
Complete Chloroplast Genomes of Three Salix Species: Genome Structures and Phylogenetic Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction and Genome Sequencing
2.3. Chloroplast Genome Assembly and Annotation
2.4. Repeat Sequence and Codon Usage Analysis
2.5. Genome Comparison and Sequence Divergence
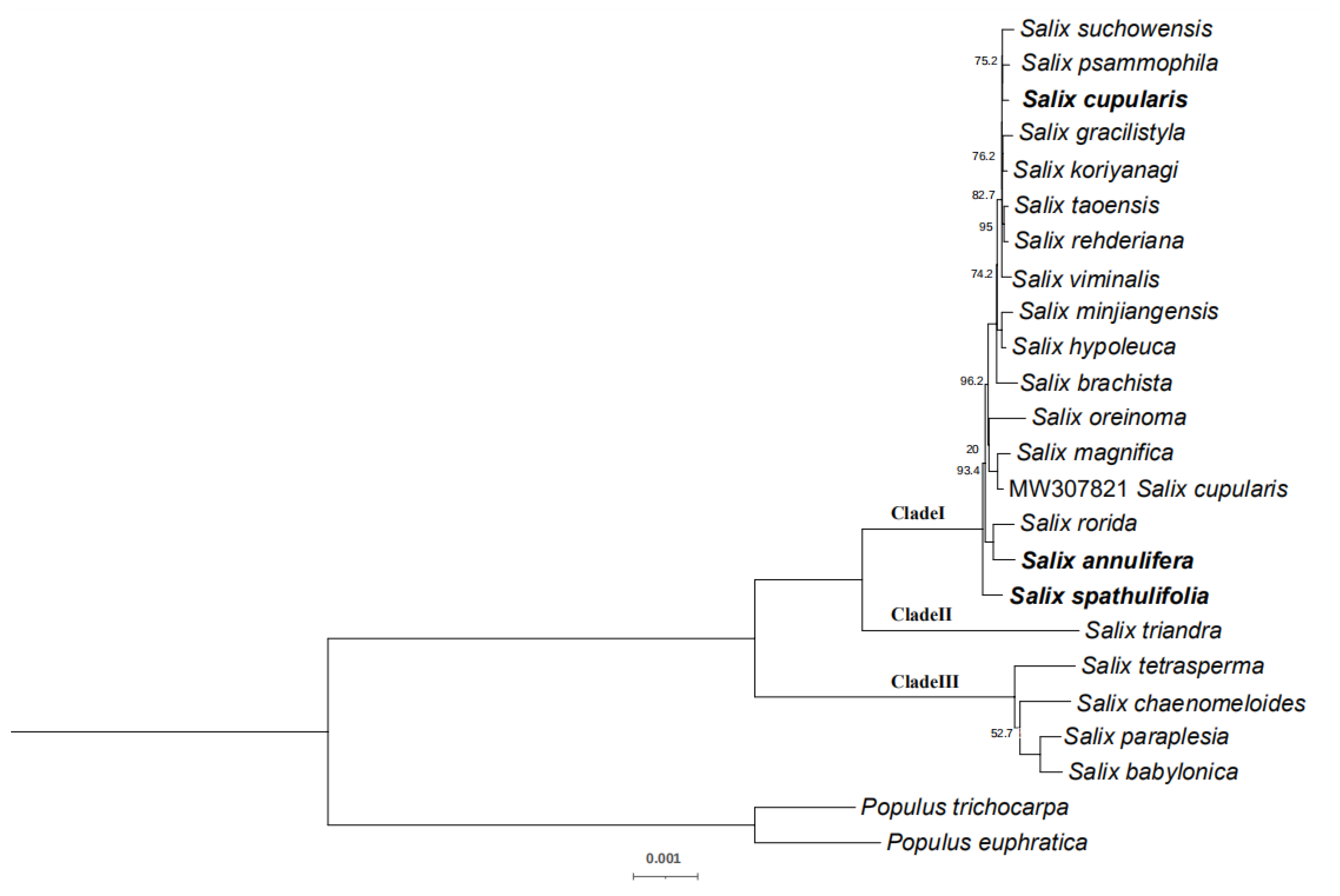
2.6. Phylogenetic Research
3. Results and Discussion
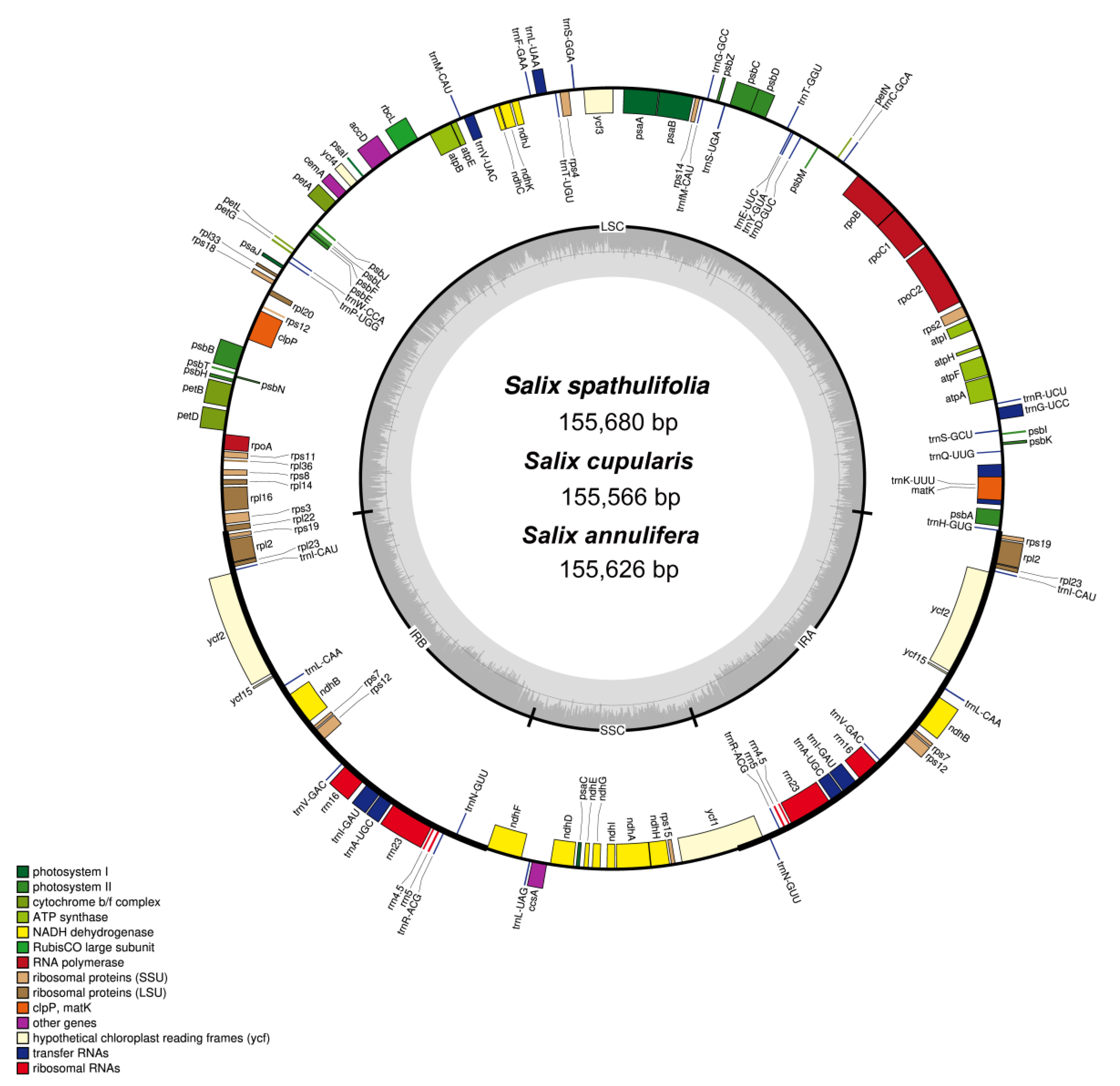
3.1. Genome Features
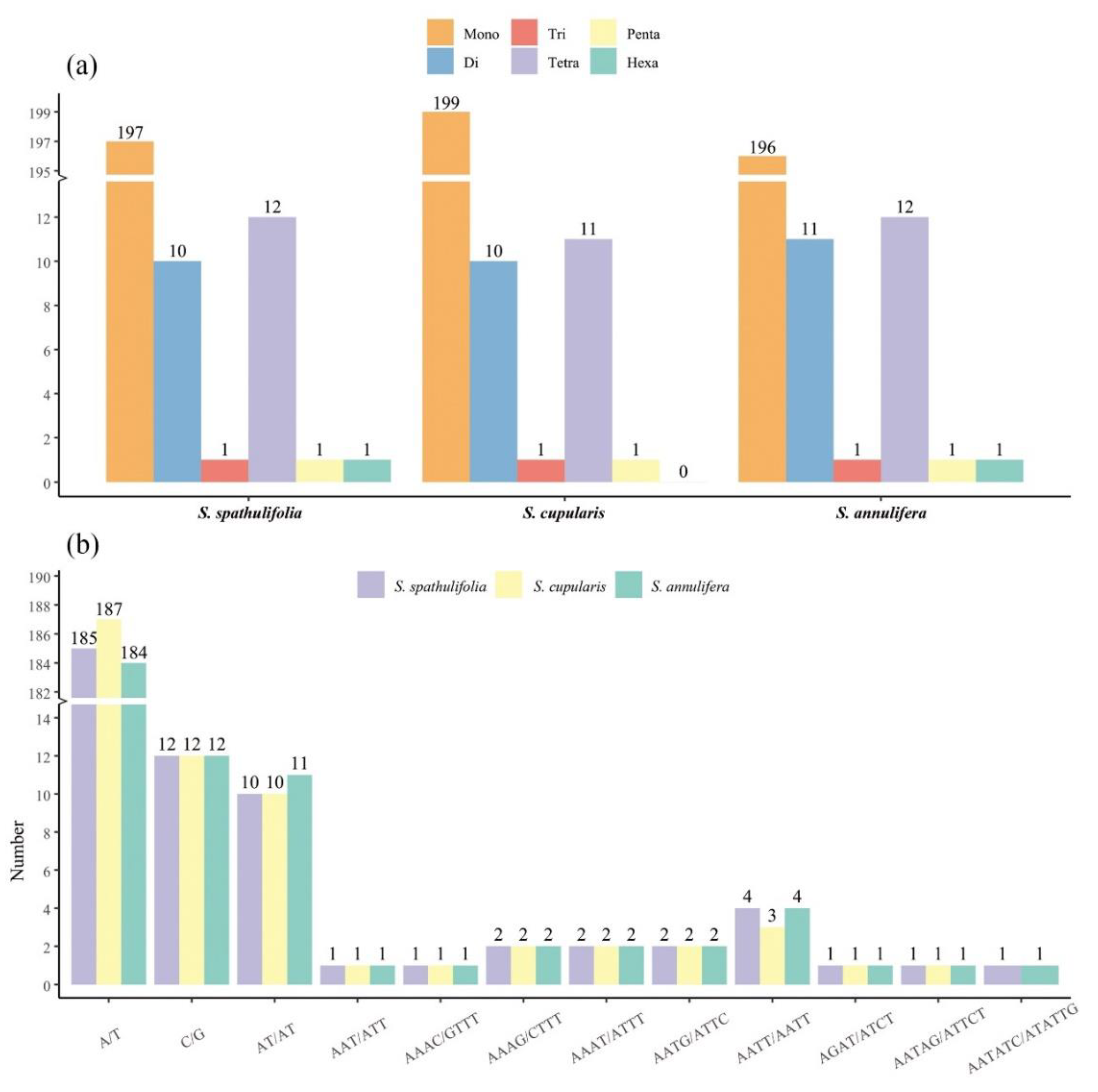
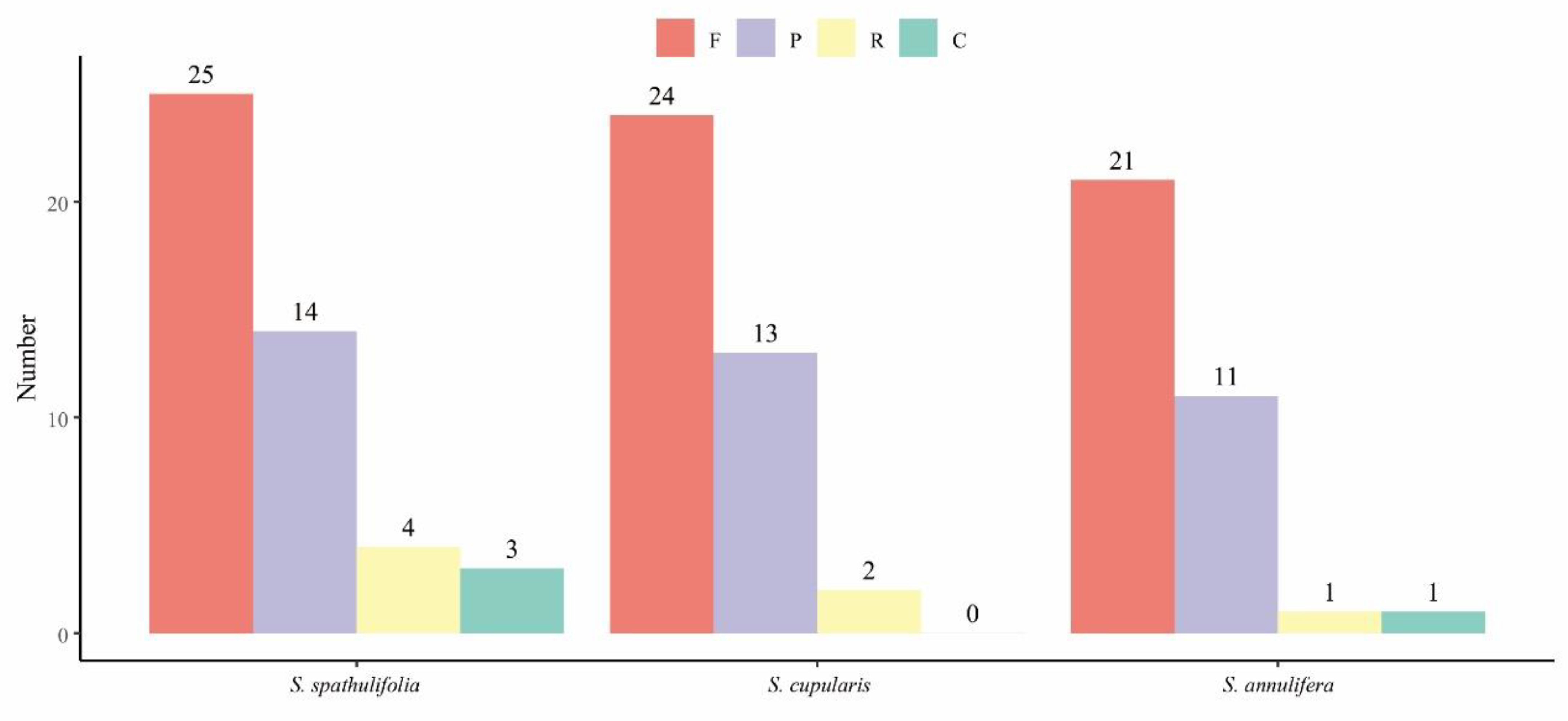
3.2. Codon Usage and Repeat Sequence Analysis
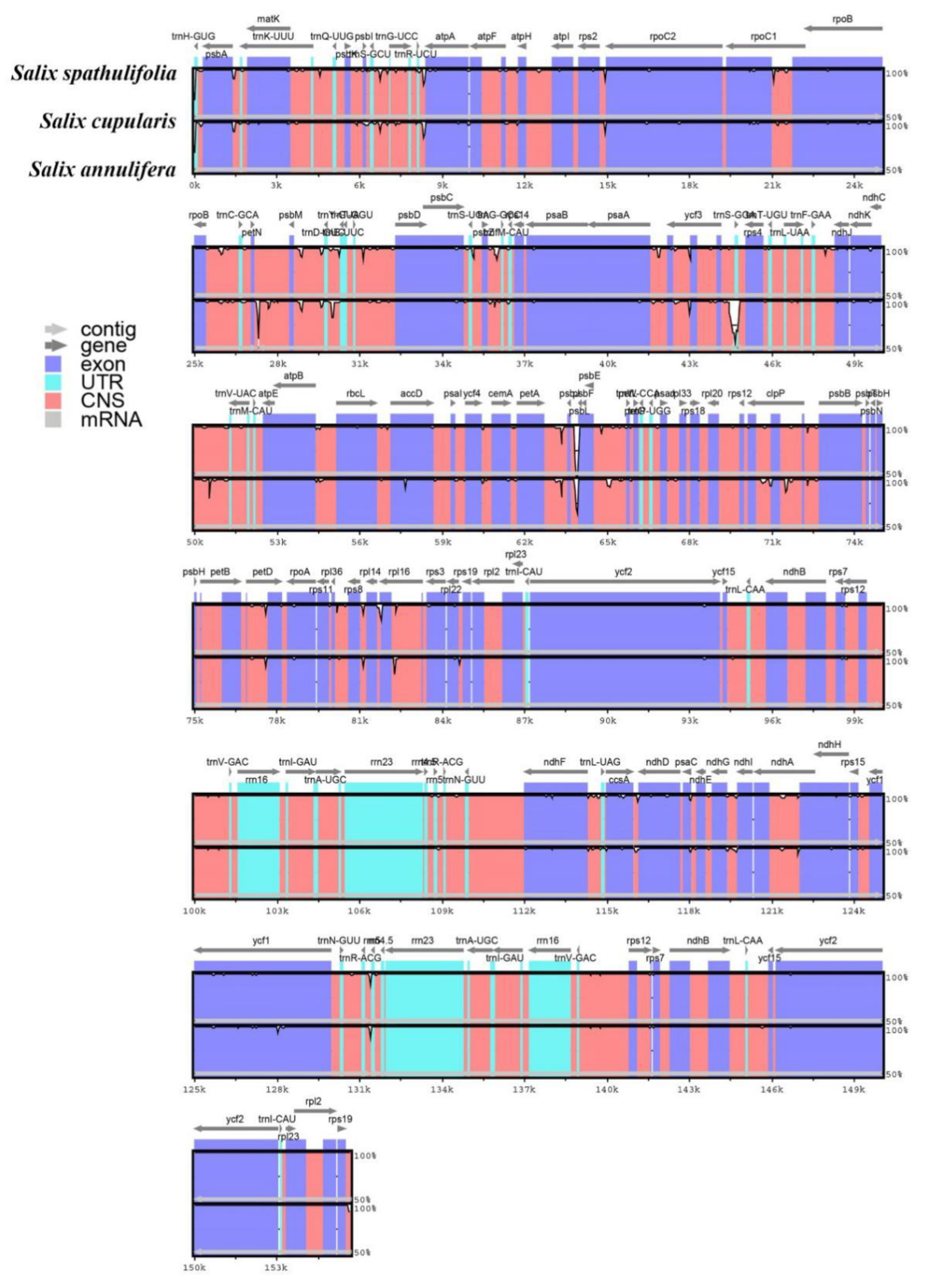
3.3. Genome Comparison
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Francisco-Ortega, J.; Wang, F.G.; Wang, Z.S.; Xing, F.W.; Liu, H.; Xu, H.; Xu, W.X.; Luo, Y.B.; Song, X.Q.; Gale, S.; et al. Endemic seed plant species from Hainan Island: A checklist. Bot. Rev. 2010, 76, 295–345. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef]
- Fan, W.B.; Wu, Y.; Yang, J.; Shahzad, K.; Li, Z.H. Comparative chloroplast genomics of dipsacales species: Insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 2018, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; de Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenetics Evol. 2014, 72, 82–89. [Google Scholar] [CrossRef]
- Wang, Y.G. Natural hybridization and speciation. Biodivers. Sci. 2017, 25, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J. The Plastids: Their Chemistry, Structure, Growth and Inheritance, 2nd ed.; JTO Kirk and RAE Tilney-Bassett, Elsevier/North-Holland Biomedical Press: Amsterdam, The Netherlands; New York, NY, USA; Oxford, UK, 1978. [Google Scholar]
- Kuroiwa, T. The replication, differentiation, and inheritance of plastids with emphasis on the concept of organelle nuclei. Int. Rev. Cytol. 1991, 128, 1–62. [Google Scholar] [CrossRef]
- Flora of North America Editorial Committee. Flora of North America: Volume 7 Magnoliophyta: Salicaceae to Brassicaceae; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Wu, Z.Y.; Raven, P.H. Flora of China; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999; Volume 4. [Google Scholar]
- Skvortsov, A.K. Willows of Russia and Adjacent Countries: Taxonomical and Geographic Revision (Translated by n. Kadis, 1999); Joensuu University: Joensuu, Finland, 1999. [Google Scholar]
- Fang, Z.F. On the distribution and origin of Salix in the world. J. Syst. Evol. 1987, 25, 307–313. [Google Scholar]
- Myers-Smith, I.H.; Forbes, B.C.; Wilmking, M.; Hallinger, M.; Lantz, T.; Blok, D.; Tape, K.D.; Macias-Fauria, M.; Sass-Klaassen, U.; Lévesque, E.; et al. Shrub expansion in tundra ecosystems: Dynamics, impacts and research priorities. Environ. Res. Lett. 2011, 6, 045509. [Google Scholar] [CrossRef] [Green Version]
- Smart, L.B.; Volk, T.A.; Lin, J.; Kopp, R.F.; Phillips, I.S.; Cameron, K.D.; White, E.H.; Abrahamson, L.P. Genetic improvement of shrub willow (Salix spp.) crops for bioenergy and environmental applications in the United States. Unasylva 2005, 56, 51. [Google Scholar]
- Argus, G.W. The genus Salix (Salicaceae) in the southeastern United States. Syst. Bot. Monogr. 1986, 9, 1–170. [Google Scholar] [CrossRef]
- Zhao, S.D. Distribution of Willows (Salix) in China. J. Syst. Evol. 1987, 25, 114–124. [Google Scholar]
- Zhang, M.L. Studies on the distribution and differentiation of Willows (Salix) in Qinling Mountain. Bull. Bot. Res. 1993, 13, 136–145. [Google Scholar]
- Chen, J.H.; Sun, H.; Wen, J.; Yang, Y.P. Molecular phylogeny of Salix L. (Salicaceae) inferred from three chloroplast datasets and its systematic implications. Taxon 2010, 59, 29–37. [Google Scholar] [CrossRef]
- Lumbsch, H.T.; Lauron-Moreau, A.; Pitre, F.E.; Argus, G.W.; Labrecque, M.; Brouillet, L. Phylogenetic relationships of American Willows (Salix L., Salicaceae). PLoS ONE 2015, 10, e0121965. [Google Scholar] [CrossRef]
- Azuma, T.; Kajita, T.; Yokoyama, J.; Ohashi, H. Phylogenetic relationships of Salix (Salicaceae) based on rbcL sequence data. Am. J. Bot. 2000, 87, 67–75. [Google Scholar] [CrossRef]
- Barkalov, V.Y.; Kozyrenko, M.M. Phylogenetic relationships of Salix L. Subg. Salix species (Salicaceae) according to sequencing data of intergenic spacers of the chloroplast genome and its rDNA. Russ. J. Genet. 2014, 50, 828–837. [Google Scholar] [CrossRef]
- Li, J.; Zhuo, Z.; Xu, D.; Yang, H.; Zhu, T. The complete chloroplast genome of Salix cupularis rehder, a sand binder in alpine hillslope, China. Mitochondrial DNA Part B 2021, 6, 2519–2520. [Google Scholar] [CrossRef]
- Hu, Y.F.; Shu, X.Y.; He, J.; Zhang, Y.L.; Xiao, H.H.; Tang, X.Y.; Gu, Y.F.; Lan, T.; Xia, J.G.; Ling, J.; et al. Storage of C, N, and P affected by afforestation with Salix cupularis in an alpine semiarid desert ecosystem. Land Degrad. Dev. 2018, 29, 188–198. [Google Scholar] [CrossRef]
- Gulyaev, S.; Cai, X.-J.; Guo, F.-Y.; Kikuchi, S.; Wendy, L.; Zhang, Z.-X.; Hörandl, E.; He, L. The phylogeny of Salix revealed by whole genome re-sequencing suggests different sex-determination systems in major groups of the genus. Ann. Bot. 2021. submitted. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- He, J.; Yao, M.; Lyu, R.D.; Lin, L.L.; Liu, H.J.; Pei, L.Y.; Yan, S.X.; Xie, L.; Cheng, J. Structural variation of the complete chloroplast genome and plastid phylogenomics of the genus Asteropyrum (Ranunculaceae). Sci. Rep. 2019, 9, 15285. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wang, J.; Yang, Y.; Fan, C.; Chen, J. Phylogenomic analysis and dynamic evolution of chloroplast genomes in Salicaceae. Front. Plant Sci. 2017, 8, 1050. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.Q. The whole chloroplast genome of shrub Willows (Salix suchowensis). Mitochondrial DNA Part A 2016, 27, 2153–2154. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.I.; Cronk, Q.C. Plann: A command-line application for annotating plastome sequences. Appl. Plant Sci. 2015, 3, 1500026. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (ogdraw) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2017, 7, 1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.F.; Zhu, G.F.; Li, D.M.; Wang, X.J. Complete chloroplast genome sequence and phylogenetic analysis of Spathiphyllum ‘Parrish’. PLoS ONE 2019, 14, e0224038. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hu, N.; Wu, H. Analyzing and characterizing the chloroplast genome of Salix wilsonii. BioMed Res. Int. 2019, 2019, 5190425. [Google Scholar] [CrossRef] [Green Version]
- Lyons-Weiler, J.; Hoelzer, G.A.; Tausch, R.J. Optimal outgroup analysis. Biol. J. Linn. Soc. 2008, 64, 493–511. [Google Scholar] [CrossRef]
- Wheeler, W.C. Nucleic acid sequence phylogeny and random outgroups. Cladistics 1990, 6, 363–367. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Bollback, J.P.; Levine, A.M. Inferring the root of a phylogenetic tree. Syst. Biol 2002, 51, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef]
- Rechinger, K.H. Salix taxonomy in Europe—Problems, interpretations, observations. Proc. R. Soc. Edinb. 1992, 98, 1–12. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Collecting Number | Longitude | Latitude | Altitude | Province | Herbaria |
|---|---|---|---|---|---|---|
| Salix spathulifolia | HLCS19_15 | 108.80° E | 33.86° N | 2456 m | Shaanxi | BJFC |
| Salix cupularis | HLCS19_45 | 107.81° E | 34.00° N | 3377 m | Shaanxi | BJFC |
| Salix annulifera | ZZX2019091207 | 94.97° E | 29.47° N | 3559 m | Tibet | BJFC |
| S. spathulifolia | S. cupularis | S. annulifera | ||
|---|---|---|---|---|
| Accession Number | HLCS19_15 | HLCS19_45 | ZZX2019091207 | |
| Genome | Length (bp) | 155,680 | 155,566 | 155,626 |
| GC (%) | 36.7 | 36.7 | 36.7 | |
| LSC | Length (bp) | 84,552 | 84,431 | 84,499 |
| GC (%) | 34.4 | 34.4 | 34.4 | |
| Length (%) | 54.31 | 54.27 | 54.30 | |
| SSC | Length (bp) | 16,206 | 16,217 | 16,221 |
| GC (%) | 31.0 | 31.0 | 30.9 | |
| Length (%) | 10.41 | 10.42 | 10.42 | |
| IR | Length (bp) | 27,461 | 27,459 | 27,453 |
| GC (%) | 41.9 | 41.9 | 41.9 | |
| Length (%) | 17.64 | 17.65 | 17.64 | |
| No. of genes (duplicated in IR) | Genes | 130 (19) | 130 (20) | 130 (21) |
| PCGs | 85 (8) | 85 (9) | 85 (10) | |
| tRNA | 37 (7) | 37 (8) | 37 (9) | |
| rRNA | 8 (4) | 8 (5) | 8 (6) | |
| With introns | 17 (5) | 17 (6) | 17 (7) | |
| Groups of Genes | Name of Genes |
|---|---|
| Transfer RNAs | trnA-UGC *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC, trnH-GUG, trnI-CAU *, trnI-GAU *, trnK-UUU, trnL-CAA *, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU *, trnP-UGG, trnQ-UUG, trnR-ACG *, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC *, trnV-UAC, trnW-CCA, trnY-GUA |
| Ribosomal RNAs | rrn4.5 *, rrn5 *, rrn16 *, rrn23 * |
| Ribosomal protein small subunit | rps2, rps3, rps4, rps7 *, rps8, rps11, rps12 *, rps14, rps15, rps18, rps19 * |
| Ribosomal protein large subunit | rpl2 *, rpl14, rpl16, rpl20, rpl22, rpl23 *, rpl33, rpl36 |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ, ycf3, ycf4 |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Cytochrome b/f complex | petA, petB, petD, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI |
| NDH complex | ndhA, ndhB *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Large subunit Rubisco | rbcL |
| Acetyl-CoA carboxylase | accD |
| Maturase | matK |
| Inner membrane protein | cemA |
| ATP-dependent protease | clpP |
| Cytochrome c biogenesis | ccsA |
| Conserved open reading frames | ycf1, ycf2 *, ycf15 * |
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Extron III (bp) |
|---|---|---|---|---|---|---|
| trnK-UUU | LSC | 37 | 2547 | 35 | ||
| rpl16 | LSC | 9 | 1120 | 399 | ||
| trnG-UCC | LSC | 23 | 693 | 48 | ||
| atpF | LSC | 145 | 741 | 398 | ||
| rpoC1 | LSC | 453 | 777 | 1617 | ||
| ycf3 | LSC | 126 | 723 | 228 | 667 | 153 |
| trnL-UAA | LSC | 35 | 586 | 50 | ||
| trnV-UAC | LSC | 39 | 609 | 35 | ||
| clpP | LSC | 71 | 837 | 292 | 585 | 228 |
| petB | LSC | 6 | 811 | 642 | ||
| petD | LSC | 8 | 788 | 490 | ||
| ndhA | SSC | 552 | 1107 | 546 | ||
| rpl2 | IR | 397 | 668 | 434 | ||
| rps12 | LSC&IR | 114 | ~ | 232 | 536 | 26 |
| ndhB | IR | 723 | 682 | 756 | ||
| trnI-GAU | IR | 37 | 949 | 35 | ||
| trnA-UGC | IR | 38 | 802 | 35 |
| Codon | Amino Acid | S. spathulifolia | S. cupularis | S. annulifera | |||
|---|---|---|---|---|---|---|---|
| Count | RSCU | Count | RSCU | Count | RSCU | ||
| UUU | Phe | 987 | 1.31 | 988 | 1.31 | 992 | 1.31 |
| UUC | Phe | 516 | 0.69 | 517 | 0.69 | 517 | 0.69 |
| UUA | Leu | 892 | 1.91 | 894 | 1.91 | 894 | 1.91 |
| UUG | Leu | 569 | 1.22 | 570 | 1.22 | 570 | 1.22 |
| CUU | Leu | 586 | 1.25 | 586 | 1.25 | 586 | 1.25 |
| CUC | Leu | 178 | 0.38 | 180 | 0.38 | 179 | 0.38 |
| CUA | Leu | 398 | 0.85 | 397 | 0.85 | 397 | 0.85 |
| CUG | Leu | 179 | 0.38 | 179 | 0.38 | 180 | 0.38 |
| AUU | Ile | 1127 | 1.49 | 1129 | 1.49 | 1130 | 1.49 |
| AUC | Ile | 432 | 0.57 | 432 | 0.57 | 430 | 0.57 |
| AUA | Ile | 708 | 0.94 | 708 | 0.94 | 711 | 0.94 |
| AUG | Met | 620 | 1 | 621 | 1 | 621 | 1 |
| GUU | Val | 493 | 1.42 | 492 | 1.42 | 491 | 1.41 |
| GUC | Val | 167 | 0.48 | 167 | 0.48 | 167 | 0.48 |
| GUA | Val | 532 | 1.53 | 532 | 1.53 | 533 | 1.53 |
| GUG | Val | 199 | 0.57 | 199 | 0.57 | 199 | 0.57 |
| UCU | Ser | 572 | 1.69 | 573 | 1.69 | 573 | 1.69 |
| UCC | Ser | 332 | 0.98 | 329 | 0.97 | 329 | 0.97 |
| UCA | Ser | 409 | 1.21 | 408 | 1.2 | 409 | 1.21 |
| UCG | Ser | 184 | 0.54 | 186 | 0.55 | 185 | 0.55 |
| CCU | Pro | 418 | 1.56 | 418 | 1.57 | 420 | 1.57 |
| CCC | Pro | 198 | 0.74 | 197 | 0.74 | 199 | 0.74 |
| CCA | Pro | 308 | 1.15 | 306 | 1.15 | 308 | 1.15 |
| CCG | Pro | 145 | 0.54 | 145 | 0.54 | 143 | 0.53 |
| ACU | Thr | 522 | 1.6 | 524 | 1.6 | 524 | 1.6 |
| ACC | Thr | 238 | 0.73 | 236 | 0.72 | 238 | 0.73 |
| ACA | Thr | 423 | 1.29 | 424 | 1.29 | 423 | 1.29 |
| ACG | Thr | 126 | 0.39 | 126 | 0.38 | 124 | 0.38 |
| GCU | Ala | 620 | 1.83 | 620 | 1.83 | 620 | 1.83 |
| GCC | Ala | 202 | 0.6 | 205 | 0.61 | 204 | 0.6 |
| GCA | Ala | 385 | 1.14 | 385 | 1.14 | 385 | 1.14 |
| GCG | Ala | 145 | 0.43 | 143 | 0.42 | 145 | 0.43 |
| UAU | Tyr | 777 | 1.64 | 780 | 1.65 | 780 | 1.64 |
| UAC | Tyr | 171 | 0.36 | 168 | 0.35 | 170 | 0.36 |
| UAA | * | 45 | 1.59 | 45 | 1.59 | 45 | 1.59 |
| UAG | * | 22 | 0.78 | 22 | 0.78 | 22 | 0.78 |
| CAU | His | 477 | 1.52 | 478 | 1.52 | 477 | 1.52 |
| CAC | His | 150 | 0.48 | 150 | 0.48 | 149 | 0.48 |
| CAA | Gln | 713 | 1.55 | 715 | 1.55 | 716 | 1.55 |
| CAG | Gln | 209 | 0.45 | 209 | 0.45 | 209 | 0.45 |
| AAU | Asn | 978 | 1.53 | 981 | 1.53 | 981 | 1.53 |
| AAC | Asn | 300 | 0.47 | 300 | 0.47 | 299 | 0.47 |
| AAA | Lys | 1051 | 1.47 | 1052 | 1.47 | 1050 | 1.47 |
| AAG | Lys | 376 | 0.53 | 376 | 0.53 | 375 | 0.53 |
| GAU | Asp | 833 | 1.58 | 832 | 1.58 | 834 | 1.58 |
| GAC | Asp | 223 | 0.42 | 223 | 0.42 | 222 | 0.42 |
| GAA | Glu | 1023 | 1.49 | 1022 | 1.49 | 1023 | 1.49 |
| GAG | Glu | 347 | 0.51 | 347 | 0.51 | 346 | 0.51 |
| UGU | Cys | 213 | 1.42 | 212 | 1.42 | 213 | 1.42 |
| UGC | Cys | 86 | 0.58 | 87 | 0.58 | 86 | 0.58 |
| UGA | * | 18 | 0.64 | 18 | 0.64 | 18 | 0.64 |
| UGG | Trp | 452 | 1 | 452 | 1 | 453 | 1 |
| CGU | Arg | 328 | 1.28 | 330 | 1.29 | 330 | 1.29 |
| CGC | Arg | 109 | 0.43 | 108 | 0.42 | 109 | 0.43 |
| CGA | Arg | 357 | 1.4 | 361 | 1.41 | 358 | 1.4 |
| CGG | Arg | 106 | 0.42 | 104 | 0.41 | 105 | 0.41 |
| AGU | Ser | 408 | 1.2 | 408 | 1.2 | 407 | 1.2 |
| AGC | Ser | 128 | 0.38 | 128 | 0.38 | 128 | 0.38 |
| AGA | Arg | 471 | 1.84 | 471 | 1.84 | 473 | 1.85 |
| AGG | Arg | 161 | 0.63 | 161 | 0.63 | 161 | 0.63 |
| GGU | Gly | 556 | 1.25 | 556 | 1.25 | 557 | 1.26 |
| GGC | Gly | 192 | 0.43 | 192 | 0.43 | 192 | 0.43 |
| GGA | Gly | 711 | 1.6 | 711 | 1.6 | 709 | 1.6 |
| GGG | Gly | 314 | 0.71 | 316 | 0.71 | 315 | 0.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-J.; Liu, K.-J.; Wang, Y.-C.; He, J.; Wu, Y.-M.; Zhang, Z.-X. Complete Chloroplast Genomes of Three Salix Species: Genome Structures and Phylogenetic Analysis. Forests 2021, 12, 1681. https://doi.org/10.3390/f12121681
Zhang X-J, Liu K-J, Wang Y-C, He J, Wu Y-M, Zhang Z-X. Complete Chloroplast Genomes of Three Salix Species: Genome Structures and Phylogenetic Analysis. Forests. 2021; 12(12):1681. https://doi.org/10.3390/f12121681
Chicago/Turabian StyleZhang, Xue-Jiao, Kang-Jia Liu, Ya-Chao Wang, Jian He, Yuan-Mi Wu, and Zhi-Xiang Zhang. 2021. "Complete Chloroplast Genomes of Three Salix Species: Genome Structures and Phylogenetic Analysis" Forests 12, no. 12: 1681. https://doi.org/10.3390/f12121681
APA StyleZhang, X.-J., Liu, K.-J., Wang, Y.-C., He, J., Wu, Y.-M., & Zhang, Z.-X. (2021). Complete Chloroplast Genomes of Three Salix Species: Genome Structures and Phylogenetic Analysis. Forests, 12(12), 1681. https://doi.org/10.3390/f12121681