
Study on the Genetic Structure Based on Geographic Populations of the Endangered Tree Species: Liriodendron chinense

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
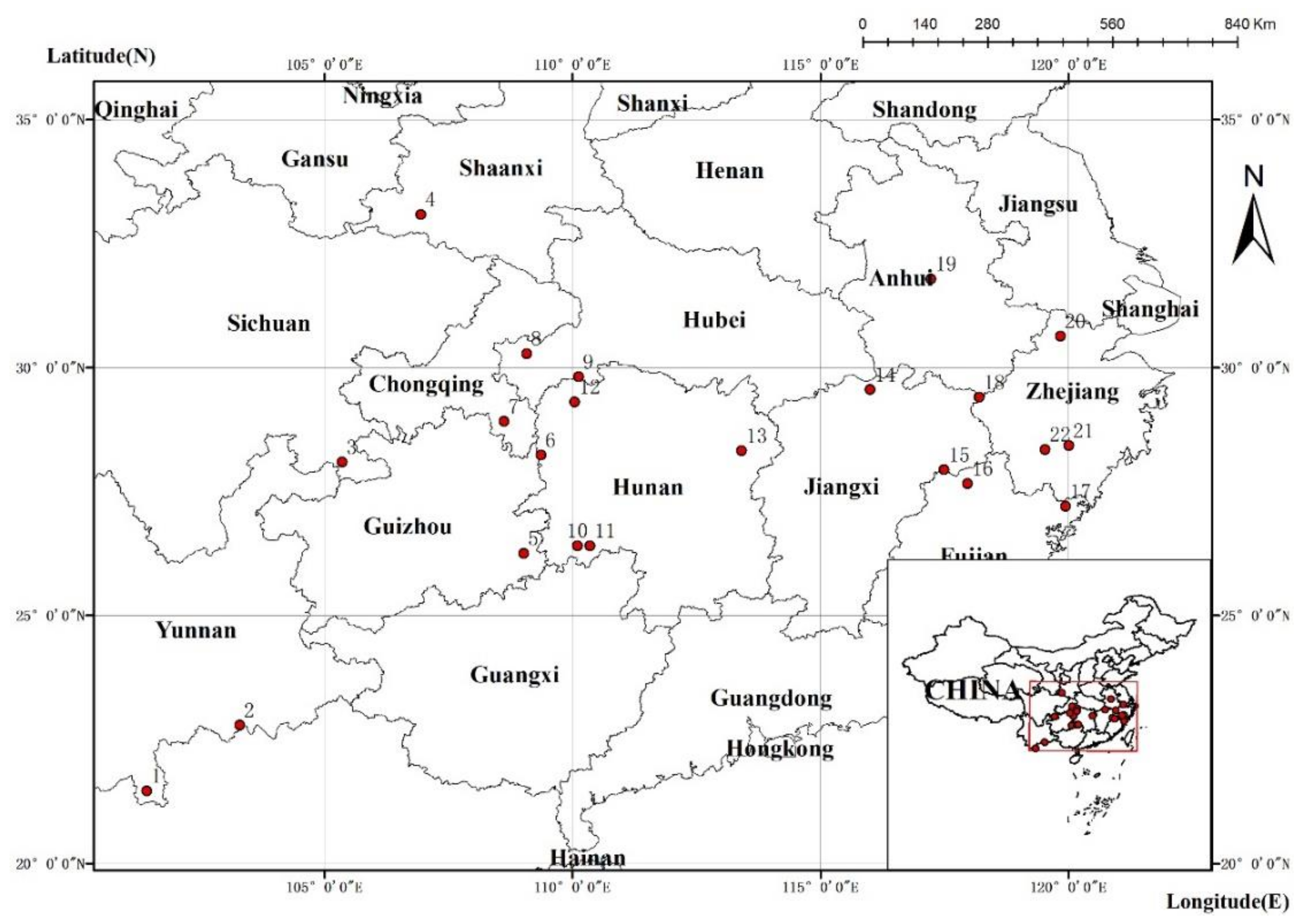
2.1. Plant Materials
2.2. DNA Extraction
2.3. EST-SSR Primers and PCR Amplification
2.4. Analysis of Data
3. Results
3.1. Genetic Diversity of Different Geographic Populations
3.2. Genetic Differentiation among Geographic Populations
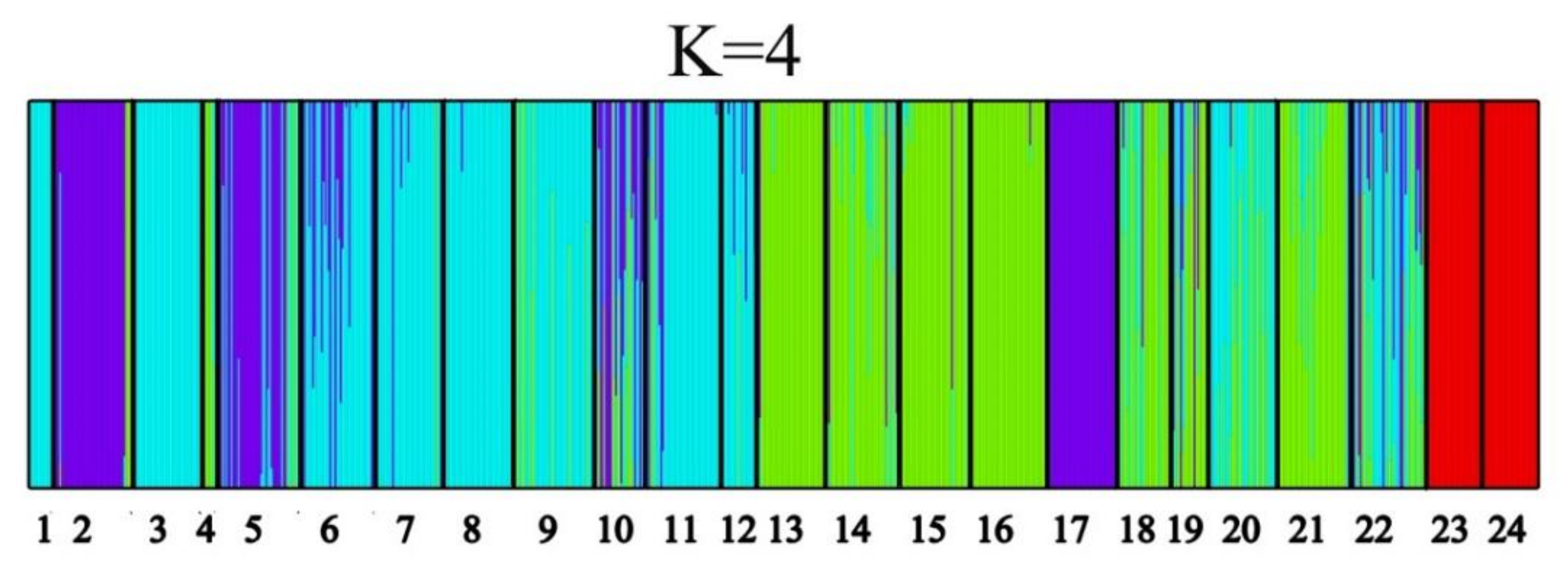
3.3. Genetic Structure of Geospatial
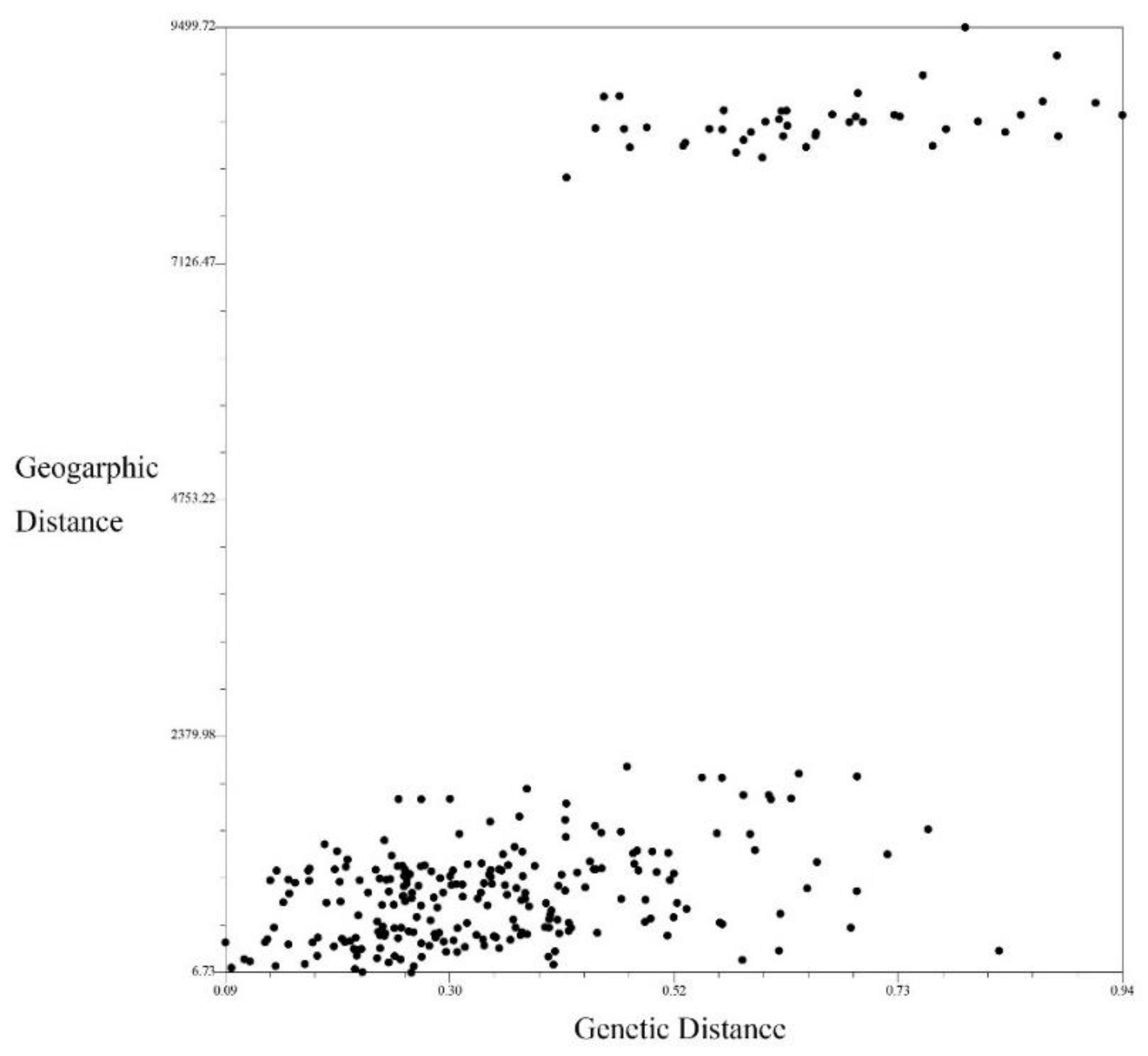
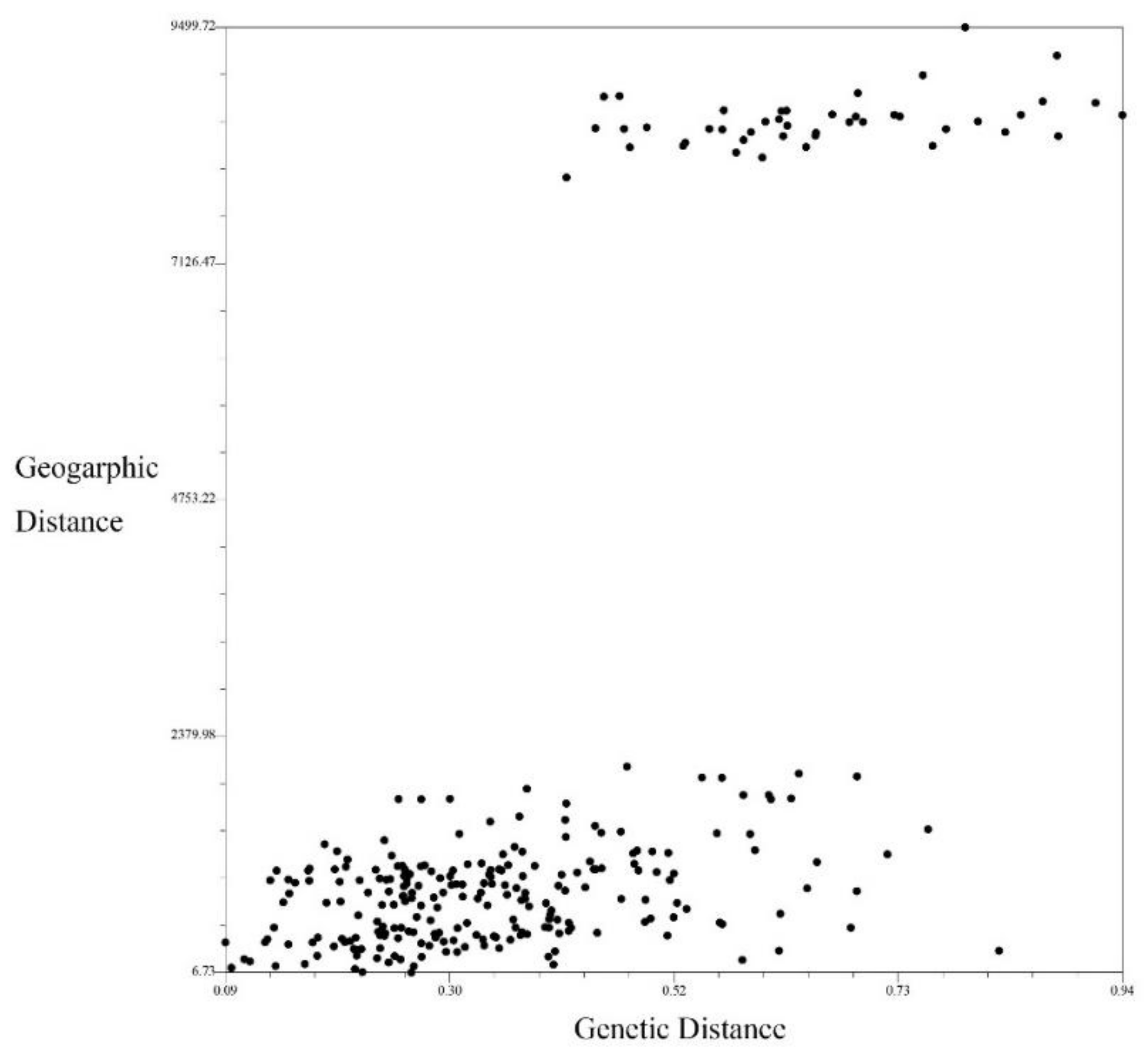
3.4. Mantel Test
4. Discussion
4.1. Genetic Diversity of Different Geographic Populations
4.2. Genetic Differentiation among Geographic Populations
4.3. Genetic Structure of Geospatial
4.4. Conservation of L. chinense Genetic Resources
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, Y.; Cheng, Y.; Li, K.; Li, H. Integrating phylogeographic analysis and geospatial methods to infer historical dispersal routes and glacial refugia of Liriodendron chinense. Forests 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.M.; Kang, H.Z.; Liu, C.J. research progress of potential plant refuges in the quaternary ice age in mainland China. Bull. Bot. Res. 2011, 31, 623–632. [Google Scholar]
- Kim, S.; Koh, J.; Yoo, M.-J.; Kong, H.Z.; Hu, Y.; Ma, H.; Soltis, P.S.; Soltis, D.E. Expression of floral MADS-box genes in basal angiosperms: Implications for the evolution of floral regulators. Plant J. 2005, 43, 724–744. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.Y.; Carlson, J.E.; Leebens-Mack, J.H.; Wall, P.K.; Muller, L.A.; Buzgo, M.; Landherr, L.L.; Hu, Y.; Di Loreto, D.S.; Ilut, D.C.; et al. An EST database for Liriodendron tulipifera L. floral buds: The first EST resource for functional and comparative genomics in Liriodendron. Tree Genet. Genomes 2008, 4, 419–433. [Google Scholar] [CrossRef]
- Parks, C.R.; Wendel, J.F. Molecular divergence between Asian and North American species of Liriodendron (Magnoliaceae) with implications for interpretation of fossil floras. Am. J. Bot. 1990, 77, 1243–1256. [Google Scholar] [CrossRef]
- Koo, B.W.; Kim, H.Y.; Park, N.; Lee, S.M.; Yeo, H.; Choi, I.G. Organosolv pretreatment of Liriodendron tulipifera and simultaneous saccharification and fermentation for bioethanol production. Biomass Bioenergy 2011, 35, 1833–1840. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, C.L.; Shi, M.; Guo, F.; Liu, J.; Li, L.Z.; Ren, Q.; Tao, S.B.; Tang, M.H.; Ye, H.Y.; et al. Ethanol extract of Liriodendron chinense (Hemsl.) Sarg barks attenuates hyperuricemic nephropathy by inhibiting renal fibrosis and inflammation in mice. J. Ethnopharmacol. 2020, 264, 113278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Y.; Zhao, Y. Advance of study on Liriodendron. J. Yunnan Agric. Univ. 2005, 20, 697–701. [Google Scholar]
- Hao, Z.Y.; Wang, X.; Zong, Y.X.; Wen, S.Y.; Cheng, Y.L.; Li, H.G. Enzymatic activity and functional analysis under multiple abiotic stress conditions of a dehydroascorbate reductase gene derived from Liriodendron Chinense. Environ. Exp. Bot. 2019, 167, 103850. [Google Scholar] [CrossRef]
- Zhang, W.W.; Niu, J.F.; Wang, X.K.; Tian, Y.; Yao, F.F.; Feng, Z.Z. Effects of ozone exposure on growth and photosynthesis of the seedlings of Liriodendron chinense (Hemsl.) Sarg, a native tree species of subtropical China. Photosynthetica 2011, 49, 29–36. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, H.; Xie, T.; Xu, Y.; Zhao, L.; Tian, W. Effects of climate change on the potentially suitable climatic geographical range of Liriodendron chinense. Forests 2017, 8, 399. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.-H.; Wei, N.; Fritsch, P.W.; Yao, X.-H. AFLP genome scanning reveals divergent selection in natural populations of Liriodendron chinense (Magnoliaceae) along a latitudinal transect. Front. Plant Sci. 2016, 7, 698. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Zhong, Y.; Liu, S.; Liu, L.; Liu, T.; Li, Y.; Yu, F. New insight into the phylogeographic pattern of Liriodendron chinense (Magnoliaceae) revealed by chloroplast DNA: East-west lineage split and genetic mixture within western subtropical China. Peer J. 2019, 7, e6355. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Chen, L.; Feng, Y.; Yao, J.; Li, B.; Xu, M.; Li, H. High genetic diversity but limited gene flow among remnant and fragmented natural populations of Liriodendron chinense Sarg. Biochem. Syst. Ecol. 2014, 54, 230–236. [Google Scholar] [CrossRef]
- Zhong, Y.; Yang, A.; Liu, S.; Liu, L.; Li, Y.; Wu, Z.; Yu, F. RAD-Seq Data Point to a Distinct Split in Liriodendron (Magnoliaceae) and Obvious East–West Genetic Divergence in Liriodendron chinense. Forests 2018, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hao, Z.; Guang, X.; Zhao, C.; Wang, P.; Xue, L.; Zhu, Q.; Yang, L.; Sheng, Y.; Zhou, Y.; et al. Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nat. Plants 2019, 5, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.M. Geographical distribution and spatial pattern of Liriodendron chinense. J. Nanjing For. Univ. 1994, 18, 13–18. [Google Scholar]
- Hao, R.M.; He, S.A.; Tang, S.J.; Wu, S.P. Natural distribution and characteristics of Liriodendron chinense in China. J. Plant. Resour. Environ. 1995, 4, 1–6. [Google Scholar]
- Zhu, Q.-W.; Li, H.-G. [Comparison of genetic diversity among five mating types of Liriodendron revealed by SSR markers]. Yi Chuan Hered. 2010, 32, 183–188. [Google Scholar] [CrossRef]
- Yang, A.; Dick, C.W.; Yao, X.; Huang, H. Impacts of biogeographic history and marginal population genetics on species range limits: A case study of Liriodendron chinense. Sci. Rep. 2016, 6, 25632. [Google Scholar] [CrossRef] [Green Version]
- Li, J.M.; Zhou, Z.C.; Wu, K.Y.; Jin, G.Q. RAPD markers to study the genetic differentiation of the geographic populations of Liriodendron chinense. For. Sci. 2002, 38, 61–66. [Google Scholar]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 2020, 53, 1–12. [Google Scholar] [CrossRef]
- Long, X.; Weng, Y.; Liu, S.; Hao, Z.; Sheng, Y.; Guan, L.; Shi, J.; Chen, J.H. Genetic Diversity and Differentiation of Relict Plant Liriodendron Populations Based on 29 Novel EST-SSR Markers. Forests 2019, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Yaghuti, A.A.; Movahedi, A.; Mohammadi, K.; Zhuge, Q.; Li, H.G. Genetic diversity among a collection of L. chinense germplasm analyzed with SSR markers. J. Biochem. Microb. Toxicol. 2017, 1, 102. [Google Scholar]
- Yaghuti, A.A.; Movahedi, A.; Mohammadi, K.; Zhuge, Q.; Li, H.G. The genetic diversity of Liriodendron tulipifera germplasm revealed by SSR markers. J. Biochem. Microb. Toxicol. 2018, 2, 1000105. [Google Scholar]
- Cota-Sánchez, J.H.; Remarchuk, K.; Ubayasena, K. Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 2006, 24, 161–167. [Google Scholar] [CrossRef]
- Nei, M. Definition and estimation of fixation indices. Evolution 1986, 40, 643–645. [Google Scholar] [CrossRef]
- Liu, K.J.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices, Version 2.9.3. 2001. Available online: http://www.unil.ch/popgen/softwares/fstat.htm (accessed on 20 June 2021).
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 64, 1567–1587. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes. 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System, Version 2.2, 2.1; Department of Ecoloy and Evolution, State University of New York: New York, NY, USA, 2005. [Google Scholar]
- Li, L.M.; Shi, X.M.; Jiang, W.X.; Bai, T.D.; Meng, Y.Y.; Tan, F.Y.; Huang, S.X. ISSR Analysis of Genetic Diversity of the Natural Populations of Liriodendron chinense in Guangxi. Guangxi Plant 2017, 37, 22–28. [Google Scholar]
- Shi, X.; Wen, Q.; Cao, M.; Guo, X.; Xu, L.A. Genetic diversity and structure of natural Quercus variabilis population in china as revealed by microsatellites markers. Forests 2017, 8, 495. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Mu, K.; Ni, Z.; Liu, X.; Li, Y.; Xu, L.-A. Analysis of genetic diversity of ancient Ginkgo populations using SSR markers. Ind. Crops Prod. 2020, 145, 111942. [Google Scholar] [CrossRef]
- Qin, Y.Y.; Han, H.R.; Kang, F.F.; Zhao, Q. Genetic diversity analysis of natural populations of Quercus wutaishansea in Shanxi Province based on SSR markers. J. Beijing For. Univ. 2012, 34, 61–65. [Google Scholar]
- Xu, X.L.; Xu, L.A.; Huang, M.R.; Wang, Z.R. Study on genetic diversity of Quercus variabilis natural population by SSR. Heredity 2004, 4, 683–688. [Google Scholar]
- Xu, L.A.; Li, X.J.; Pan, H.X.; Zou, P.Y.; Yin, T.M.; Huang, M.R. Study on the genetic structure of Castanopsis fargesii population by SSR. Chin. Bull. Bot. 2001, 43, 409–412. [Google Scholar]
- Feng, Y.H.; Li, H.G.; Yang, Z.Q.; Wu, D.S. Study on the genetic structure of natural populations of Pinus massoniana provenance. Guangxi Plant 2016, 36, 1275–1275, 1395. [Google Scholar]
- Huang, S.Q.; Guo, Y.H. Variation of pollination and resource limitation in a low seed-set tree, Liriodendron chinense (Magnoliaceae). Bot. J. Linn. Soc. 2002, 140, 31–38. [Google Scholar] [CrossRef] [Green Version]
- He, S.A.; Hao, R.M.; Tang, S.J. Study on the Ecological Factors of Liriodendron chinense Endangering. J. Plant. Resour. Environ. 1996, 5, 1–8. [Google Scholar]
- Li, J.M. Geographical Genetic Variations and Selection of Superior Provenances of Liriodendron chinense. For. Sci. 2001, 37, 41–49. [Google Scholar]
- Luo, G.Z.; Shi, J.S.; Yin, T.M.; Huang, M.R.; Wang, M.X. Analysis of genetic diversity between Liriodendron chinense and Liriodendron tulipifera using RAPD markers. J. Plant Resour. Environ. 2000, 2, 9–13. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Locus | Na | Ne | He | PIC | I | Fst | Nm |
|---|---|---|---|---|---|---|---|
| LCEST-41 | 4 | 2.899 | 0.655 | 0.602 | 1.202 | 0.273 | 0.666 |
| LCEST-53 | 4 | 1.442 | 0.307 | 0.292 | 0.650 | 0.166 | 1.255 |
| LCEST-57 | 3 | 1.830 | 0.454 | 0.371 | 0.719 | 0.267 | 0.687 |
| LCEST-60 | 3 | 2.951 | 0.662 | 0.587 | 1.090 | 0.302 | 0.579 |
| LCEST-61 | 5 | 3.772 | 0.735 | 0.687 | 1.411 | 0.165 | 1.262 |
| LCEST-62 | 4 | 1.616 | 0.382 | 0.349 | 0.702 | 0.222 | 0.875 |
| LCEST-68 | 5 | 2.775 | 0.640 | 0.576 | 1.183 | 0.256 | 0.727 |
| LCEST-75 | 5 | 2.231 | 0.552 | 0.505 | 1.032 | 0.372 | 0.423 |
| LCEST-80 | 4 | 3.519 | 0.716 | 0.662 | 1.306 | 0.298 | 0.588 |
| LCEST-97 | 2 | 1.018 | 0.017 | 0.017 | 0.050 | 0.052 | 4.550 |
| LCEST-103 | 4 | 1.169 | 0.145 | 0.138 | 0.318 | 0.125 | 1.756 |
| LCEST-104 | 4 | 3.489 | 0.714 | 0.664 | 1.318 | 0.355 | 0.453 |
| LCEST-107 | 4 | 2.946 | 0.661 | 0.591 | 1.146 | 0.430 | 0.332 |
| LCEST-119 | 3 | 2.903 | 0.656 | 0.582 | 1.083 | 0.196 | 1.023 |
| LCEST-131 | 4 | 3.019 | 0.669 | 0.616 | 1.232 | 0.259 | 0.715 |
| LCEST-169 | 4 | 2.689 | 0.629 | 0.553 | 1.056 | 0.402 | 0.372 |
| LCEST-181 | 4 | 2.910 | 0.657 | 0.584 | 1.113 | 0.362 | 0.442 |
| LCEST-184 | 4 | 2.763 | 0.639 | 0.564 | 1.059 | 0.361 | 0.442 |
| LCEST-186 | 5 | 3.398 | 0.706 | 0.649 | 1.290 | 0.220 | 0.889 |
| LCEST-195 | 3 | 2.304 | 0.566 | 0.478 | 0.920 | 0.520 | 0.231 |
| Mean | 3.9 | 2.582 | 0.558 | 0.503 | 0.994 | 0.302 | 0.578 |
| Species | Population Code | Na | Ne | H | Ho | He | AR | F |
|---|---|---|---|---|---|---|---|---|
| L. chinense (Hemsl.) Sarg. | YML | 1.600 | 1.333 | 0.170 | 0.100 | 0.177 | 1.555 | 0.434 |
| YJP | 2.150 | 1.362 | 0.226 | 0.125 | 0.229 | 1.900 | 0.453 | |
| SXY | 2.600 | 1.910 | 0.372 | 0.320 | 0.377 | 2.286 | 0.150 | |
| SHZ | 2.650 | 1.862 | 0.402 | 0.222 | 0.425 | 2.650 | 0.477 | |
| GLP | 3.050 | 2.046 | 0.429 | 0.280 | 0.434 | 2.668 | 0.354 | |
| GMN | 2.550 | 1.745 | 0.332 | 0.223 | 0.336 | 2.207 | 0.337 | |
| CYY | 3.150 | 2.178 | 0.480 | 0.310 | 0.486 | 2.849 | 0.362 | |
| HLC | 2.800 | 2.025 | 0.426 | 0.471 | 0.431 | 2.497 | −0.093 | |
| HHF | 2.800 | 1.908 | 0.409 | 0.217 | 0.413 | 2.542 | 0.476 | |
| HSN | 2.750 | 2.066 | 0.430 | 0.183 | 0.437 | 2.617 | 0.581 | |
| HCB | 3.150 | 2.391 | 0.532 | 0.384 | 0.539 | 2.884 | 0.288 | |
| HSZ | 3.250 | 2.251 | 0.508 | 0.295 | 0.521 | 2.985 | 0.433 | |
| HLY | 2.550 | 1.768 | 0.343 | 0.290 | 0.347 | 2.213 | 0.164 | |
| JLS | 2.700 | 1.870 | 0.375 | 0.223 | 0.379 | 2.365 | 0.412 | |
| JWY | 2.550 | 1.893 | 0.399 | 0.241 | 0.404 | 2.377 | 0.403 | |
| FWY | 2.300 | 1.702 | 0.345 | 0.278 | 0.350 | 2.064 | 0.206 | |
| FZR | 1.550 | 1.362 | 0.194 | 0.179 | 0.196 | 1.495 | 0.090 | |
| AHS | 3.250 | 2.165 | 0.471 | 0.294 | 0.478 | 2.938 | 0.386 | |
| ADB | 2.950 | 2.067 | 0.472 | 0.213 | 0.484 | 2.766 | 0.561 | |
| ZAJ | 3.200 | 2.141 | 0.481 | 0.285 | 0.487 | 2.796 | 0.415 | |
| ZLS | 2.700 | 1.907 | 0.407 | 0.330 | 0.412 | 2.456 | 0.198 | |
| ZSY | 3.300 | 1.987 | 0.437 | 0.301 | 0.442 | 2.890 | 0.318 | |
| mean | 2.707 | 1.906 | 0.393 | 0.262 | 0.399 | 2.455 | 0.337 | |
| L. tulipifera L. | LA | 3.250 | 2.279 | 0.473 | 0.363 | 0.481 | 2.914 | 0.245 |
| MO | 3.000 | 2.203 | 0.420 | 0.358 | 0.427 | 2.744 | 0.161 | |
| mean | 3.125 | 2.241 | 0.446 | 0.360 | 0.454 | 2.829 | 0.203 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.-Y.; Hui, L.-X.; Huang, S.-J.; Ni, Z.-X.; Yu, F.-X.; Xu, L.-A. Study on the Genetic Structure Based on Geographic Populations of the Endangered Tree Species: Liriodendron chinense. Forests 2021, 12, 917. https://doi.org/10.3390/f12070917
Zhou P-Y, Hui L-X, Huang S-J, Ni Z-X, Yu F-X, Xu L-A. Study on the Genetic Structure Based on Geographic Populations of the Endangered Tree Species: Liriodendron chinense. Forests. 2021; 12(7):917. https://doi.org/10.3390/f12070917
Chicago/Turabian StyleZhou, Peng-Yan, Li-Xing Hui, Shu-Jing Huang, Zhou-Xian Ni, Fa-Xin Yu, and Li-An Xu. 2021. "Study on the Genetic Structure Based on Geographic Populations of the Endangered Tree Species: Liriodendron chinense" Forests 12, no. 7: 917. https://doi.org/10.3390/f12070917
APA StyleZhou, P.-Y., Hui, L.-X., Huang, S.-J., Ni, Z.-X., Yu, F.-X., & Xu, L.-A. (2021). Study on the Genetic Structure Based on Geographic Populations of the Endangered Tree Species: Liriodendron chinense. Forests, 12(7), 917. https://doi.org/10.3390/f12070917