Bacterial Community Changes Associated with Land Use Type in the Forest Montane Region of Northeast China

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Research Area
2.2. Sample Unit Selection, Sample Collection
2.2.1. Soil Chemical Properties
2.2.2. Soil DNA Extraction
2.3. 16S rDNA High-Throughput Sequencing
2.4. Bioinformatics and Statistical Analyses
3. Results
3.1. Chemical Soil Characteristics
3.2. Diversity of Soil Bacterial Communities under Different Land Use Patterns
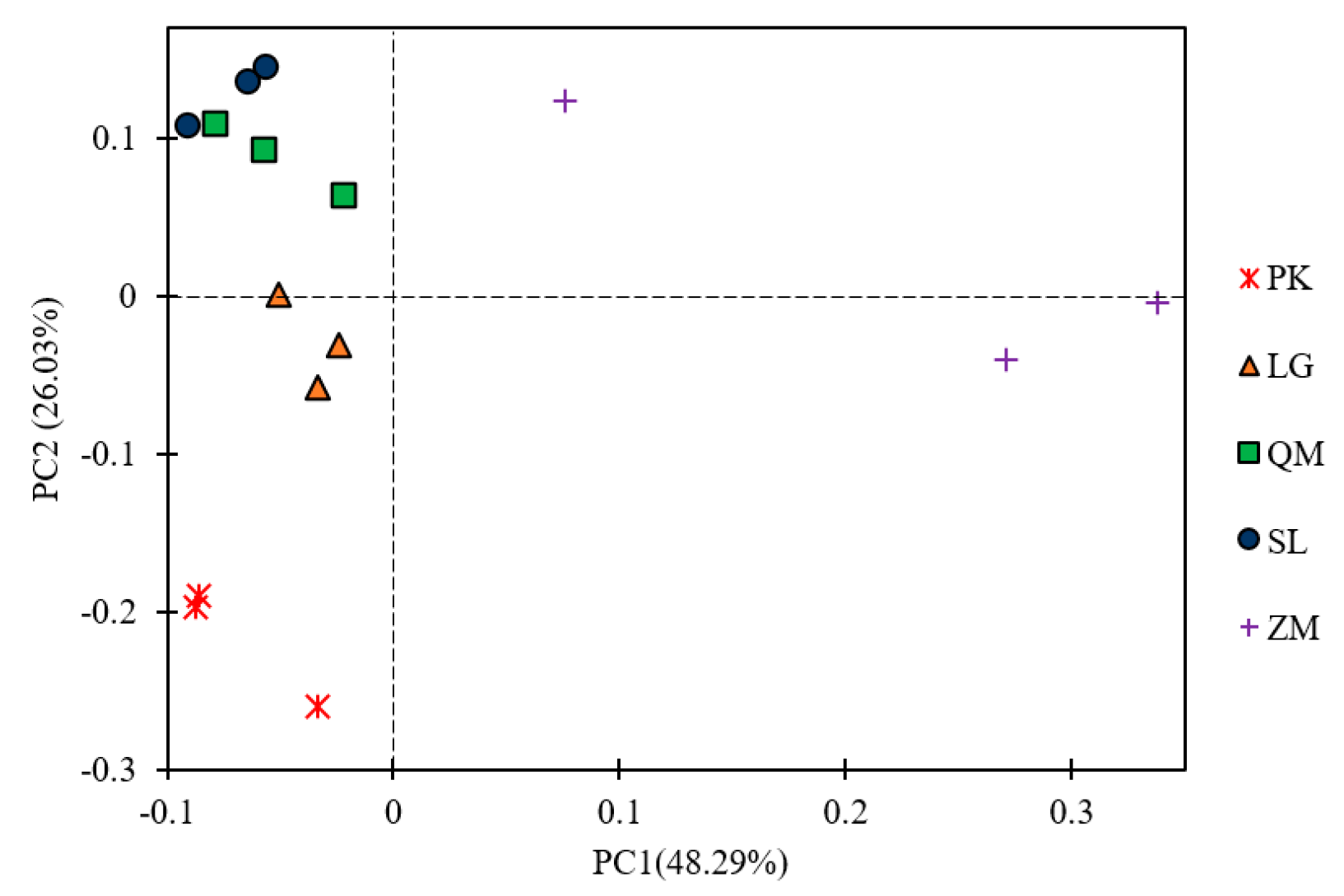
3.3. Soil Bacterial Community Compositions under Different Land Use Patterns
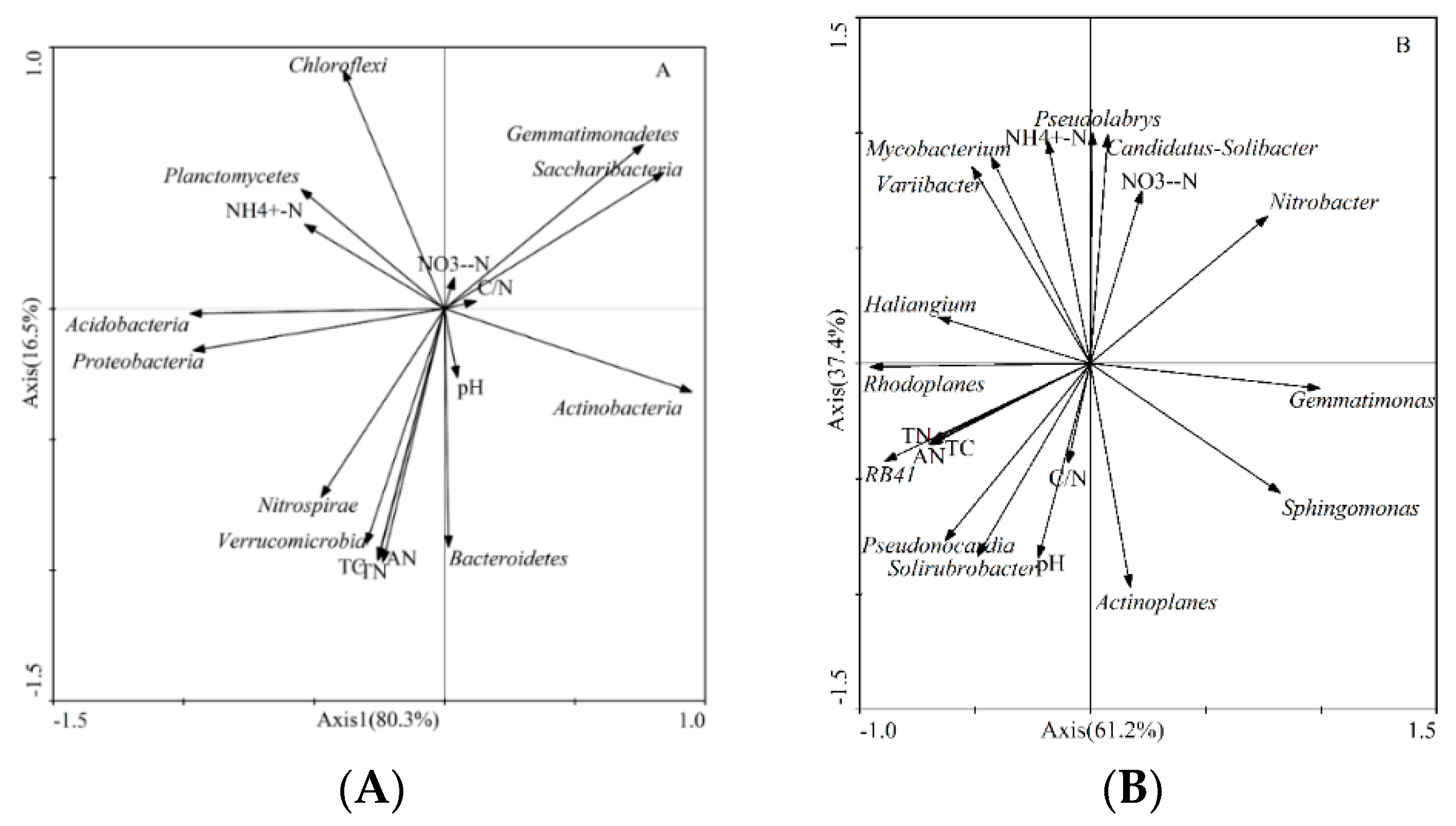
3.4. Relationships between the Microbial Community Composition and Soil Environmental Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, D.; Keiblinger, K.M.; Leitner, S.; Wegner, U.; Zimmermann, M.; Fuchs, S.; Lassek, C.; Riedel, K.; Zechmeister-Boltenstern, S. Response of microbial communities and their metabolic functions to drying–rewetting stress in a temperate forest soil. Microorganisms 2019, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, N.L.; Jankowski, K.J.; Tuchman, N.C.; Kelly, J.J. Effects of an invasive cattail species (Typha×glauca) on sediment nitrogen and microbial community composition in a freshwater wetland. FEMS Microbiol. Lett. 2006, 263, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Averill, C.; Hawkes, C.V. Ectomycorrhizal fungi slow soil carbon cycling. Ecol. Lett. 2016, 19, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Kogelknabner, I. The macromolecular organic composition of plant and microbial residues as inputs to soil organic matter. Soil Biol. Biochem. 2002, 34, 139–162. [Google Scholar] [CrossRef]
- Khan, K.S.; Mack, R.; Castillo, X.; Kaiser, M.; Joergensena, R.G. Microbial biomass, fungal and bacterial residues, and their relationships to the soil organic matter C/N/P/S ratios. Geoderma 2016, 271, 115–123. [Google Scholar] [CrossRef]
- Bastida, F.; Zsolnay, A.; Hernández, T.; García, C. Past, present and future of soil quality indices: A biological perspective. Geoderma 2008, 147, 159–171. [Google Scholar] [CrossRef]
- Gyaneshwar, P.; Kumar, G.N.; Parekh, L.J.; Poole, P.S. Role of soil microorganisms in improving P nutrition of plants. Plant Soil. 2002, 245, 83–93. [Google Scholar] [CrossRef]
- Cruz-Paredes, C.; Wallander, H.; Kjøller, R.; Rousk, J. Using community trait-distributions to assign microbial responses to pH changes and Cd in forest soils treated with wood ash. Soil Biol. Biochem. 2017, 112, 153–164. [Google Scholar] [CrossRef]
- Berg, G. Plant–microbe interactions promoting plant growth and health: Perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef]
- Yin, C.; Jones, K.L.; Peterson, D.E.; Garrett, K.A.; Hulbert, S.H.; Paulitz, T.C. Members of soil bacterial communities sensitive to tillage and crop rotation. Soil Biol. Biochem. 2010, 42, 2111–2118. [Google Scholar] [CrossRef]
- Guo, X.; Chen, H.Y.H.; Meng, M.; Biswas, S.R.; Ye, L.; Zhang, J. Effects of land use change on the composition of soil microbial communities in a managed subtropical forest. For. Ecol. Manag. 2016, 373, 93–99. [Google Scholar] [CrossRef]
- Wang, Q.; He, T.; Wang, S.; Liu, L. Carbon input manipulation affects soil respiration and microbial community composition in a subtropical coniferous forest. Agric. For. Meteorol. 2013, 178, 152–160. [Google Scholar] [CrossRef]
- Rosenzweig, S.T.; Carson, M.A.; Baer, S.G.; Blair, J.M. Changes in soil properties, microbial biomass, and fluxes of C and N in soil following post-agricultural grassland restoration. Appl. Soil Ecol. 2016, 100, 186–194. [Google Scholar] [CrossRef]
- Jangid, K.; Williams, M.A.; Franzluebbers, A.J.; Schmidt, T.M.; Coleman, D.C.; Whitman, W.B. Land-use history has a stronger impact on soil microbial community composition than aboveground vegetation and soil properties. Soil Biol. Biochem. 2011, 43, 2184–2193. [Google Scholar] [CrossRef]
- Brant, J.B.; Myrold, D.D.; Sulzman, E.W. Root controls on soil microbial community composition in forest soils. Oecologia 2006, 148, 650–659. [Google Scholar] [CrossRef]
- Peng, M.; Jia, H.; Wang, Q. The Effect of Land Use on Bacterial Communities in Saline-Alkali Soil. Curr. Microbiol. 2017, 74, 325–333. [Google Scholar] [CrossRef]
- Ding, G.C.; Piceno, Y.M.; Heuer, H.; Weinert, N.; Dohrmann, A.B.; Carrillo, A.; Andersen, G.L.; Castellanos, T.; Tebbe, C.C.; Smalla, K. Changes of Soil Bacterial Diversity as a Consequence of Agricultural Land Use in a Semi-Arid Ecosystem. PLoS ONE 2013, 8, e59497. [Google Scholar] [CrossRef]
- Bissett, A.; Richardson, A.E.; Baker, G.; Thralla, P.H. Long-term land use effects on soil microbial community structure and function. Appl. Soil Ecol. 2011, 51, 66–78. [Google Scholar] [CrossRef]
- Acosta-Martínez, V.; Dowd, S.; Sun, Y.; Allen, V. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 2008, 40, 2762–2770. [Google Scholar] [CrossRef]
- Da C Jesus, E.; Marsh, T.L.; Tiedje, J.M.; de S Moreira, F.M. Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J. 2009, 3, 1004–1011. [Google Scholar] [CrossRef]
- Moscatelli, M.C.; Secondi, L.; Marabottini, R.; Papp, R.; Stazi, S.R.; Mania, E.; Marinari, S. Assessment of soil microbial functional diversity: Land use and soil properties affect CLPP-MicroResp and enzymes responses. Pedobiologia 2018, 66, 36–42. [Google Scholar] [CrossRef]
- Bossio, D.A.; Girvan, M.S.; Verchot, L.; Bullimore, J.; Borelli, T.; Albrecht, A.; Scow, K.M.; Ball, A.S.; Pretty, J.N.; Osborn, A.M. Soil Microbial Community Response to Land Use Change in an Agricultural Landscape of Western Kenya. Microb. Ecol. 2005, 49, 50–62. [Google Scholar] [CrossRef]
- Lagerlöf, J.; Adolfsson, L.; Börjesson, G.; Ehlers, K.; Vinyoles, G.P.; Sundh, I. Land-use intensification and agroforestry in the Kenyan highland: Impacts on soil microbial community composition and functional capacity. Appl. Soil Ecol. 2014, 82, 93–99. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, X.X.; Guo, X.; Wang, D.; Chu, H. Bacterial diversity in soils subjected to long-term chemical fertilization can be more stably maintained with the addition of livestock manure than wheat straw. Soil Biol. Biochem. 2015, 88, 9–18. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Lauber, C.L.; Knight, R.; Bradford, M.A.; Fierer, N. Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology 2010, 91, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Coolon, J.D.; Jones, K.L.; Todd, T.C.; Blair, J.M.; Herman, M.A. Long-Term Nitrogen Amendment Alters the Diversity and Assemblage of Soil Bacterial Communities in Tallgrass Prairie. PLoS ONE 2013, 8, e67884. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.; Liu, F. Seasonal variations of soil microbial biomass and its influence on soil microbial respiration in secondary forest communities in montane region of Eastern Liaoning Province. J. Northeast For. Univ. 2014, 42, 99–102. [Google Scholar]
- Qi, J.H. Contents of soil organic carbon and its relations with physicochemical properties of secondary natural oak forests in Eastern Mountain Area of Liaoning Province. J. Soil Water Conserv. 2017, 31, 135–140. [Google Scholar]
- Deng, J.J.; Yin, Y.; Luo, J.Y.; Zhu, W.X.; Zhou, Y.B. Different revegetation types alter soil physical-chemical characteristics and fungal community in the Baishilazi Nature Reserve. Peer J 2019, 6, e6251. [Google Scholar] [CrossRef]
- Deng, J.J.; Yin, Y.; Zhu, W.X.; Zhou, Y.B. Variations in soil bacterial community diversity and structures among different revegetation types. Front. Microbiol. 2018, 9, 2874. [Google Scholar] [CrossRef]
- Cassán, F.; Diaz-Zorita, M. Azospirillum sp. in current agriculture: From the laboratory to the field. Soil Biol. Biochem. 2016, 103, 117–130. [Google Scholar] [CrossRef]
- Roesch, L.F.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A. What is microbial community ecology? ISME J. 2009, 3, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Tian, Q.; Taniguchi, T.; Shi, W.Y.; Li, G.; Yamanaka, N.; Du, S. Land-use types and soil chemical properties influence soil microbial communities in the semiarid Loess Plateau region in China. Sci. Rep. 2017, 7, 45289. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, Y.; Lin, X.; Zhang, H.; Zeng, J.; Hou, J.; Yang, Y.; Yao, T.; Knight, R.; Chu, H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Mander, C.; Wakelin, S.; Young, S.; Condron, L.; O’Callaghana, M. Incidence and diversity of phosphate-solubilising bacteria are linked to phosphorus status in grassland soils. Soil Biol. Biochem. 2012, 44, 93–101. [Google Scholar] [CrossRef]
- Huang, J.; Hu, B.; Qi, K.; Chen, W.; Pang, X.; Bao, W.; Tian, G. Effects of phosphorus addition on soil microbial biomass and community composition in a subalpine spruce plantation. Eur. J. Soil. Biol. 2016, 72, 35–41. [Google Scholar] [CrossRef]
- Navarrete, I.A.; Tsutsuki, K. Land-use impact on soil carbon, nitrogen, neutral sugar composition and related chemical properties in a degraded Ultisol in Leyte, Philippines. Soil Sci. Plant Nutr. 2008, 54, 321–331. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, P.C.; Tian, H.M.; Xiao, Q.Q.; Jiang, H.K. Pyrosequencing-based assessment of soil microbial community structure and analysis of soil properties with vegetable planted at different years under greenhouse conditions. Soil Till. Res. 2019, 187, 1–10. [Google Scholar] [CrossRef]
- Sauvadet, M.; Chauvat, M.; Cluzeau, D.; Maron, P.A.; Villenave, C.; Bertrand, I. The dynamics of soil micro-food web structure and functions vary according to litter quality. Soil Biol. Biochem. 2016, 95, 262–274. [Google Scholar] [CrossRef]
- Ushio, M.; Wagai, R.; Balser, T.C.; Kitayama, K. Variations in the soil microbial community composition of a tropical montane forest ecosystem: Does tree species matter. Soil Biol. Biochem. 2008, 40, 2699–2702. [Google Scholar] [CrossRef]
- Sauheitl, L.; Glaser, B.; Dippold, M.; Leiber, K.; Weigelt, A. Amino acid fingerprint of a grassland soil reflects changes in plant species richness. Plant Soil. 2010, 334, 353–363. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Kim, M.; Singh, D.; Lee-Cruz, L.; Lai-Hoe, A.; Ainuddin, A.N.; Go, R.; Rahim, R.A.; Husni, M.H.; Chun, J.; et al. Tropical soil bacterial communities in Malaysia: pH dominates in the equatorial tropics too. Microb. Ecol. 2012, 64, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-Based Assessment of Soil pH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microbiol. 2009, 75, 51115120. [Google Scholar] [CrossRef]
- Hartman, W.H.; Richardson, C.J.; Vilgalys, R.; Bruland, G.L. Environmental and anthropogenic controls over bacterial communities in wetland soils. Proc. Natl. Acad. Sci. USA 2008, 105, 17842–17847. [Google Scholar] [CrossRef]
- Meng, H.; Li, K.; Nie, M.; Wan, J.R.; Quan, Z.X.; Fang, C.M.; Chen, J.K.; Gu, J.D.; Li, B. Responses of bacterial and fungal communities to an elevation gradient in a subtropical montane forest of China. Appl. Microbiol. Biotechnol. 2013, 97, 2219–2230. [Google Scholar] [CrossRef]
- Eichorst, S.A.; Trojan, D.; Roux, S.; Herbold, C.; Rattei, T.; Woebken, D. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 2018, 20, 1041–1063. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.F.; Castro, A.P.D.; Costa, M.M.; Togawa, R.C.; Júnior, G.J.; Quirino, B.F.; Bustamante, M.M.; Williamson, L.; Handelsman, J.; Krüger, R.H. Characterization of Soil Bacterial Assemblies in Brazilian Savanna-Like Vegetation Reveals Acidobacteria Dominance. Microb. Ecol. 2012, 64, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Nemergut, D.R.; Cleveland, C.C.; Wieder, W.R.; Washenberger, C.L.; Townsendad, A.R. Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol. Biochem. 2011, 42, 2153–2160. [Google Scholar] [CrossRef]
- Hogberg, M.; Hogberg, P.; Myrold, D. Is microbial community composition in boreal forest soils determined by pH, C-to-N ratio, the trees, or all three? Oecologia 2007, 150, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.S.; Lin, X.G.; Shi, W.M.; Min, J.; Gao, N.; Zhang, H.; Yin, R.; He, X. Higher rates of nitrogen fertilization decrease soil enzyme activities, microbial functional diversity and nitrification capacity in a Chinese polytunnel greenhouse vegetable land. Plant Soil. 2010, 337, 137–150. [Google Scholar] [CrossRef]
- Allison, S.D.; Hanson, C.A.; Treseder, K.K. Nitrogen fertilization reduces diversity and alters community structure of active fungi in boreal ecosystems. Soil Biol. Biochem. 2007, 39, 1878–1887. [Google Scholar] [CrossRef]
- Pester, M.; Maixner, F.; Berry, D.; Rattei, T.; Koch, H.; Lücker, S.; Nowka, B.; Richter, A.; Spieck, E.; Lebedeva, E.; et al. NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira. Environ. Microbiol. 2014, 16, 3055–3071. [Google Scholar] [CrossRef]
- Gruberdorninger, C.; Pester, M.; Kitzinger, K.; Savio, D.F.; Loy, A.; Rattei, T.; Wagner, M.; Daims, H. Functionally relevant diversity of closely related Nitrospira in activated sludge. ISME J. 2015, 9, 643–655. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Land Use Pattern | Forest Age (Year) | Slope (°) | Stand Density (ind·hm−2) | Crown Density | Mean DBH (cm) | Herb Coverage | Main Herb Under the Forest |
|---|---|---|---|---|---|---|---|
| QM | 61 | 32 | 2357 | 90% | 20.18 | 60% | Vicia unijuga, Gueldenstaedtia verna, Atractylodes Lancea, Schisandra chinensis, Asparagus oligoclonos, Corylus mandshurica, Celastrus orbiculatus, Lespedeza bicolor |
| LG | 28 | 36 | 2100 | 80% | 12.68 | 90% | Rubus crataegifolius, Rubus idaeus, Asparagus oligoclonos, Schisandra chinensis, Athyrium brevifrons, Menispermum dauricum |
| PK | 61 | 27 | 1800 | 70% | 21.94 | 30% | Polygonatum odoratum, Vitis amurensis, Athyrium brevifrons, Menispermum dauricum, Vicia unijuga, Asparagus oligoclonos, Gueldenstaedtia verna, Atractylodes Lancea, Schisandra chinensis, Asparagus oligoclonos |
| SL | 32 | 70% | 60% | Rubus crataegifolius, Schisandra chinensis, Rubus idaeus, Asparagus oligoclonos, Atractylodes lancea, Athyrium brevifrons, Menispermum dauricum | |||
| ZM | 32 | 3333 |
| Land Use Pattern | pH | Total C (g·kg−1) | Total N (g·kg−1) | C/N Ratio | AN (mg·kg−1) | NH4+−N (mg·kg−1) | NO3—N (mg·kg−1) |
|---|---|---|---|---|---|---|---|
| QM | 5.99 ± 0.15ab | 57.74 ± 15.68 a | 4.40 ± 1.11 a | 13.09 ± 0.39 a | 33.63 ± 7.19 a | 4.42 ± 0.50 b | 17.09 ± 3.69 ab |
| LG | 5.57 ± 0.24c | 52.24 ± 3.36 ab | 3.89 ± 0.27 a | 13.43 ± 0.20 a | 32.05 ± 3.61 a | 6.33 ± 1.44 a | 26.44 ± 9.88 a |
| PK | 5.54 ± 0.11c | 20.08 ± 4.01 c | 1.53 ± 0.32 c | 13.24 ± 1.88 a | 12.96 ± 2.56 c | 7.68 ± 1.45 a | 24.17 ± 2.81 a |
| SL | 6.29 ± 0.16a | 38.27 ± 5.49 b | 2.79 ± 0.42 b | 13.71 ± 0.31 a | 23.50 ± 5.87 b | 3.78 ± 0.15 b | 9.58 ± 0.68 b |
| ZM | 5.85 ± 0.16bc | 17.46 ± 2.33 c | 1.31 ± 0.20 c | 13.39 ± 0.28 a | 11.90 ± 1.33 c | 3.94 ± 0.77 b | 19.53 ± 6.47 ab |
| Stand Type | No. of Sequences | OTU Number(P) | Shannon Index | Chao1 Index | ACE Index | Simpson Index |
|---|---|---|---|---|---|---|
| QM | 60,778 | 2940 | 9.37 ± 0.06 b | 2863.65 ± 187.86 b | 2958.54 ± 266.53 b | 0.9923 ± 0.0007 c |
| LG | 58,098 | 2773 | 10.04 ± 0.13 a | 3321.32 ± 275.92 a | 3548.18 ± 311.62 a | 0.9964 ± 0.0008 ab |
| PK | 63,395 | 2418 | 10.00 ± 0.09 a | 3288.14 ± 109.13 ab | 3495.91 ± 135.43 a | 0.9967 ± 0.0004 a |
| SL | 63,671 | 2871 | 9.79 ± 0.19 a | 3172.18 ± 333.52 ab | 3385.38 ± 366.98 ab | 0.9953 ± 0.0007 ab |
| ZM | 57,095 | 1965 | 9.37 ± 0.36 b | 2072.43 ± 265.19 c | 2114.55 ± 292.22 c | 0.9949 ± 0.0014 b |
| pH | TC | TN | C/N | AN | NH4+−N | NO3−−N | |
|---|---|---|---|---|---|---|---|
| Simpson | 0.688 ** | 0.593 * | 0.583 * | 0.071 | 0.585 * | −0.723 ** | −0.484 |
| Chao1 | 0.140 | 0.686 ** | 0.683 ** | 0.043 | 0.691 ** | 0.147 | −0.229 |
| ACE | 0.142 | 0.710 ** | 0.703 ** | 0.105 | 0.716 ** | 0.139 | −0.224 |
| Shannon | 0.513 | 0.729 ** | 0.724 ** | 0.051 | 0.727 ** | −0.316 | −0.386 |
| Bacteria Group | pH | TC | TN | AN | C/N | NH4+–N | NO3−–N |
|---|---|---|---|---|---|---|---|
| Phylum | |||||||
| Proteobacteria | −0.171 | 0.341 | 0.352 | 0.299 | −0.105 | 0.515 * | 0.016 |
| Actinobacteria | 0.120 | 0.061 | 0.059 | 0.119 | 0.005 | −0.471 | 0.075 |
| Acidobacteria | 0.108 | 0.236 | 0.242 | 0.198 | –0.103 | 0.366 | −0.161 |
| Chloroflexi | −0.129 | −0.491 | −0.518 * | −0.442 | 0.334 | 0.296 | 0.137 |
| Gemmatimonadetes | −0.281 | −0.712 ** | −0.714 ** | −0.680 ** | 0.071 | 0.020 | 0.199 |
| Verrucomicrobia | 0.335 | 0.612 * | 0.612 * | 0.590 * | −0.063 | −0.184 | 0.060 |
| Bacteroidetes | 0.401 | 0.676 ** | 0.676 ** | 0.622 ** | 0.002 | −0.456 | −0.434 |
| Planctomycetes | 0.221 | −0.180 | −0.142 | −0.276 | −0.486 | −0.106 | −0.318 |
| Saccharibacteria | −0.218 | −0.501 | −0.496 | −0.511 | −0.035 | −0.203 | −0.079 |
| Nitrospirae | 0.415 | 0.590 * | 0.548 * | 0.582 * | 0.473 | −0.174 | −0.033 |
| Genus | |||||||
| Nitrobacter | −0.473 | 0.019 | 0.032 | 0.024 | –0.124 | 0.806 ** | 0.414 |
| Gemmatimonas | −0.196 | −0.565 * | −0.558 * | −0.549 * | –0.041 | −0.156 | 0.099 |
| Sphingomonas | 0.176 | −0.420 | −0.416 | −0.433 | −0.019 | −0.493 | −0.276 |
| Rhodoplanes | 0.232 | 0.491 | 0.469 | 0.514 * | 0.211 | 0.168 | −0.300 |
| Variibacter | −0.521 * | 0.034 | 0.041 | 0.048 | −0.041 | 0.800 ** | 0.341 |
| RB41 | 0.522 * | 0.527 * | 0.521 * | 0.526 * | 0.064 | −0.213 | −0.506 |
| Pseudolabrys | −0.739 ** | −0.270 | −0.259 | −0.276 | −0.121 | 0.807 ** | 0.638 ** |
| Mycobacterium | −0.486 | 0.032 | 0.032 | 0.053 | −0.019 | 0.670 ** | 0.431 |
| Actinoplanes | 0.643 ** | 0.021 | 0.014 | 0.055 | 0.111 | −0.723 ** | −0.603 * |
| Solirubrobacter | 0.622 * | 0.507 | 0.499 | 0.537 * | 0.087 | −0.506 | −0.374 |
| Haliangium | −0.237 | 0.514 | 0.511 | 0.532 * | −0.041 | 0.189 | 0.213 |
| Candidatus-Solibacter | −0.632 * | −0.394 | −0.356 | −0.409 | −0.478 | 0.722 ** | 0.481 |
| Pseudonocardia | 0.568 * | 0.511 | 0.495 | 0.559 * | 0.138 | −0.490 | −0.460 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.-J.; Deng, J.-J.; Yin, Y.; Qin, S.-J.; Zhu, W.-X.; Zhou, Y.-B.; Wang, B.; Ruan, H.; Jin, L. Bacterial Community Changes Associated with Land Use Type in the Forest Montane Region of Northeast China. Forests 2020, 11, 40. https://doi.org/10.3390/f11010040
Wu S-J, Deng J-J, Yin Y, Qin S-J, Zhu W-X, Zhou Y-B, Wang B, Ruan H, Jin L. Bacterial Community Changes Associated with Land Use Type in the Forest Montane Region of Northeast China. Forests. 2020; 11(1):40. https://doi.org/10.3390/f11010040
Chicago/Turabian StyleWu, Shi-Jun, Jiao-Jiao Deng, You Yin, Sheng-Jin Qin, Wen-Xu Zhu, Yong-Bin Zhou, Bing Wang, Honghua Ruan, and Long Jin. 2020. "Bacterial Community Changes Associated with Land Use Type in the Forest Montane Region of Northeast China" Forests 11, no. 1: 40. https://doi.org/10.3390/f11010040
APA StyleWu, S.-J., Deng, J.-J., Yin, Y., Qin, S.-J., Zhu, W.-X., Zhou, Y.-B., Wang, B., Ruan, H., & Jin, L. (2020). Bacterial Community Changes Associated with Land Use Type in the Forest Montane Region of Northeast China. Forests, 11(1), 40. https://doi.org/10.3390/f11010040