
Impact of Gene Flow and Introgression on the Range Wide Genetic Structure of Quercus robur (L.) in Europe

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
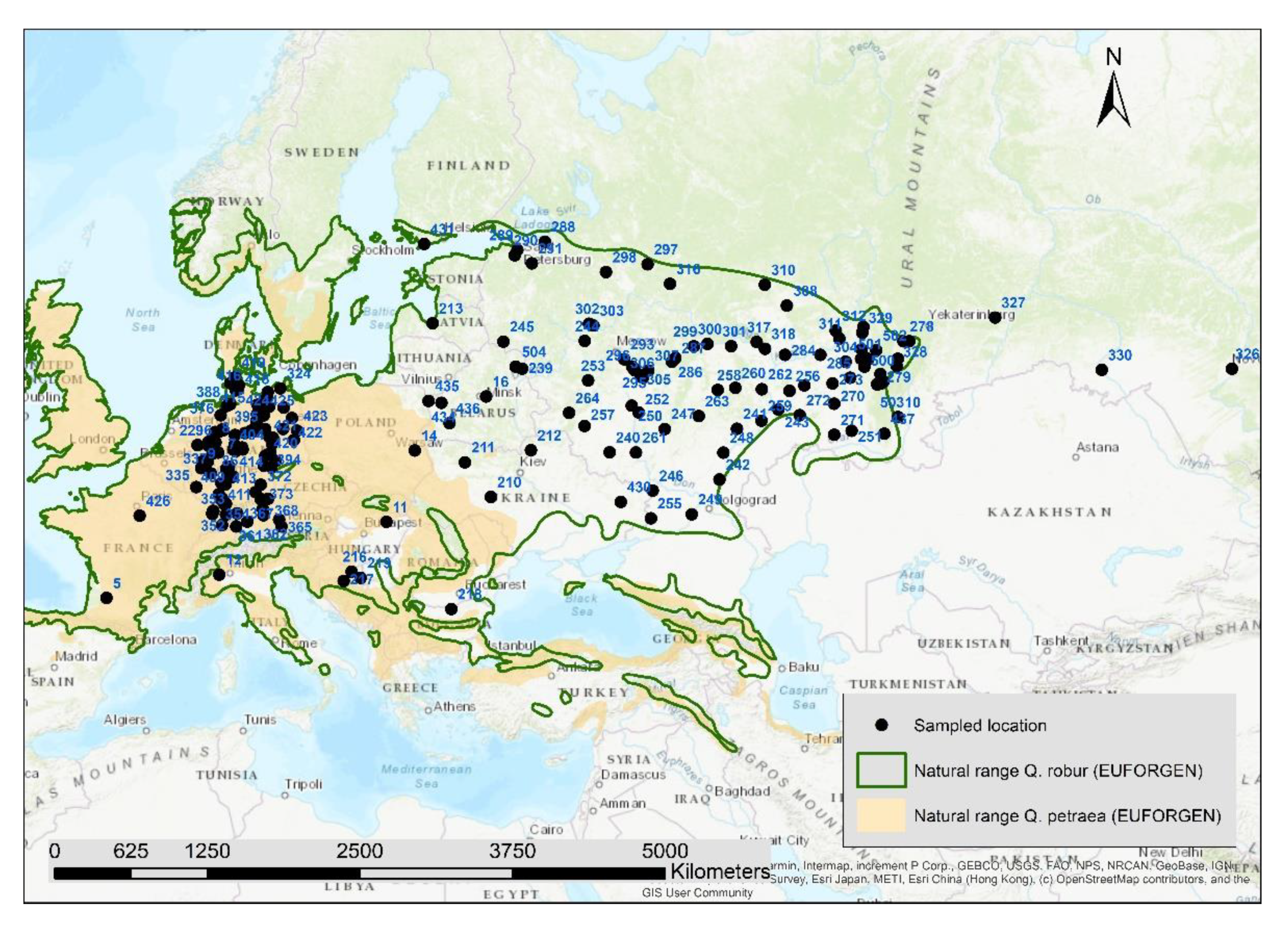
2.1. Sampling
2.2. DNA Extraction and Genotyping
2.3. Statistical Analysis
2.3.1. Haplotypes
2.3.2. Genetic Differentiation
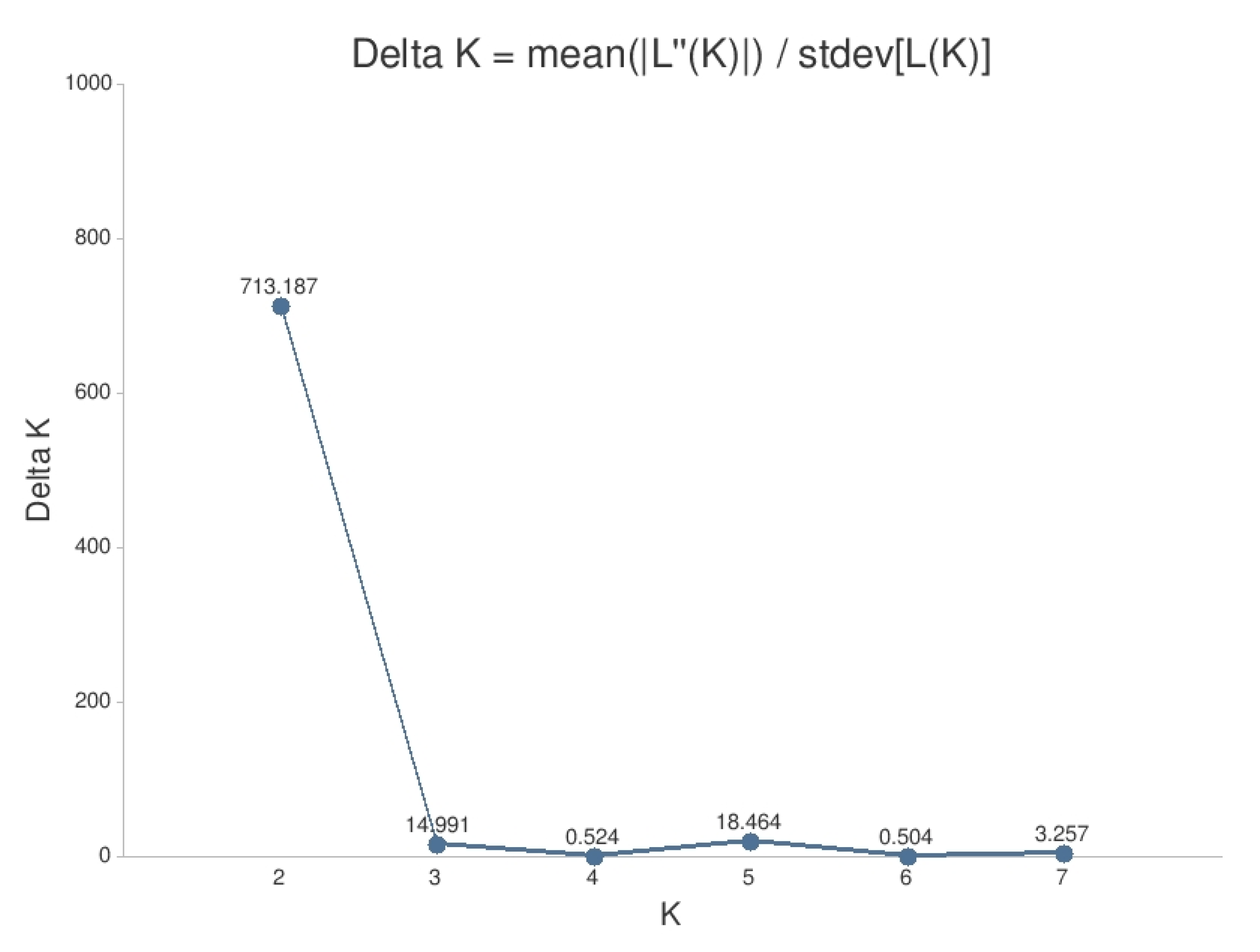
2.3.3. Bayesian Clustering Analysis
2.3.4. Spatial Structure and Maps
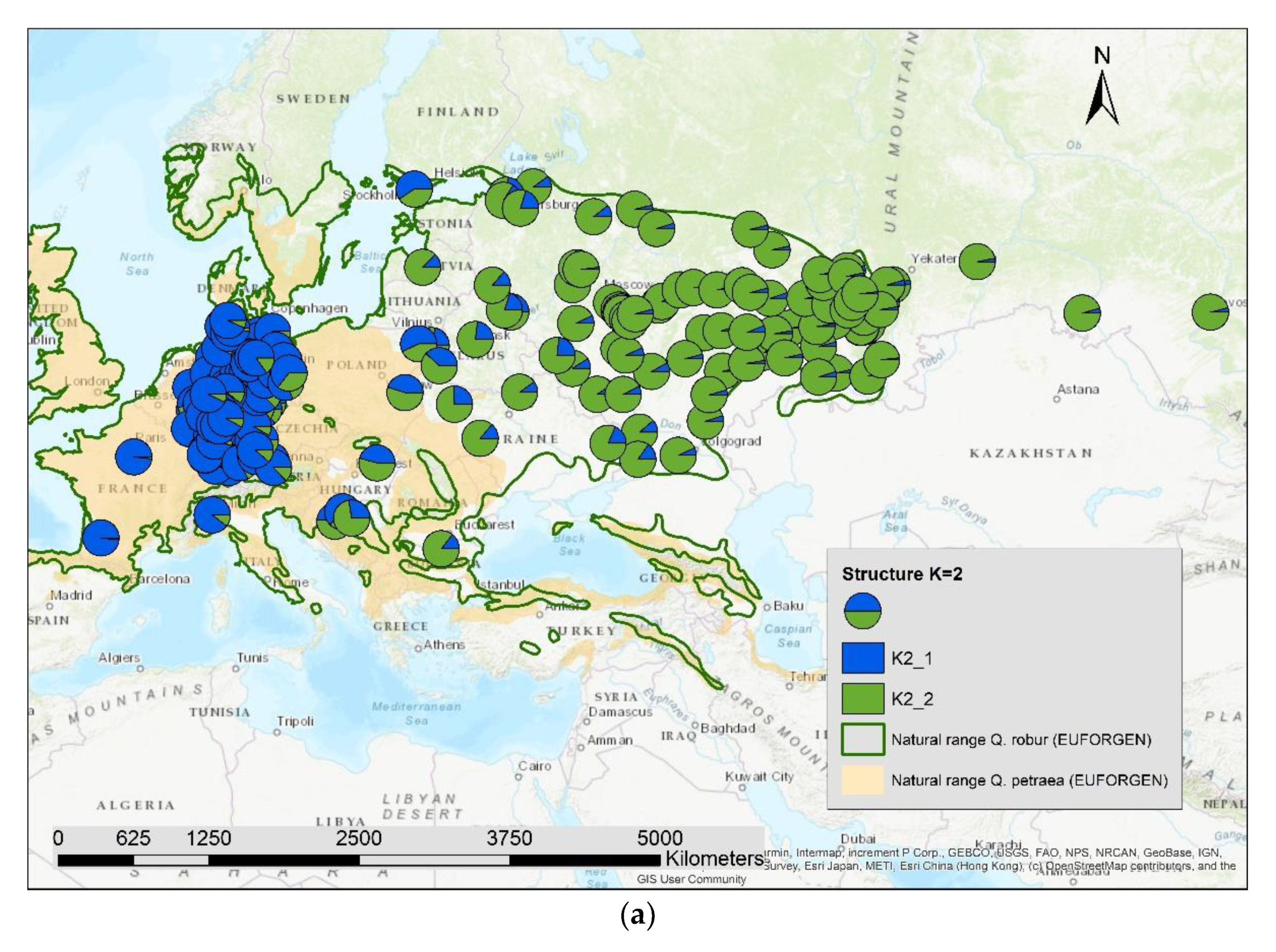
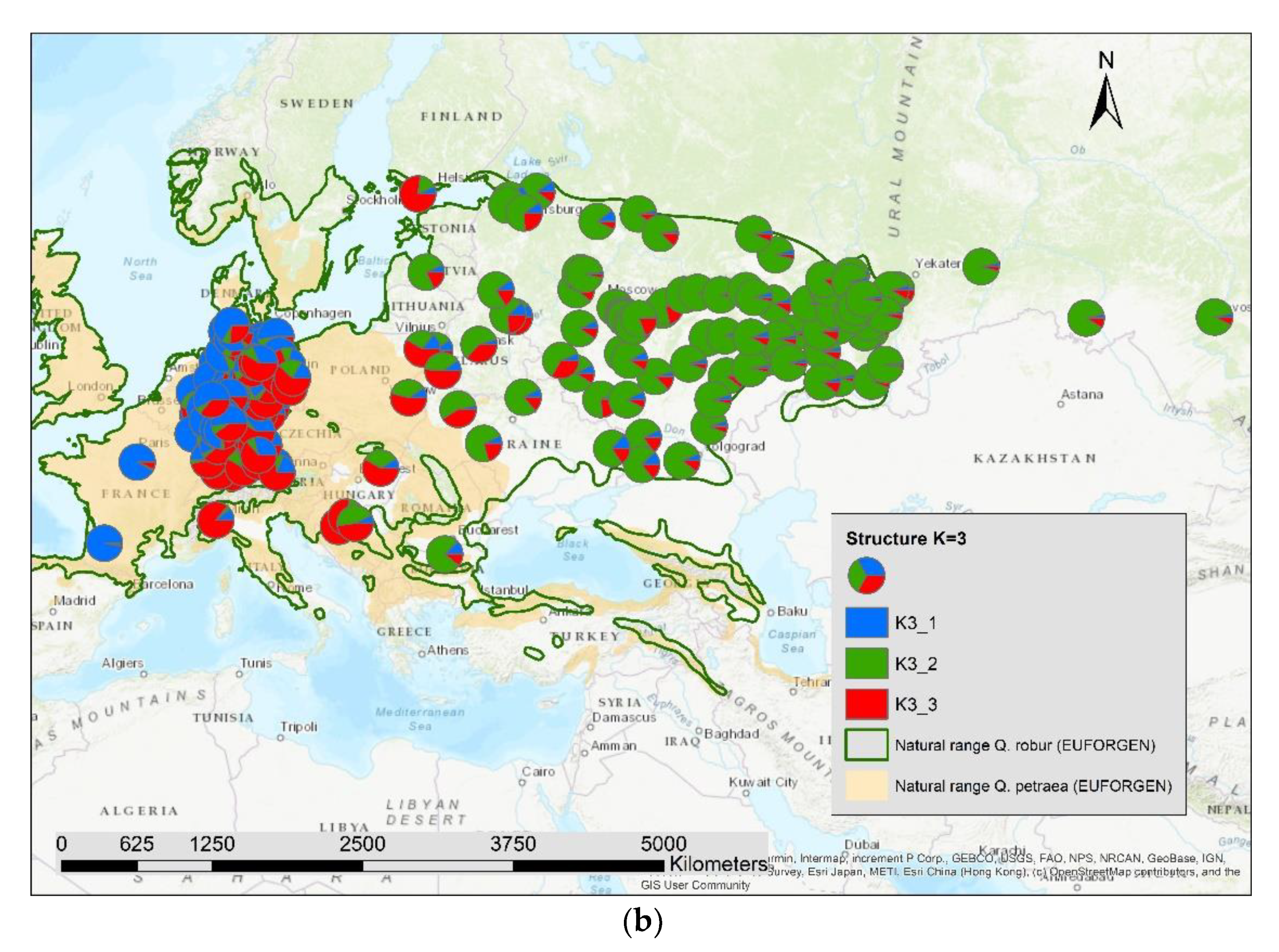
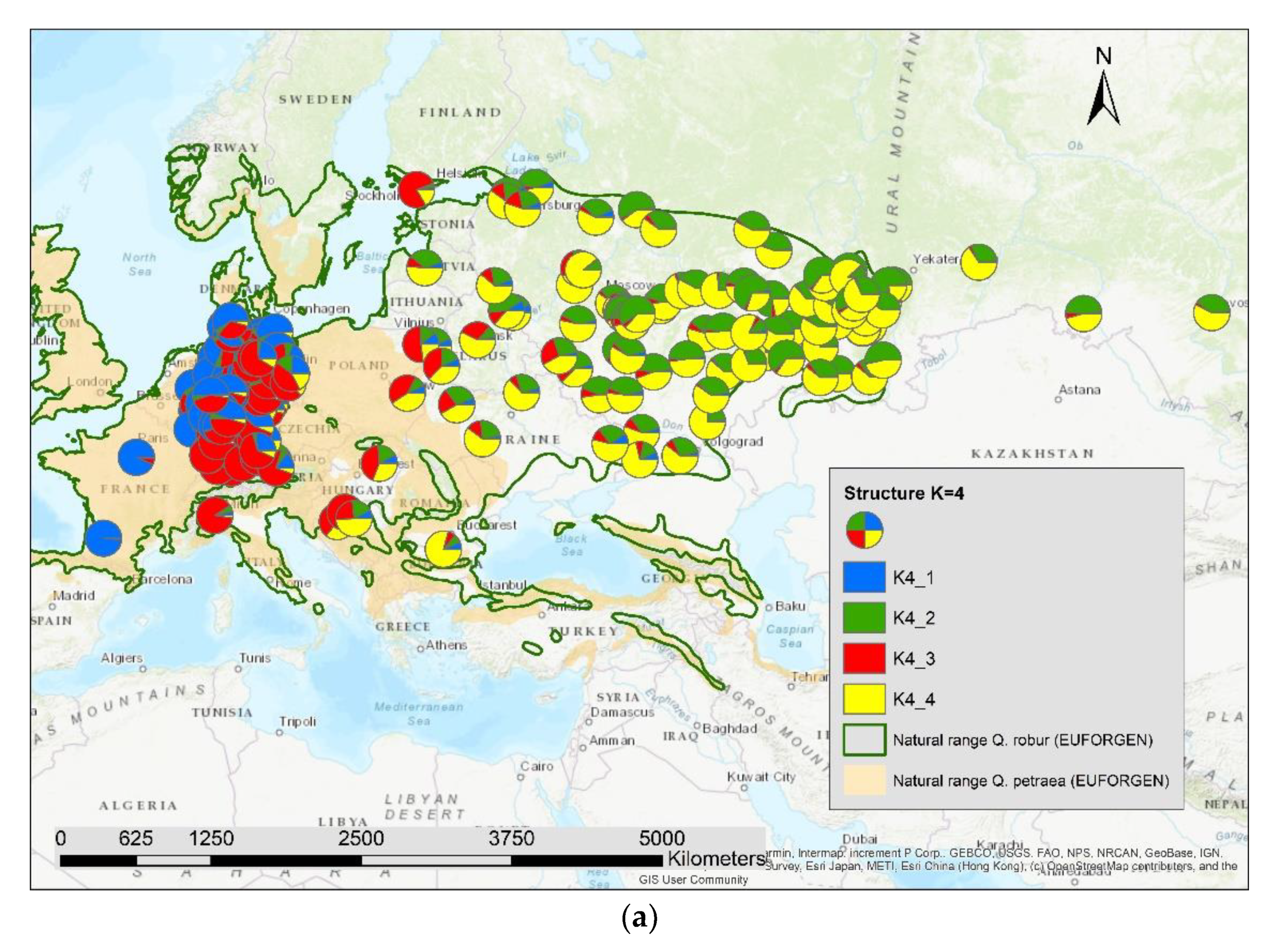
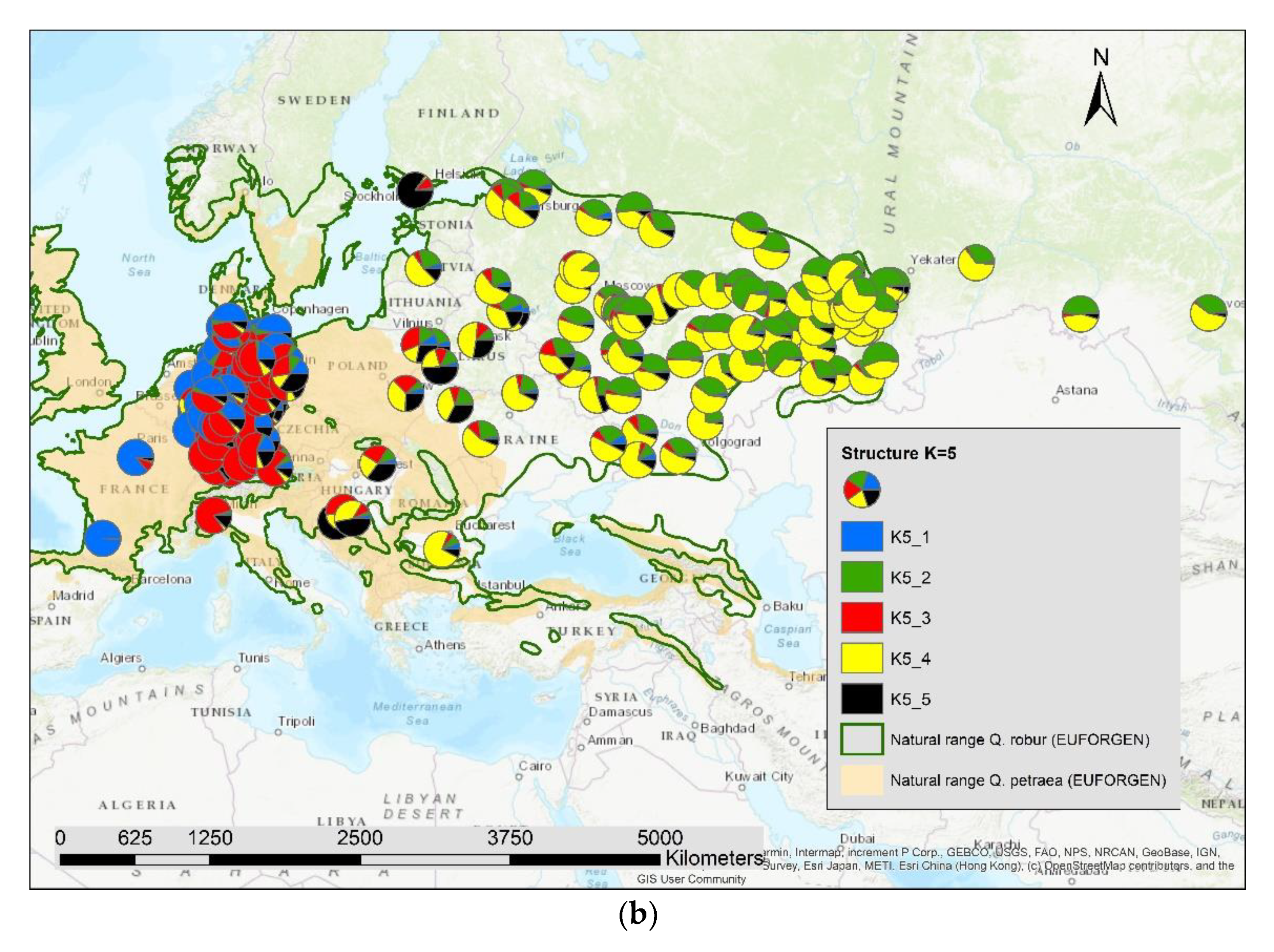
3. Results
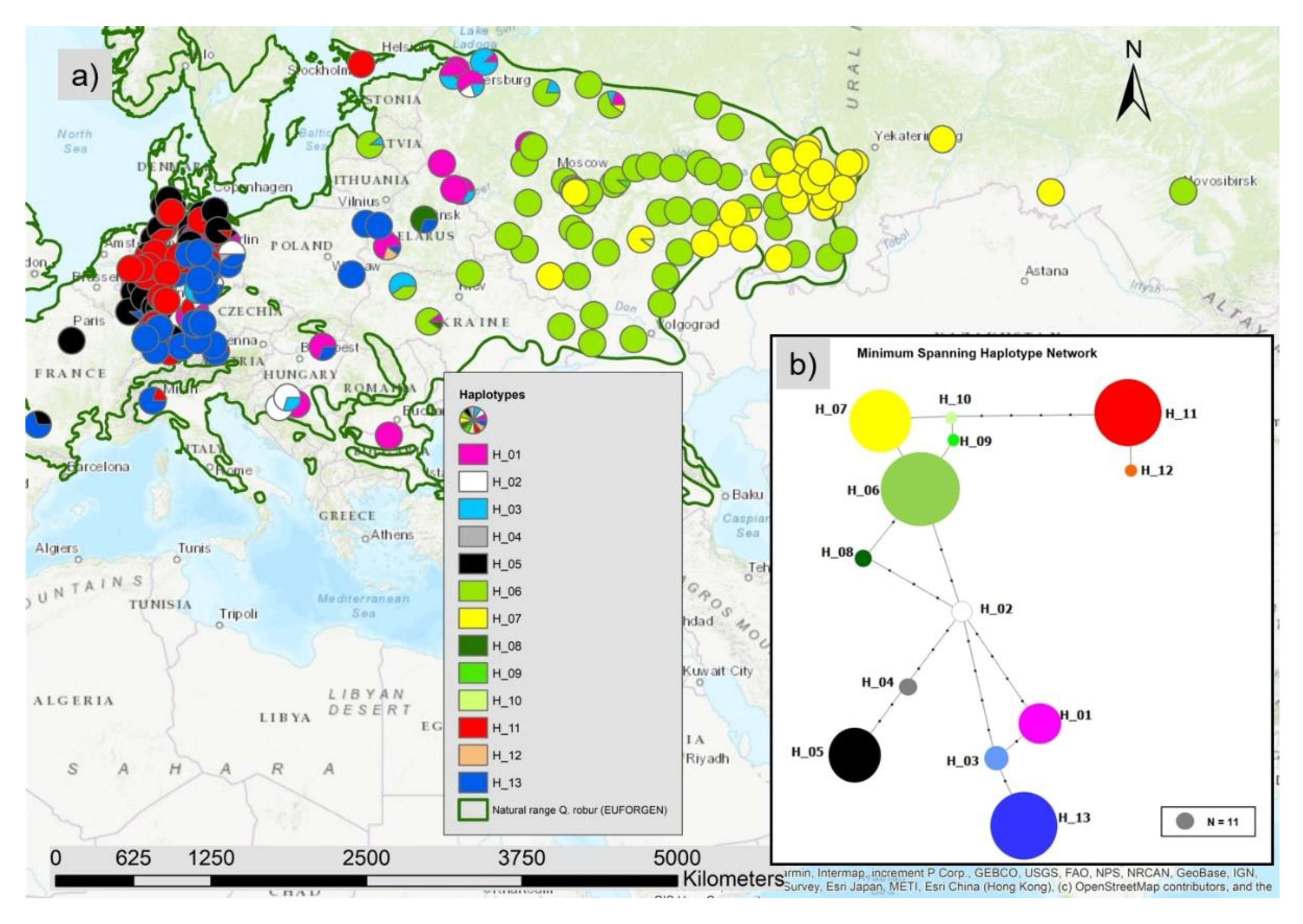
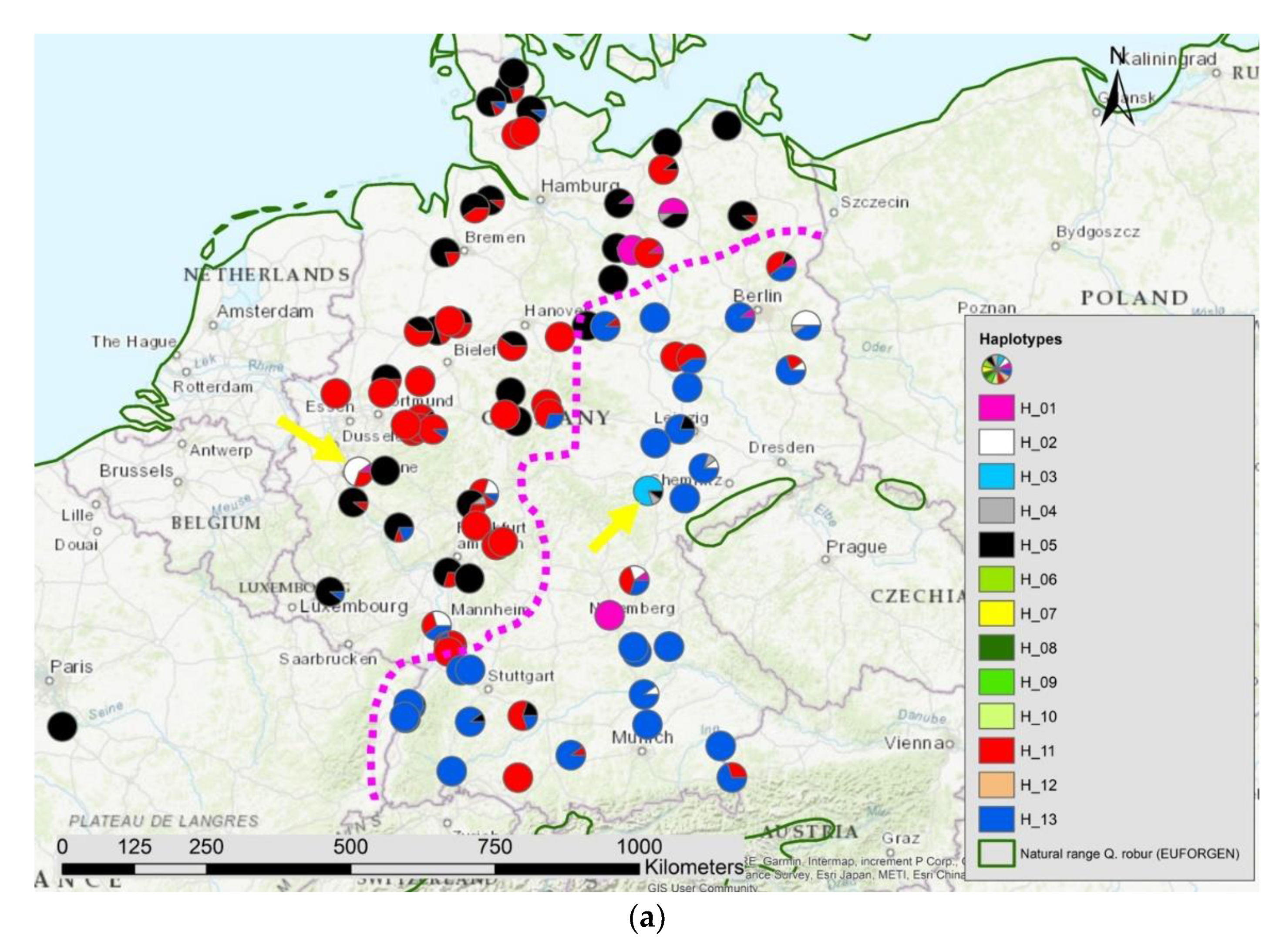
3.1. Haplotypes
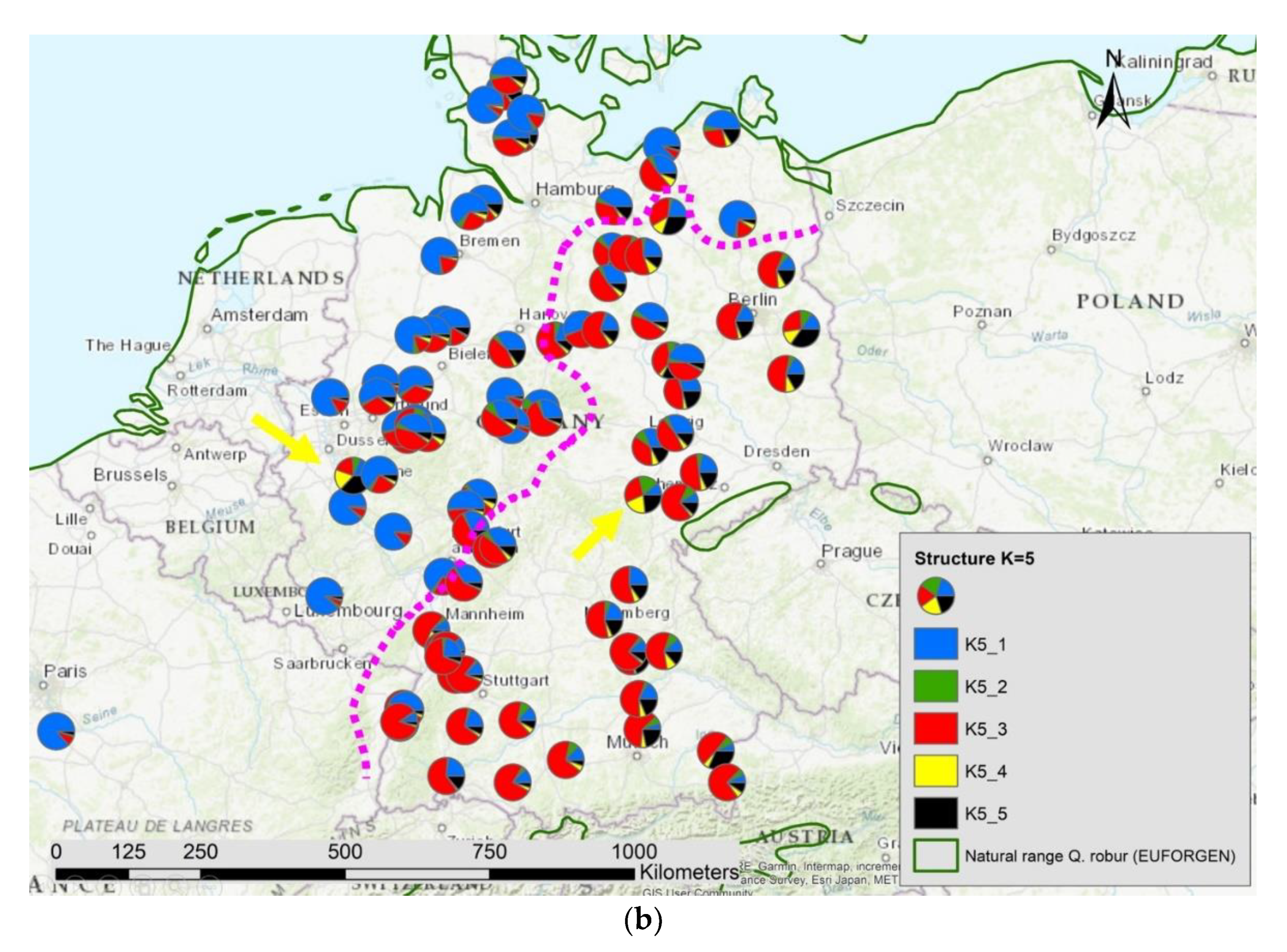
3.2. Nuclear Markers
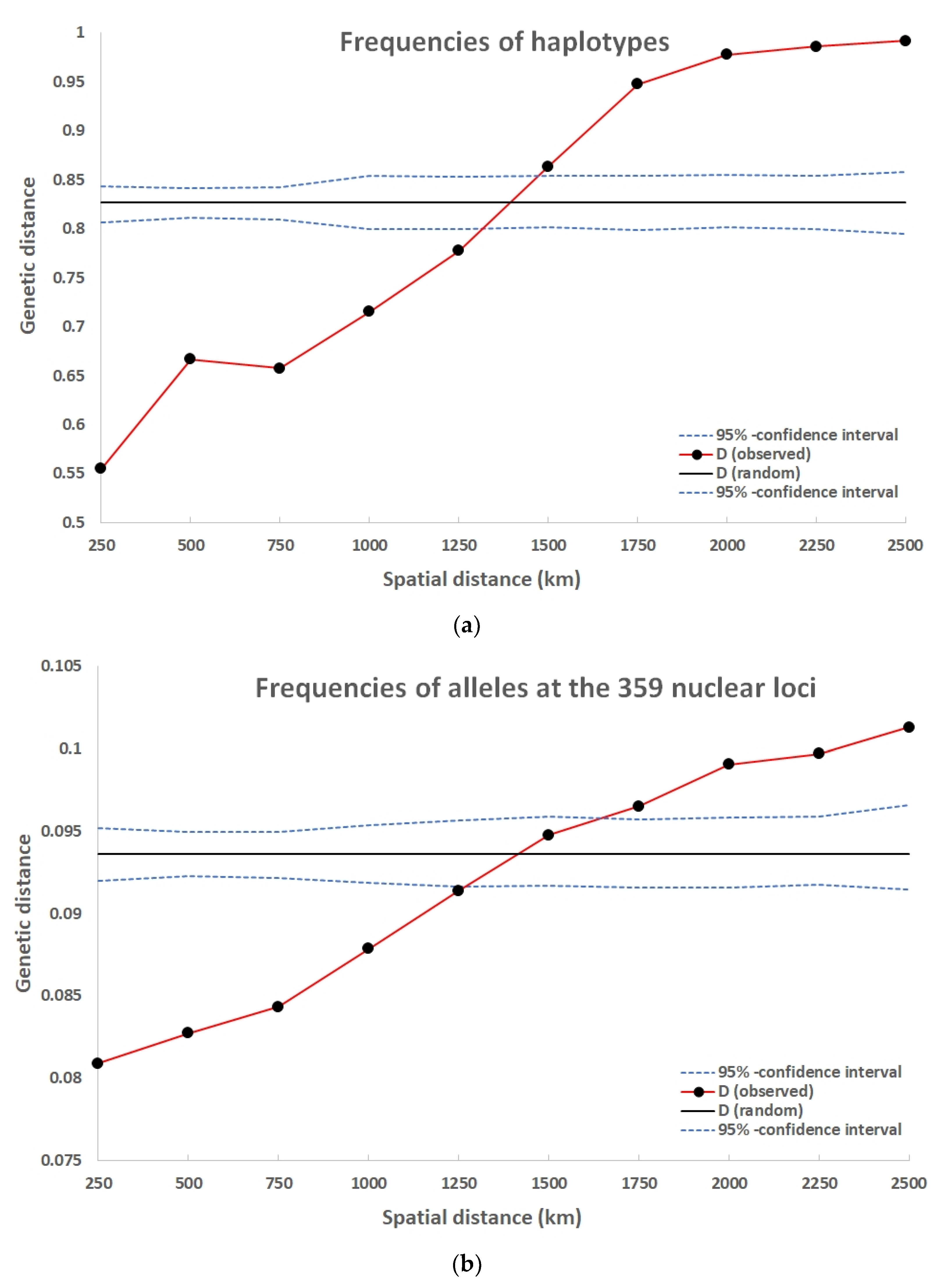
3.3. Genetic Correlation among Nuclear and Organelle Genome
4. Discussion
4.1. Impact of Seed and Pollen Mediated Gene Flow
4.2. Hybridization and Introgression
4.3. Genetic Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petit, R.J.; Csaikl, U.M.; Bordács, S.; Burg, K.; Coart, E.; Cottrell, J.; van Dam, B.; Deans, J.D.; Lapegue, S.; Fineschi, S.; et al. Chloroplast DNA variation in European white oaks-Phylogeography and patterns of diversity based on data from over 2600 populations. For. Ecol. Manag. 2002, 156, 5–26. [Google Scholar] [CrossRef]
- Semerikova, S.A.; Isakov, I.Y.; Semerikov, V.L. Chloroplast DNA variation and phylogeography of pedunculate oak Quercus robur L. in the eastern part of the range. Russ. J. Genet. 2021, 57, 47–60. [Google Scholar] [CrossRef]
- Deguilloux, M.-F.; Pemonge, M.-H.; Petit, R.J. Use of chloroplast microsatellites to differentiate oak populations. Ann. For. Sci. 2004, 61, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Petit, R.J.; Brewer, S.; Sándorbordácsde, S.; Burge, K.; Cheddadibc, R.; Coartf, E.; Cottrellg, J.; Csaikl, U.M.; van Dam, B.; Deans, J.D.; et al. Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For. Ecol. Manag. 2002, 156, 49–74. [Google Scholar] [CrossRef]
- Degen, B.; Thuenen-Institute of Forest Genetics, Grosshansdorf, Germany; Yanbaev, Y.; Bashkir State Agrarian University, Ufa, Russia; Ianbaev, R.; Bashkir State Agrarian University, Ufa, Russia; Blanc-Jolivet, C.; Thuenen-Institute of Forest Genetics, Grosshansdorf, Germany; Mader, M.; Thuenen-Institute of Forest Genetics, Grosshansdorf, Germany; Bakhtina, S.; Bashkir State Agrarian University, Ufa, Russia. Targeted sequencing reveals large scale genetic structure of Quercus robur in Russia and neighbouring countries. Personal communication, 2021. [Google Scholar]
- Gomory, D.; Zhelev, P.; Brus, R. The Balkans: A genetic hotspot but not a universal colonization source for trees. Plant. Syst. Evol. 2020, 306, 9. [Google Scholar] [CrossRef]
- Beatty, G.E.; Montgomery, W.I.; Spaans, F.; Tosh, D.G.; Provan, J. Pure species in a continuum of genetic and morphological variation: Sympatric oaks at the edge of their range. Ann. Bot. 2016, 117, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Konig, A.O.; Ziegenhagen, B.; van Dam, B.C.; Csaikl, U.M.; Coart, E.; Degen, B.; Burg, K.; de Vries, S.M.G.; Petit, R.J. Chloroplast DNA variation of oaks in western Central Europe and genetic consequences of human influences. For. Ecol. Manag. 2002, 156, 147–166. [Google Scholar] [CrossRef]
- Neophytou, C.; Aravanopoulos, F.A.; Fink, S.; Dounavi, A. Detecting interspecific and geographic differentiation patterns in two interfertile oak species (Quercus petraea (Matt.) Liebl. and Q. robur L.) using small sets of microsatellite markers. For. Ecol. Manag. 2010, 259, 2026–2035. [Google Scholar] [CrossRef] [Green Version]
- Degen, B.; Yanbaev, R.; Yanbaev, Y. Genetic differentiation of Quercus robur in the South-Ural. Silvae Genet. 2019, 68, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Pohjanmies, T.; Elshibli, S.; Pulkkinen, P.; Rusanen, M.; Vakkari, P.; Korpelainen, H.; Roslin, T. Fragmentation-related patterns of genetic differentiation in pedunculate oak (Quercus robur) at two hierarchical scales. Silva. Fenn. 2016, 50, 15. [Google Scholar] [CrossRef] [Green Version]
- Degen, B.; Streiff, R.; Ziegenhagen, B. Comparative study of genetic variation and differentiation of two pedunculate oak (Quercus robur) stands using microsatellite and allozyme loci. Heredity 1999, 83, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorius, H.R.; Degen, B.; Konig, A. Problems in the analysis of genetic differentiation among populations—A case study in Quercus robur. Silvae Genet. 2007, 56, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Leroy, T.; Louvet, J.M.; Lalanne, C.; Le Provost, G.; Labadie, K.; Aury, J.M.; Delzon, S.; Plomion, C.; Kremer, A. Adaptive introgression as a driver of local adaptation to climate in European white oaks. New Phytol. 2020, 226, 1171–1182. [Google Scholar] [CrossRef]
- Rellstab, C.; Zoller, S.; Walthert, L.; Lesur, I.; Pluess, A.R.; Graf, R.; Bodenes, C.; Sperisen, C.; Kremer, A.; Gugerli, F. Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol. Ecol. 2016, 25, 5907–5924. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.N.; Wang, W.T.; Zhang, D.Y. Contrasts between the phylogeographic patterns of chloroplast and nuclear DNA highlight a role for pollen-mediated gene flow in preventing population divergence in an East Asian temperate tree. Mol. Phylogenet. Evol. 2014, 81, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Dick, C.W.; Pascoini, A.L.; Dayanandan, S. Despite introgressive hybridization, North American birches (Betula spp.) maintain strong differentiation at nuclear microsatellite loci. Tree Genet. Genomes 2015, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Curtu, A.L.; Gailing, O.; Finkeldey, R. Evidence for hybridization and introgression within a species-rich oak (Quercus spp.) community. BMC Evol. Biol. 2007, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chybicki, I.J.; Burczyk, J. Realized gene flow within mixed stands of Quercus robur L. and Q-petraea (Matt.) L. revealed at the stage of naturally established seedling. Mol. Ecol. 2010, 19, 2137–2151. [Google Scholar] [CrossRef]
- Olrik, D.C.; Hauser, T.P.; Kjaer, E.D. Natural colonisation of an open area by Quercus robur L-From where did the vectors disperse the seed? Scand. J. For. Res. 2012, 27, 350–360. [Google Scholar] [CrossRef]
- Buschbom, J.; Yanbaev, Y.; Degen, B. Efficient long-distance gene flow into an isolated relict oak stand. J. Hered. 2011, 102, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Schueler, S.; Schlunzen, K.; Scholz, F. Viability and sunlight sensitivity of oak pollen and its implications for pollen-mediated gene flow. Trees 2005, 19, 154–161. [Google Scholar] [CrossRef]
- Degen, B.; Blanc-Jolivet, C.; Bakhtina, S.; Ianbaev, R.; Yanbaev, Y.; Mader, M.; Nürnberg, S.; Schröder, H. Applying targeted genotyping by sequencing with a new set of nuclear and plastid SNP and indel loci for Quercus robur and Quercus petraea. Conserv. Genet. Resour. 2021, 13, 345–347. [Google Scholar] [CrossRef]
- Dumolin, S.; Demesure, B.; Petit, R.J. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor. Appl. Genet. 1995, 91, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. GENEPOP’ 007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Kruskal, J.B. On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Gregorius, H.R. A unique genetic distance. Biom. J. 1984, 26, 13–18. [Google Scholar] [CrossRef]
- Gregorius, H.R. The relationship between the concepts of genetic diversity and differentiation. Theor. Appl. Genet. 1987, 74, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution and the Genetics of Populations: A Treatise in Four Volumes: Variability within and Among Natural Populations; University of Chicago Press: Chicargo, IL, USA, 1978; Volume 4. [Google Scholar]
- Degen, B. GDA-NT 2021: Genetic Data Analysis and Numercical Tests. Available online: https://www.thuenen.de/en/fg/software/gda-nt-2021/ (accessed on 1 August 2021).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Degen, B. Spatial Genetic Software Version 2–2021. Available online: https://www.thuenen.de/en/fg/software/sgs/ (accessed on 1 August 2021).
- Degen, B.; Petit, R.J.; Kremer, A. SGS-Spatial genetic software: A computer program for analysis of spatial genetic and phenotypic structures of individuals and populations. J. Hered. 2001, 92, 447–449. [Google Scholar] [CrossRef] [Green Version]
- Brewer, S.; Cheddadi, R.; de Beaulieu, J.L.; Reille, M. The spread of deciduous Quercus throughout Europe since the last glacial period. For. Ecol. Manag. 2002, 156, 27–48. [Google Scholar] [CrossRef]
- Kozharinov, A.V.; Borisov, P.V. Distribution of oak forests in Eastern Europe over the last 13000 years. Contemp. Probl. Ecol. 2013, 6, 755–760. [Google Scholar] [CrossRef]
- Magyari, E.O.K.; Kunes, P.; Jakab, G.; Sumegi, P.; Pelankova, B.; Schabitz, F.; Braun, M.; Chytry, M. Late Pleniglacial vegetation in eastern-central Europe: Are there modern analogues in Siberia? Quat. Sci. Rev. 2014, 95, 60–79. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [Green Version]
- Gailing, O.; Wachter, H.; Heyder, J.; Schmittz, H.P.; Finkeldey, R. Chloroplast DNA analysis in oak stands (Quercus robur L.) in North Rhine-Westphalia with presumably Slavonian origin: Is there an association between geographic origin and bud phenology? J. Appl. Bot. Food Qual. 2007, 81, 165–171. [Google Scholar]
- Jansen, S.; Konrad, H.; Geburek, T. The extent of historic translocation of Norway spruce forest reproductive material in Europe. Ann. For. Sci. 2017, 74, 1–17. [Google Scholar] [CrossRef]
- Eusemann, P.; Liesebach, H. Small-scale genetic structure and mating patterns in an extensive sessile oak forest (Quercus petraea (Matt.) Liebl.). Ecol. Evol. 2021, 11, 7796–7809. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, A.; Brzeziecki, B. Role of jay (Garrulus glandarius) in initializing successional changes in forest communities with the participation of oak (Quercus sp.). Sylwan 2019, 163, 479–488. [Google Scholar] [CrossRef]
- Chybicki, I.J.; Burczyk, J. Seeing the forest through the trees: Comprehensive inference on individual mating patterns in a mixed stand of Quercus robur and Q-petraea. Ann. Bot. 2013, 112, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streiff, R.; Ducousso, A.; Lexer, C.; Steinkellner, H.; Gloessl, J.; Kremer, A. Pollen dispersal inferred from paternity analysis in a mixed oak stand of Quercus robur L-and Q-petraea (Matt.) Liebl. Mol. Ecol. 1999, 8, 831–841. [Google Scholar] [CrossRef]
- Gerber, S.; Chadoeuf, J.; Gugerli, F.; Lascoux, M.; Buiteveld, J.; Cottrell, J.; Dounavi, A.; Fineschi, S.; Forrest, L.L.; Fogelqvist, J.; et al. High rates of gene flow by pollen and seed in oak populations across Europe. PLoS ONE 2014, 9, e85130. [Google Scholar] [CrossRef]
- Moracho, E.; Moreno, G.; Jordano, P.; Hampe, A. Unusually limited pollen dispersal and connectivity of Pedunculate oak (Quercus robur) refugial populations at the species’ southern range margin. Mol. Ecol. 2016, 25, 3319–3331. [Google Scholar] [CrossRef] [PubMed]
- Buschbom, J.; Gimmerthal, S.; Kirschner, P.; Michalczyk, I.M.; Sebbenn, A.; Schueler, S.; Schlunzen, K.H.; Degen, B. Spatial composition of pollen-mediated gene flow in sessile oak. Forstarchiv 2012, 83, 12–18. [Google Scholar]
- Schueler, S.; Schlunzen, K.H. Modeling of oak pollen dispersal on the landscape level with a mesoscale atmospheric model. Environ. Modeling Assess. 2006, 11, 179–194. [Google Scholar] [CrossRef]
- Homolka, A.; Schueler, S.; Burg, K.; Fluch, S.; Kremer, A. Insights into drought adaptation of two European oak species revealed by nucleotide diversity of candidate genes. Tree Genet. Genomes 2013, 9, 1179–1192. [Google Scholar] [CrossRef]
- Savolainen, O.; Pyhajarvi, T.; Knurr, T. Gene flow and local adaptation in trees. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 595–619. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114. [Google Scholar] [CrossRef] [PubMed]
- Lepais, O.; Petit, R.J.; Guichoux, E.; Lavabre, J.E.; Alberto, F.; Kremer, A.; Gerber, S. Species relative abundance and direction of introgression in oaks. Mol. Ecol. 2009, 18, 2228–2242. [Google Scholar] [CrossRef] [PubMed]
- Guichoux, E.; Garnier-Gere, P.; Lagache, L.; Lang, T.; Boury, C.; Petit, R.J. Outlier loci highlight the direction of introgression in oaks. Mol. Ecol. 2013, 22, 450–462. [Google Scholar] [CrossRef]
- Petit, R.J.; Bodenes, C.; Ducousso, A.; Roussel, G.; Kremer, A. Hybridization as a mechanism of invasion in oaks. New Phytol. 2004, 161, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.; Larsen, A.; Nielsen, L.R.; Cottrell, J. Hybridization between Quercus robur and Q. petraea in a mixed oak stand in Denmark. Ann. For. Sci. 2009, 66, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Thomson, A.M.; Dick, C.W.; Dayanandan, S. A similar phylogeographical structure among sympatric North American birches (Betula) is better explained by introgression than by shared biogeographical history. J. Biogeogr. 2015, 42, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Leroy, T.; Roux, C.; Villate, L.; Bodenes, C.; Romiguier, J.; Paiva, J.A.P.; Dossat, C.; Aury, J.M.; Plomion, C.; Kremer, A. Extensive recent secondary contacts between four European white oak species. New Phytol. 2017, 214, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Plomion, C.; Aury, J.M.; Amselem, J.; Leroy, T.; Murat, F.; Duplessis, S.; Faye, S.; Francillonne, N.; Labadie, K.; Le Provost, G.; et al. Oak genome reveals facets of long lifespan. Nat. Plants 2018, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degen, B.; Yanbaev, Y.; Mader, M.; Ianbaev, R.; Bakhtina, S.; Schroeder, H.; Blanc-Jolivet, C. Impact of Gene Flow and Introgression on the Range Wide Genetic Structure of Quercus robur (L.) in Europe. Forests 2021, 12, 1425. https://doi.org/10.3390/f12101425
Degen B, Yanbaev Y, Mader M, Ianbaev R, Bakhtina S, Schroeder H, Blanc-Jolivet C. Impact of Gene Flow and Introgression on the Range Wide Genetic Structure of Quercus robur (L.) in Europe. Forests. 2021; 12(10):1425. https://doi.org/10.3390/f12101425
Chicago/Turabian StyleDegen, Bernd, Yulai Yanbaev, Malte Mader, Ruslan Ianbaev, Svetlana Bakhtina, Hilke Schroeder, and Celine Blanc-Jolivet. 2021. "Impact of Gene Flow and Introgression on the Range Wide Genetic Structure of Quercus robur (L.) in Europe" Forests 12, no. 10: 1425. https://doi.org/10.3390/f12101425
APA StyleDegen, B., Yanbaev, Y., Mader, M., Ianbaev, R., Bakhtina, S., Schroeder, H., & Blanc-Jolivet, C. (2021). Impact of Gene Flow and Introgression on the Range Wide Genetic Structure of Quercus robur (L.) in Europe. Forests, 12(10), 1425. https://doi.org/10.3390/f12101425