Genome-Wide Identification and Coexpression Network Analysis of DNA Methylation Pathway Genes and Their Differentiated Functions in Ginkgo biloba L.

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of DNA Methylation Pathway Gene Families in G. biloba
2.2. Phylogenetic Analysis and Classification of DNA Methylation Pathway Gene Families in G. biloba
2.3. Chromosomal Localization and Gene Tandem Duplication
2.4. Plant Tissues and Abiotic Stress Treatments for the Gene Expression Analysis
2.5. Transcriptome Sequencing and Gene Expression Analysis of DNA Methylation Pathway Genes
2.6. Coexpression Analysis of DNA Methylation Pathway Genes
3. Results
3.1. Identification and Structural Analysis of DNA Methylation Pathway Gene Families
3.2. Phylogenetic Analysis of DNA Methylation Pathway Genes in G. biloba
3.3. Chromosomal Location and Tandem Duplication of DNA Methylation Pathway Genes in the G. biloba Genome
3.4. Expression Patterns of DNA Methylation Pathway Genes in Various Tissues of G. biloba
3.5. Expression Changes of DNA Methylation Pathway Genes under Different Abiotic Stresses in G. biloba
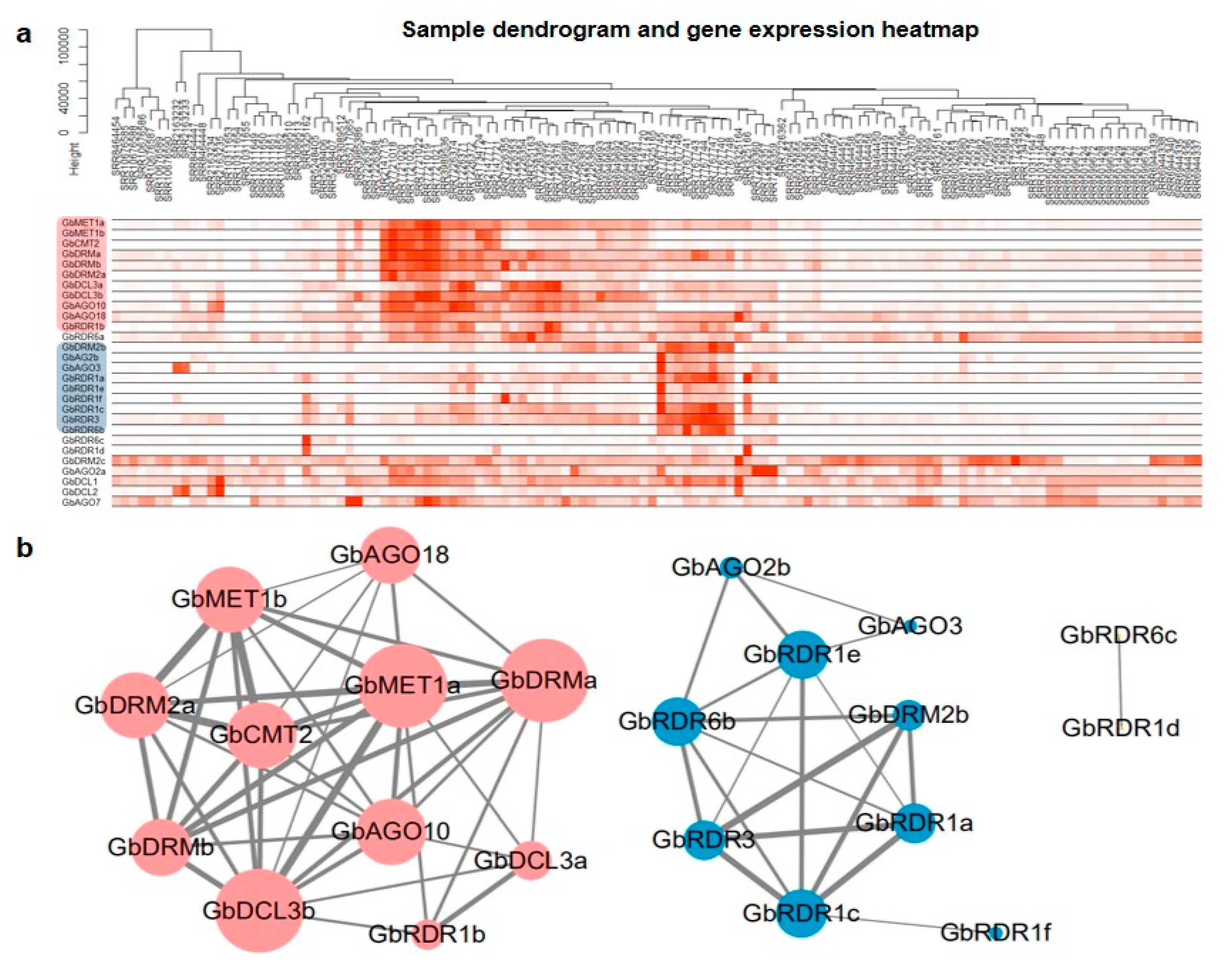
3.6. Coexpression Analysis of DNA Methylation Pathway Genes in G. biloba
3.7. Weighted Gene Co-Expression Network Analysis (WGCNA) of DNA Methylation Pathway Genes
3.8. Key Genes Coexpressed with DNA Methylation Pathway Genes
4. Discussion
4.1. The Structure of DNA Methylation Pathway Genes
4.2. The Expression Pattern of DNA Methylation Pathway Genes in Various Tissues and under Abiotic Stresses
4.3. The Differentiated Functions of DNA Methylation Pathway Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dinh, T.T.; O’Leary, M.; Won, S.Y.; Li, S.; Arroyo, L.; Liu, X.; Defries, A.; Zheng, B.; Cutler, S.R.; Chen, X. Generation of a luciferase-based reporter for CHH and CG DNA methylation in Arabidopsis thaliana. Silence 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Liu, H.; Liu, J.; Hua, W.; Xu, S.; Li, J. Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses. Int. J. Mol. Sci. 2020, 21, 953. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zhang, S.; Zhou, C.; Chen, L.; Fu, H.; Li, X.; Lin, Y.; Lai, Z.; Guo, Y. Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant (Camellia sinensis) under abiotic stress and withering processing. PEERJ 2020, 8, e8432. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Arıkan, B.; Özden, S.; Turgut-Kara, N. DNA methylation related gene expression and morphophysiological response to abiotic stresses in Arabidopsis thaliana. Environ. Exp. Bot. 2018, 149, 17–26. [Google Scholar] [CrossRef]
- Yaari, R.; Katz, A.; Domb, K.; Harris, K.D.; Zemach, A.; Ohad, N. RdDM-independent de novo and heterochromatin DNA methylation by plant CMT and DNMT3 orthologs. Nat. Commun. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Sabbione, A.; Daurelio, L.; Vegetti, A.; Talón, M.; Tadeo, F.; Dotto, M. Genome-wide analysis of AGO, DCL and RDR gene families reveals RNA-directed DNA methylation is involved in fruit abscission in Citrus sinensis. BMC Plant Biol. 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Yadav, C.B.; Muthamilarasan, M.; Pandey, G.; Prasad, M. Identification, Characterization and Expression Profiling of Dicer-Like, Argonaute and RNA-Dependent RNA Polymerase Gene Families in Foxtail Millet. Plant Mol. Biol. Rep. 2015, 33, 43–55. [Google Scholar] [CrossRef]
- Kapoor, M.; Arora, R.; Lama, T.; Nijhawan, A.; Khurana, J.P.; Tyagi, A.K.; Kapoor, S. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genom. 2008, 9, 451. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Sung, S. Mechanisms Underlying Epigenetic Regulation in Arabidopsis thaliana. Integr. Comp. Biol. 2014, 54, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Daccord, N.; Celton, J.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Bitonti, M.B.; Cozza, R.; Chiappetta, A.; Giannino, D.; Castiglione, M.R.; Dewitte, W.; Mariotti, D.; Onckelen, H.V.; Innocenti, A.M. Distinct nuclear organization, DNA methylation pattern and cytokinin distribution mark juvenile, juvenile-like and adult vegetative apical meristems in peach (Prunus persica (L.) Batsch). J. Exp. Bot. 2002, 53, 1047–1054. [Google Scholar] [CrossRef]
- Palmgren, G.; Mattsson, O.; Okkels, F.T. Specific Levels of DNA Methylation in Various Tissues, Cell Lines, and Cell Types of Daucus carota. Plant Physiol. 1991, 95, 174–178. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.; Pearson, J.; Hsieh, T.; An, Y.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Bai, M.; Yang, G.; Chen, W.; Mao, Z.; Kang, H.; Chen, G.; Yang, Y.; Xie, B. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene 2012, 501, 52–62. [Google Scholar] [CrossRef]
- Qian, Y.; Cheng, Y.; Cheng, X.; Jiang, H.; Zhu, S.; Cheng, B. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 2011, 30, 1347–1363. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, H.; Chen, Z.; Feng, L.; Ren, J.; Cai, R.; Xiang, Y. The Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in Populus trichocarpa: Gene structure, gene expression, phylogenetic analysis and evolution. J. Genet. 2015, 94, 317–321. [Google Scholar] [CrossRef]
- Das, S.; Meher, P.K.; Rai, A.; Bhar, L.M.; Mandal, B.N. Statistical Approaches for Gene Selection, Hub Gene Identification and Module Interaction in Gene Co-Expression Network Analysis: An Application to Aluminum Stress in Soybean (Glycine max L.). PLoS ONE 2017, 12, e169605. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.L.; Davidson, R.M.; Buell, C.R. Gene coexpression network analysis as a source of functional annotation for rice genes. PLoS ONE 2011, 6, e22196. [Google Scholar] [CrossRef] [PubMed]
- Weston, D.J.; Karve, A.A.; Gunter, L.E.; Jawdy, S.S.; Yang, X.; Allen, S.M.; Wullschleger, S.D. Comparative physiology and transcriptional networks underlying the heat shock response in Populus trichocarpa, Arabidopsis thaliana and Glycine max. Plant Cell Environ. 2011, 34, 1488–1506. [Google Scholar] [CrossRef]
- Downs, G.S.; Bi, Y.; Colasanti, J.; Wu, W.; Chen, X.; Zhu, T.; Rothstein, S.J.; Lukens, L.N. A Developmental Transcriptional Network for Maize Defines Coexpression Modules. Plant Physiol. 2013, 161, 1830–1843. [Google Scholar] [CrossRef]
- Ficklin, S.P.; Feltus, F.A. Gene Coexpression Network Alignment and Conservation of Gene Modules between Two Grass Species: Maize and Rice. Plant Physiol. 2011, 156, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Jara, A.B.; Souliman-Youssef, S.; Novo-Uzal, E.; Almagro, L.; Belchí-Navarro, S.; Pedreño, M.A. Biotechnological approaches to enhance the biosynthesis of ginkgolides and bilobalide in Ginkgo biloba. Phytochem. Rev. 2013, 12, 191–205. [Google Scholar] [CrossRef]
- Ye, J.; Cheng, S.; Zhou, X.; Chen, Z.; Kim, S.U.; Tan, J.; Zheng, J.; Xu, F.; Zhang, W.; Liao, Y.; et al. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis. Ind. Crop. Prod. 2019, 139, 111547. [Google Scholar] [CrossRef]
- Guo, J.; Wu, Y.; Wang, G.; Wang, T.; Cao, F. Integrated analysis of the transcriptome and metabolome in young and mature leaves of Ginkgo biloba L. Ind. Crop. Prod. 2020, 143, 111906. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, X.; Wang, T.; Wang, G.; Cao, F. Regulation of flavonoid metabolism in ginkgo leaves in response to different day-night temperature combinations. Plant Physiol. Biochem. 2020, 147, 133–140. [Google Scholar] [CrossRef]
- Wang, L.; Cui, J.; Jin, B.; Zhao, J.; Xu, H.; Lu, Z.; Li, W.; Li, X.; Li, L.; Liang, E.; et al. Multifeature analyses of vascular cambial cells reveal longevity mechanisms in old Ginkgo biloba trees. Proc. Natl. Acad. Sci. USA 2020, 117, 2201–2210. [Google Scholar] [CrossRef]
- He, B.; Gu, Y.; Xu, M.; Wang, J.; Cao, F.; Xu, L.A. Transcriptome analysis of Ginkgo biloba kernels. Front. Plant Sci. 2015, 6, 819. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, D.; Li, S.; Guo, N.; Gao, J.; Sun, X.; Cai, Y. Transcriptomic profiling identifies differentially expressed genes associated with programmed cell death of nucellar cells in Ginkgo biloba L. BMC Plant Biol. 2019, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, L.; Wu, Q.; Men, X.; Yao, L.; Xing, S. Transcriptome profile analysis reveals the ontogenesis of rooted chichi in Ginkgo biloba L. Gene 2018, 669, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Roodt, D.; Lohaus, R.; Sterck, L.; Swanepoel, R.L.; van de Peer, Y.; Mizrachi, E. Evidence for an ancient whole genome duplication in the cycad lineage. PLoS ONE 2017, 12, e184454. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, L.; Pang, S.; Jia, Z.; Wang, L.; Li, W.; Jin, B. UV-B promotes flavonoid synthesis in Ginkgo biloba leaves. Ind. Crop. Prod. 2020, 151, 112483. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, H.; Xu, W.; You, Q.; Yan, H.; Gao, Z.; Su, Z. Co-expression Gene Network Analysis and Functional Module Identification in Bamboo Growth and Development. Front. Genet. 2018, 9, 574. [Google Scholar] [CrossRef]
- Niu, S.; Liu, C.; Yuan, H.; Li, P.; Li, Y.; Li, W. Identification and expression profiles of sRNAs and their biogenesis and action-related genes in male and female cones of Pinus tabuliformis. BMC Genom. 2015, 16, 693. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, K.; Wang, J.; Chen, X.; Chen, Z.; Cai, R.; Xiang, Y. Comprehensive Analysis of Dicer-Like, Argonaute, and RNA-dependent RNA Polymerase Gene Families in Grapevine (Vitis Vinifera). J. Plant Growth Regul. 2015, 34, 108–121. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, D.; Wang, R.; Kong, N.; Zhang, C.; Yang, C.; Wu, W.; Ma, H.; Chen, Q. Genome-wide analysis of the potato Hsp20 gene family: Identification, genomic organization and expression profiles in response to heat stress. BMC Genom. 2018, 19, 61. [Google Scholar] [CrossRef]
- Wang, L.; Guo, K.; Li, Y.; Tu, Y.; Hu, H.; Wang, B.; Cui, X.; Peng, L. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universial RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: http://www.R-project.org/ (accessed on 25 September 2020).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 2003, 3, 1. [Google Scholar] [CrossRef]
- Song, Y.; Ma, K.; Ci, D.; Chen, Q.; Tian, J.; Zhang, D. Sexual dimorphic floral development in dioecious plants revealed by transcriptome, phytohormone, and DNA methylation analysis in Populus tomentosa. Plant Mol. Biol. 2013, 83, 559–576. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Kossida, S. Plant cytosine-5 DNA methyltransferases: Structure, function, and molecular evolution. Genomics 2007, 90, 530–541. [Google Scholar] [CrossRef]
- Ahmad, F.; Huang, X.; Lan, H.X.; Huma, T.; Bao, Y.M.; Huang, J.; Zhang, H.S. Comprehensive gene expression analysis of the DNA (cytosine-5) methyltransferase family in rice (Oryza sativa L.). Genet. Mol. Res. 2014, 13, 5159–5172. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Xi, Y.; Cheng, B.; Zhu, S. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant Cell Rep. 2014, 33, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hatlen, A.; Kelly, L.J.; Becher, H.; Wang, W.; Kovarik, A.; Leitch, I.J.; Leitch, A.R. Angiosperms Are Unique among Land Plant Lineages in the Occurrence of Key Genes in the RNA-Directed DNA Methylation (RdDM) Pathway. Genome Biol. Evol. 2015, 7, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Qi, Y. RNAi in Plants: An Argonaute-Centered View. Plant Cell 2016, 28, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Chen, F.; Huan, X.; Machida, S.; Song, J.; Yuan, Y.A. Structure of the Arabidopsis thaliana DCL4 DUF283 domain reveals a noncanonical double-stranded RNA-binding fold for protein-protein interaction. RNA 2010, 16, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.O.; Fausto, A.K.S.; Fanelli, A.; Guedes, F.A.D.F.; Silva, T.D.F.; Romanel, E.; Vaslin, M.F.S. Genome-wide identification of the Dicer-like family in cotton and analysis of the DCL expression modulation in response to biotic stress in two contrasting commercial cultivars. BMC Plant Biol. 2019, 19, 503. [Google Scholar] [CrossRef]
- Qin, L.; Mo, N.; Muhammad, T.; Liang, Y. Genome-Wide Analysis of DCL, AGO, and RDR Gene Families in Pepper (Capsicum Annuum L.). Int. J. Mol. Sci. 2018, 19, 1038. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Guo, X.; Wang, X.; Zhang, X. Arabidopsis AGO3 predominantly recruits 24-nt small RNAs to regulate epigenetic silencing. Nat. Plants 2016, 2, 16049. [Google Scholar] [CrossRef]
- Polydore, S.; Axtell, M.J. Analysis ofRDR1/RDR2/RDR6-independent small RNAs in Arabidopsis thaliana improves MIRNA annotations and reveals unexplained types of short interfering RNA loci. Plant J. 2018, 94, 1051–1063. [Google Scholar] [CrossRef]
- Willmann, M.R.; Endres, M.W.; Cook, R.T.; Gregory, B.D. The Functions of RNA-Dependent RNA Polymerases in Arabidopsis. Arab. Book 2011, 9, e146. [Google Scholar] [CrossRef]
- Du, S.; Sang, Y.; Liu, X.; Xing, S.; Li, J.; Tang, H.; Sun, L. Transcriptome Profile Analysis from Different Sex Types of Ginkgo biloba L. Front. Plant Sci. 2016, 7, 871. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, S.E.; Gross, S.M.; Hollick, J.B. Maize sex determination and abaxial leaf fates are canalized by a factor that maintains repressed epigenetic states. Dev. Biol. 2007, 308, 462–473. [Google Scholar] [CrossRef]
- Baubec, T.; Finke, A.; Mittelsten, S.O.; Pecinka, A. Meristem-specific expression of epigenetic regulators safeguards transposon silencing in Arabidopsis. EMBO Rep. 2014, 15, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Teng, F.; Zheng, K.; Xiao, J.; Deng, W.; Sun, W. Expression analysis of Argonaute genes in maize (Zea mays L.) in response to abiotic stress. Hereditas 2019, 156, 27. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Kumari, R.; Tiwari, S.; Goyal, S. Genomic survey, gene expression analysis and structural modeling suggest diverse roles of DNA methyltransferases in legumes. PLoS ONE 2014, 9, e88947. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, E104. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.F.; Xue, H.W. Coexpression analysis identifies Rice Starch Regulator1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiol. 2010, 154, 927–938. [Google Scholar] [CrossRef]
- Yang, D.L.; Zhang, G.; Tang, K.; Li, J.; Yang, L.; Huang, H.; Zhang, H.; Zhu, J.K. Dicer-independent RNA-directed DNA methylation in Arabidopsis. Cell Res. 2016, 26, 66–82. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Family | Gene Name | Gene ID | Chr. | Coordinates (5′–3′) | ORF (bp) | Protein (aa) | PI | MW (KDa) | No. of Introns |
|---|---|---|---|---|---|---|---|---|---|
| MET | GbMET1a | Gb_34186 | 1 | 204,603,561–204,649,352 | 45,792 | 1519 | 6.21 | 171.27 | 9 |
| GbMET1b | Gb_24436 | 6 | 118,058,848–118,066,891 | 8044 | 1536 | 5.78 | 168.15 | 8 | |
| CMT | GbCMT2 | Gb_13672 | 2 | 64,359,582–64,366,200 | 6619 | 902 | 8.27 | 101.47 | 21 |
| DRM | GbDRMa | Gb_25633 | 3 | 282,894,262–282,895,575 | 1314 | 437 | 9.46 | 49.32 | 0 |
| GbDRMb | Gb_33731 | 12 | 405,552,612–405,778,186 | 225,575 | 650 | 5.66 | 73.73 | 7 | |
| GbDRM2a | Gb_24437 | 6 | 118,086,495–118,087,159 | 665 | 144 | 9.33 | 16.38 | 2 | |
| GbDRM2b | Gb_03787 | 11 | 101,816,084–101,817,173 | 1090 | 150 | 9.18 | 16.89 | 3 | |
| GbDRM2c | Gb_23570 | 7 | 55,632,811–55,743,991 | 111,181 | 202 | 5.55 | 22.97 | 4 | |
| DNMT3 | GbDNMT3 | Gb_23575 | 9 | 613,883,922–613,885,240 | 1319 | 140 | 5.10 | 15.18 | 3 |
| DCL | GbDCL1 | Gb_25982 | 4 | 88,761,181–88,828,642 | 67,462 | 2164 | 6.07 | 244.87 | 19 |
| GbDCL2 | Gb_32044 | 5 | 539,277,719–539,892,110 | 614,392 | 1464 | 6.76 | 165.28 | 22 | |
| GbDCL3a | Gb_34767 | 1 | 433,398,375-433,857,929 | 459,555 | 1815 | 6.66 | 203.09 | 25 | |
| GbDCL3b | Gb_13632 | 1 | 428,462,217–428,704,798 | 459,555 | 1890 | 7.57 | 212.83 | 25 | |
| AGO | GbAGO2a | Gb_13708 | 9 | 218,695,767–218,699,026 | 3260 | 1025 | 9.48 | 113.92 | 2 |
| GbAGO2b | Gb_13719 | 9 | 219,330,477–219,333,276 | 2800 | 896 | 9.78 | 100.94 | 1 | |
| GbAGO3 | Gb_21789 | 9 | 220,234,425–220,237,707 | 3283 | 1025 | 8.91 | 115.16 | 2 | |
| GbAGO7 | Gb_15866 | 11 | 513,379,788–513,384,603 | 4816 | 1199 | 9.07 | 134.38 | 4 | |
| GbAGO10 | Gb_34642 | 1 | 919,522,451–919,531,642 | 9192 | 1061 | 9.33 | 119.28 | 23 | |
| GbAGO18 | Gb_34718 | 11 | 144,054,917–144,136,109 | 81,193 | 741 | 9.26 | 83.67 | 19 | |
| RDR | GbRDR1a | Gb_17278 | 2 | 146,510,397–146,518,861 | 8465 | 915 | 8.06 | 103.57 | 9 |
| GbRDR1b | Gb_28437 | 2 | 147,783,139–147,785,354 | 2216 | 618 | 7.45 | 69.79 | 3 | |
| GbRDR1c | Gb_28433 | 2 | 149,061,893–149,088,988 | 27,096 | 897 | 8.49 | 102.07 | 2 | |
| GbRDR1d | Gb_28429 | 2 | 148,784,524–148,786,257 | 1734 | 577 | 9.62 | 65.37 | 0 | |
| GbRDR1e | Gb_28426 | 2 | 148,423,209–148,425,400 | 2192 | 649 | 9.58 | 73.35 | 1 | |
| GbRDR1f | Gb_28427 | 2 | 148,451,204–148,455,736 | 4533 | 491 | 6.50 | 55.78 | 2 | |
| GbRDR3 | Gb_32086 | 1 | 607,044,984–607,566,259 | 521,276 | 1086 | 5.88 | 123.15 | 18 | |
| GbRDR6a | Gb_12326 | 3 | 382,504,304–382,559,266 | 54,963 | 1186 | 8.47 | 136.02 | 1 | |
| GbRDR6b | Gb_08298 | 3 | 383,759,636–383,761,795 | 2160 | 624 | 8.90 | 71.62 | 1 | |
| GbRDR6c | Gb_08306 | 3 | 382,920,448–382,945,656 | 25,209 | 1187 | 7.71 | 135.78 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Deng, M.; Yang, X.; Yu, W.; Cai, J.; Shi, Y.; Zhu, Z.; Zhou, T.; Xue, L.; Cao, F.; et al. Genome-Wide Identification and Coexpression Network Analysis of DNA Methylation Pathway Genes and Their Differentiated Functions in Ginkgo biloba L. Forests 2020, 11, 1076. https://doi.org/10.3390/f11101076
Gao C, Deng M, Yang X, Yu W, Cai J, Shi Y, Zhu Z, Zhou T, Xue L, Cao F, et al. Genome-Wide Identification and Coexpression Network Analysis of DNA Methylation Pathway Genes and Their Differentiated Functions in Ginkgo biloba L. Forests. 2020; 11(10):1076. https://doi.org/10.3390/f11101076
Chicago/Turabian StyleGao, Caiyun, Miao Deng, Xiaoming Yang, Wanwen Yu, Jinfeng Cai, Yuanbao Shi, Zhibo Zhu, Tingting Zhou, Liangjiao Xue, Fuliang Cao, and et al. 2020. "Genome-Wide Identification and Coexpression Network Analysis of DNA Methylation Pathway Genes and Their Differentiated Functions in Ginkgo biloba L." Forests 11, no. 10: 1076. https://doi.org/10.3390/f11101076
APA StyleGao, C., Deng, M., Yang, X., Yu, W., Cai, J., Shi, Y., Zhu, Z., Zhou, T., Xue, L., Cao, F., Wang, G., & Fu, F.-F. (2020). Genome-Wide Identification and Coexpression Network Analysis of DNA Methylation Pathway Genes and Their Differentiated Functions in Ginkgo biloba L. Forests, 11(10), 1076. https://doi.org/10.3390/f11101076