdar, A.; Kubo, T.; Nyachiro, J.; Juskiw, P.; Capettini, F.; Slaski, J.J.; Ramirez, G.H.; Beattie, A.D. Genetic diversity and population structure assessment of Western Canadian barley cooperative trials. Front. Plant Sci. 2022, 13, 1006719. [Google Scholar] [CrossRef]
- Varshney, R.K.; Chabane, K.; Hendre, P.S.; Aggarwal, R.K.; Graner, A. Comparative assessment of EST-SSR, EST-SNP and AFLP markers for evaluation of genetic diversity and conservation of genetic resources using wild, cultivated and elite barleys. Plant Sci. 2007, 173, 638–649. [Google Scholar] [CrossRef]
- Yang, X.H.; Xu, Y.B.; Shah, T.; Li, H.H.; Han, Z.H.; Li, J.S.; Yan, J.B. Comparison of SSRs and SNPs in assessment of genetic relatedness in maize. Genetica 2011, 139, 1045–1054. [Google Scholar] [CrossRef]
- Amar, M.H.; Biswas, M.K.; Zhang, Z.W.; Guo, W.W. Exploitation of SSR, SRAP and CAPS-SNP markers for genetic diversity of Citrus germplasm collection. Sci. Hortic. 2011, 128, 220–227. [Google Scholar] [CrossRef]
- Singh, N.; Choudhury, D.R.; Singh, A.K.; Kumar, S.; Srinivasan, K.; Tyagi, R.K.; Singh, N.K.; Singh, R. Comparison of SSR and SNP markers in estimation of genetic diversity and population structure of Indian rice varieties. PLoS ONE 2013, 8, e84136. [Google Scholar] [CrossRef]
- Garcia, C.; Guichoux, E.; Hampe, A. A comparative analysis between SNPs and SSRs to investigate genetic variation in a juniper species (Juniperus phoenicea ssp. turbinata). Tree Genet. Genomes 2018, 14, 87. [Google Scholar] [CrossRef]
- Kimaro, D.; Melis, R.; Sibiya, J.; Shimelis, H.; Shayanowako, A. Analysis of genetic diversity and population structure of pigeonpea [Cajanus cajan (L.) Millsp] accessions using SSR markers. Plants 2020, 9, 1643. [Google Scholar] [CrossRef]
- Zavinon, F.; Adoukonou-Sagbadja, H.; Keilwagen, J.; Lehnert, H.; Ordon, F.; Perovic, D. Genetic diversity and population structure in Beninese pigeon pea Cajanus cajan (L.) Huth landraces collection revealed by SSR and genome wide SNP markers. Genet. Resour. Crop Evol. 2020, 67, 191–208. [Google Scholar] [CrossRef]
- Duan, H.J.; Hu, D.H.; Li, Y.; Zheng, H.Q. Characterization of a collection of Chinese fir elite genotypes using sequence-related amplified polymorphism markers. J. For. Res. 2016, 27, 1105–1110. [Google Scholar] [CrossRef]
- Fung, L.E. Wood properties of New Zealand-grown Cunninghamia lanceolata. N. Z. J. For. Sci. 1993, 23, 324–338. [Google Scholar]
- Tran, B.D.; Le, T.N.H. A review of Cunninghamia lanceolata (Lamb.) Hook: A recent update and potential application in Vietnam. Vietnam. J. Agric. Sci. 2020, 3, 892–902. [Google Scholar] [CrossRef]
- Farooq, T.H.; Yan, W.; Rashid, M.H.U.; Tigabu, M.; Gilani, M.M.; Zou, X.H.; Wu, P.F. Chinese fir (Cunninghamia Lanceolata) a green gold of China with continues decline in its productivity over the successive rotations: A review. Appl. Ecol. Environ. Res. 2019, 17, 11055–11067. [Google Scholar] [CrossRef]
- Li, M.; Chen, X.Z.; Huang, M.S.; Wu, P.F.; Ma, X.Q. Genetic diversity and relationships of ancient Chinese fir (Cunninghamia lanceolata) genotypes revealed by sequence-related amplified polymorphism markers. Genet. Resour. Crop Evol. 2017, 64, 1087–1099. [Google Scholar] [CrossRef]
- Lin, E.P.; Zhuang, H.B.; Yu, J.J.; Liu, X.Y.; Huang, H.H.; Zhu, M.Y.; Tong, Z.K. Genome survey of Chinese fir (Cunninghamia lanceolata): Identification of genomic SSRs and demonstration of their utility in genetic diversity analysis. Sci. Rep. 2020, 10, 4698. [Google Scholar] [CrossRef]
- Duan, H.J.; Hu, R.Y.; Wu, B.; Chen, D.X.; Huang, K.Y.; Dai, J.; Chen, Q.; Wei, Z.C.; Cao, S.; Sun, Y.H.; et al. Genetic characterization of red-colored heartwood genotypes of Chinese fir using simple sequence repeat (SSR) markers. Genet. Mol. Res. 2015, 14, 18552–18561. [Google Scholar] [CrossRef]
- Zheng, H.Q.; Duan, H.J.; Hu, D.H.; Wei, R.P.; Li, Y. Sequence-related amplified polymorphism primer screening on Chinese fir (Cunninghamia lanceolata (Lamb.) Hook). J. For. Res. 2015, 26, 101–106. [Google Scholar] [CrossRef]
- Duan, H.J.; Cao, S.; Zheng, H.Q.; Hu, D.H.; Lin, J.; Cui, B.B.; Lin, H.Z.; Hu, R.Y.; Wu, B.; Sun, Y.H.; et al. Genetic characterization of Chinese fir from six provinces in Southern China and construction of a core collection. Sci. Rep. 2017, 7, 13814. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Q.; Hu, D.H.; Zheng, H.Q.; Liu, W.X.; Lü, Y.Z. Preliminary study on anthracnose resistance of Chinese fir third-generation seed orchard parents. Subtrop. Plant Sci. 2020, 49, 196–199. [Google Scholar] [CrossRef]
- Wang, R.H.; Hu, D.H.; Wei, R.P.; Yan, S.; Zheng, H.Q. Early evaluation and selection of the Chinese fir breeding parents based on growth and cone production traits. J. Trop. Subtrop. Bot. 2022, 30, 195–201. [Google Scholar] [CrossRef]
- Wen, Y.; Ueno, S.; Han, W.; Tsumura, Y. Development and characterization of 28 polymorphic EST-SSR markers for Cunninghamia lanceolata (Taxodiaceae) based on transcriptome sequences. Silvae Genet. 2013, 62, 137–141. [Google Scholar] [CrossRef]
- Sun, X.W.; Liu, D.Y.; Zhang, X.F.; Li, W.B.; Liu, H.; Hong, W.G.; Jiang, C.B.; Guan, N.; Ma, C.X.; Zeng, H.P.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data, P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Lynch, M.; Ritland, K. Estimation of pairwise relatedness with molecular markers. Genetics 1999, 152, 1753–1766. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Liu, K.J.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Yang, J.A.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef]
- Bessega, C.; Saidman, B.O.; Darquier, M.R.; Ewens, M.; Felker, P.; Vilardi, J.C. Accuracy of dominant markers for estimation of relatedness and heritability in an experimental stand of Prosopis alba (Leguminosae). Tree Genet. Genomes 2011, 7, 103–115. [Google Scholar] [CrossRef]
- Jia, S.Z.; Yan, Z.M.; Wang, Y.H.; Wei, Y.; Xie, Z.Q.; Zhang, F. Genetic diversity and relatedness among ornamental purslane (Portulaca L.) accessions unraveled by SRAP markers. 3 Biotech 2017, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.Q.; Liu, R.; Xue, J.Q.; Wang, S.L.; Zhang, X.X. Genetic diversity and relatedness analysis of nine wild species of tree peony based on simple sequence repeats markers. Hortic. Plant J. 2021, 7, 579–588. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Emanuelli, F.; Lorenzi, S.; Grzeskowiak, L.; Catalano, V.; Stefanini, M.; Troggio, M.; Myles, S.; Martinez-Zapater, J.M.; Zyprian, E.; Moreira, F.M.; et al. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 2013, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Paczos-Grzeda, E. Pedigree, RAPD and simplified AFLP-based assessment of genetic relationships among Avena sativa L. cultivars. Euphytica 2004, 138, 13–22. [Google Scholar] [CrossRef]
- Huang, R.; Zeng, W.S.; Deng, H.Y.; Hu, D.H.; Wang, R.H.; Zheng, H.Q. Inbreeding in Chinese fir: Insight into the rare self-fertilizing event from a genetic view. Genes 2022, 13, 2105. [Google Scholar] [CrossRef]
- Govindaraj, M.; Vetriventhan, M.; Srinivasan, M. Importance of genetic diversity assessment in crop plants and its recent advances: An overview of its analytical perspectives. Genet. Res. Int. 2015, 2015, 431487. [Google Scholar] [CrossRef]
- Chen, X.B.; Sun, X.M.; Dong, L.M.; Zhang, S.G. Mating patterns and pollen dispersal in a Japanese larch (Larix kaempferi) clonal seed orchard: A case study. Sci. China Life Sci. 2018, 61, 1011–1023. [Google Scholar] [CrossRef]
- Huang, R.; Hu, D.H.; Deng, H.Y.; Wang, R.H.; Wei, R.P.; Yan, S.; Zheng, H.Q. SNPs-Based Assessment of Genetic Diversity and Genetic Structure in Elite Chinese Fir. Mol. Plant Breed. 2021. Available online: https://kns.cnki.net/kcms/detail/46.1068.S.20211130.1703.012.html (accessed on 1 December 2021).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, W.; Su, Y.; Huang, R.; Hu, D.; Huang, S.; Zheng, H. Insight into the Complex Genetic Relationship of Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) Advanced Parent Trees Based on SSR and SNP Datasets. Forests 2023, 14, 347. https://doi.org/10.3390/f14020347
Zeng W, Su Y, Huang R, Hu D, Huang S, Zheng H. Insight into the Complex Genetic Relationship of Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) Advanced Parent Trees Based on SSR and SNP Datasets. Forests. 2023; 14(2):347. https://doi.org/10.3390/f14020347
Chicago/Turabian StyleZeng, Weishan, Yan Su, Rong Huang, Dehuo Hu, Shaowei Huang, and Huiquan Zheng. 2023. "Insight into the Complex Genetic Relationship of Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) Advanced Parent Trees Based on SSR and SNP Datasets" Forests 14, no. 2: 347. https://doi.org/10.3390/f14020347
APA StyleZeng, W., Su, Y., Huang, R., Hu, D., Huang, S., & Zheng, H. (2023). Insight into the Complex Genetic Relationship of Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) Advanced Parent Trees Based on SSR and SNP Datasets. Forests, 14(2), 347. https://doi.org/10.3390/f14020347