Identification of Reference Genes for Quantitative Gene Expression Studies in Pinus massoniana and Its Introgression Hybrid

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
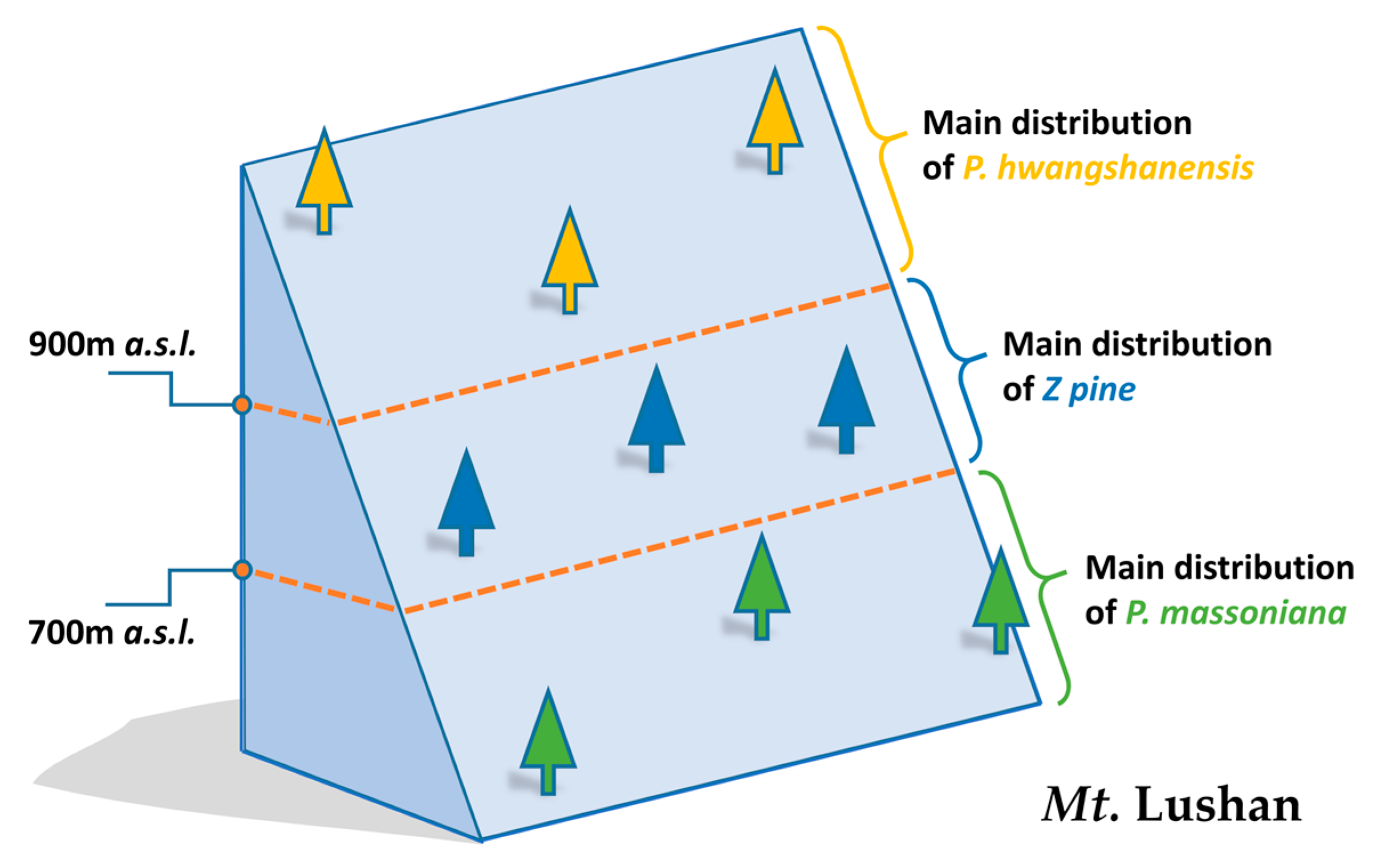
2.1. Preparation of Samples
2.2. Candidate Reference Gene Selection and Primer Design
2.3. RNA Extraction, cDNA Template Preparation, and Quantitative Real-Time PCR (qRT-PCR)
2.4. Stability Evaluation of Candidate Reference Genes
3. Results
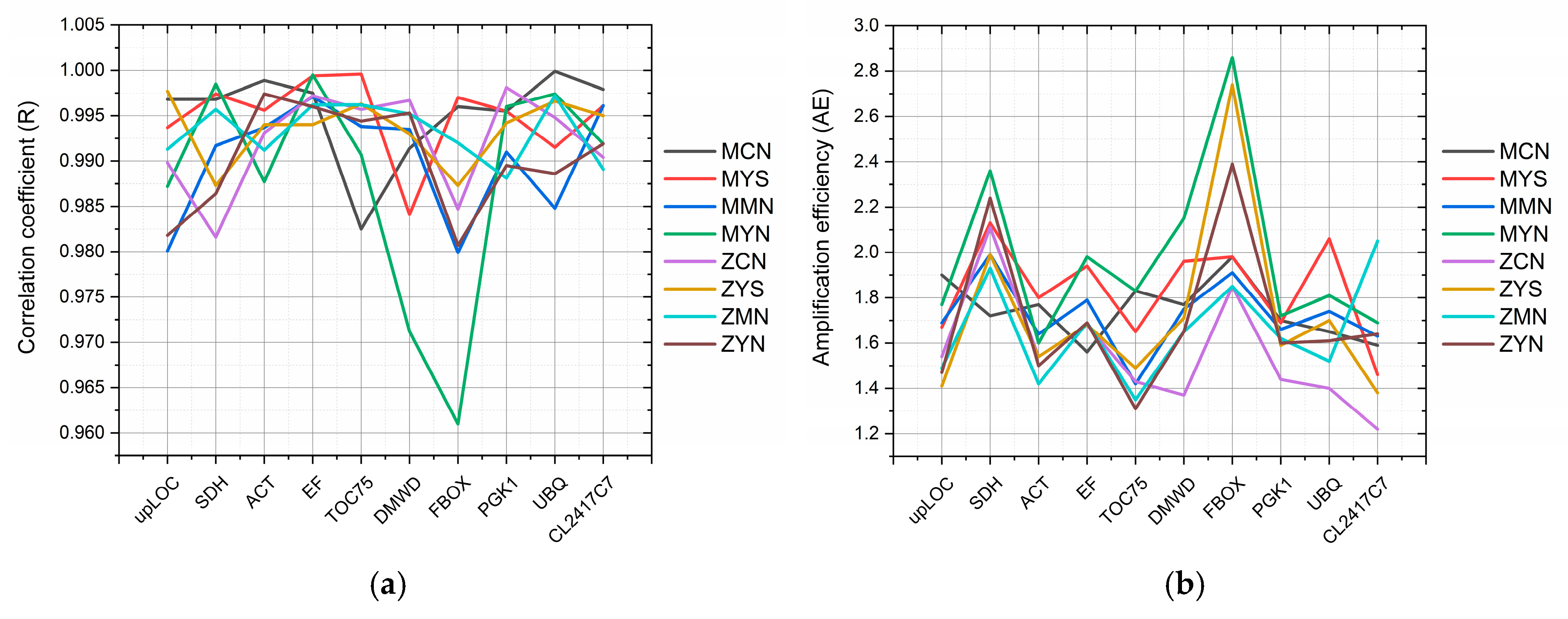
3.1. Reference Gene Evaluation
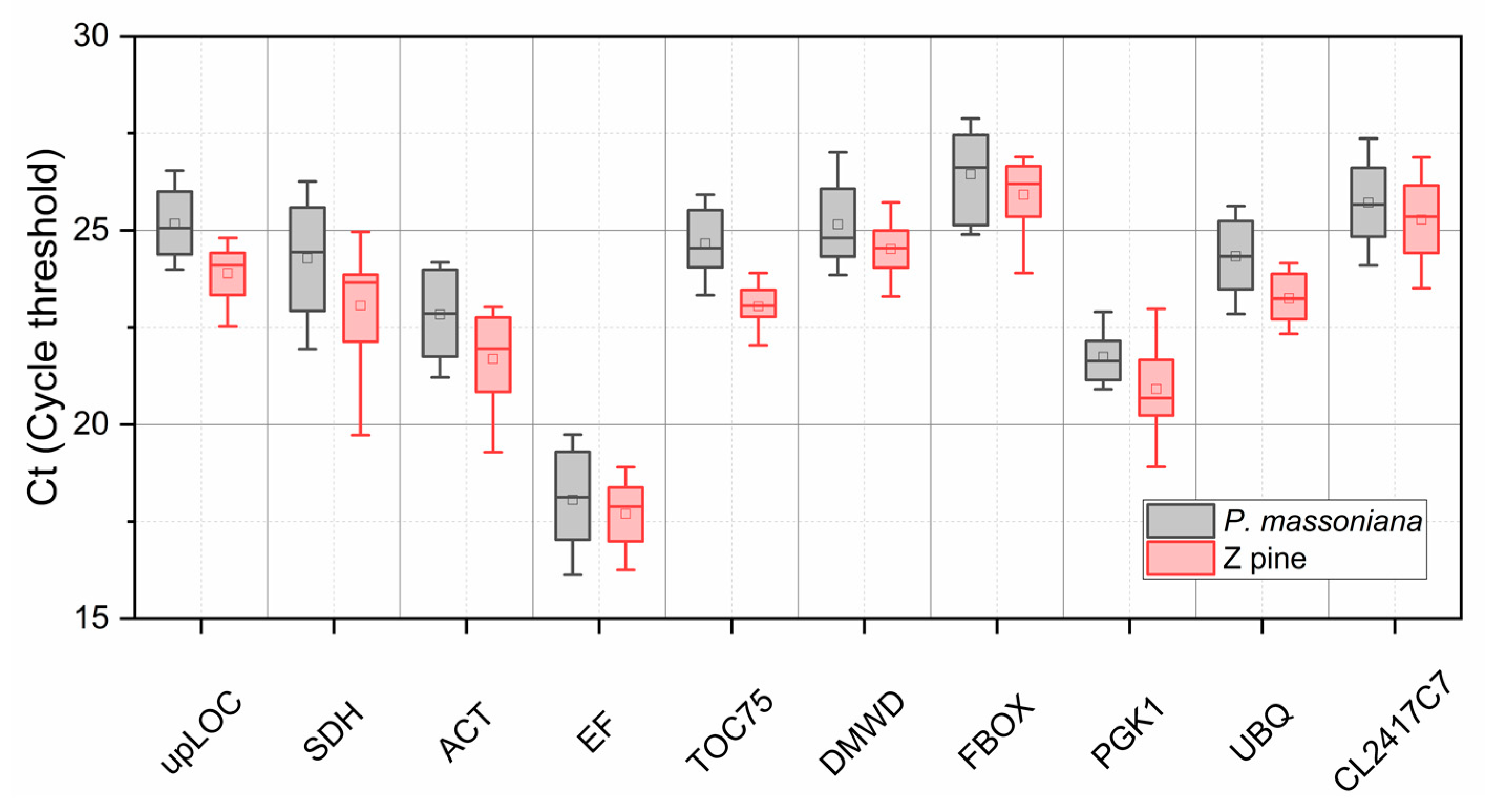
3.2. Z pine Has a Higher Expression Abundance than P. massoniana Across Ten Candidate Genes
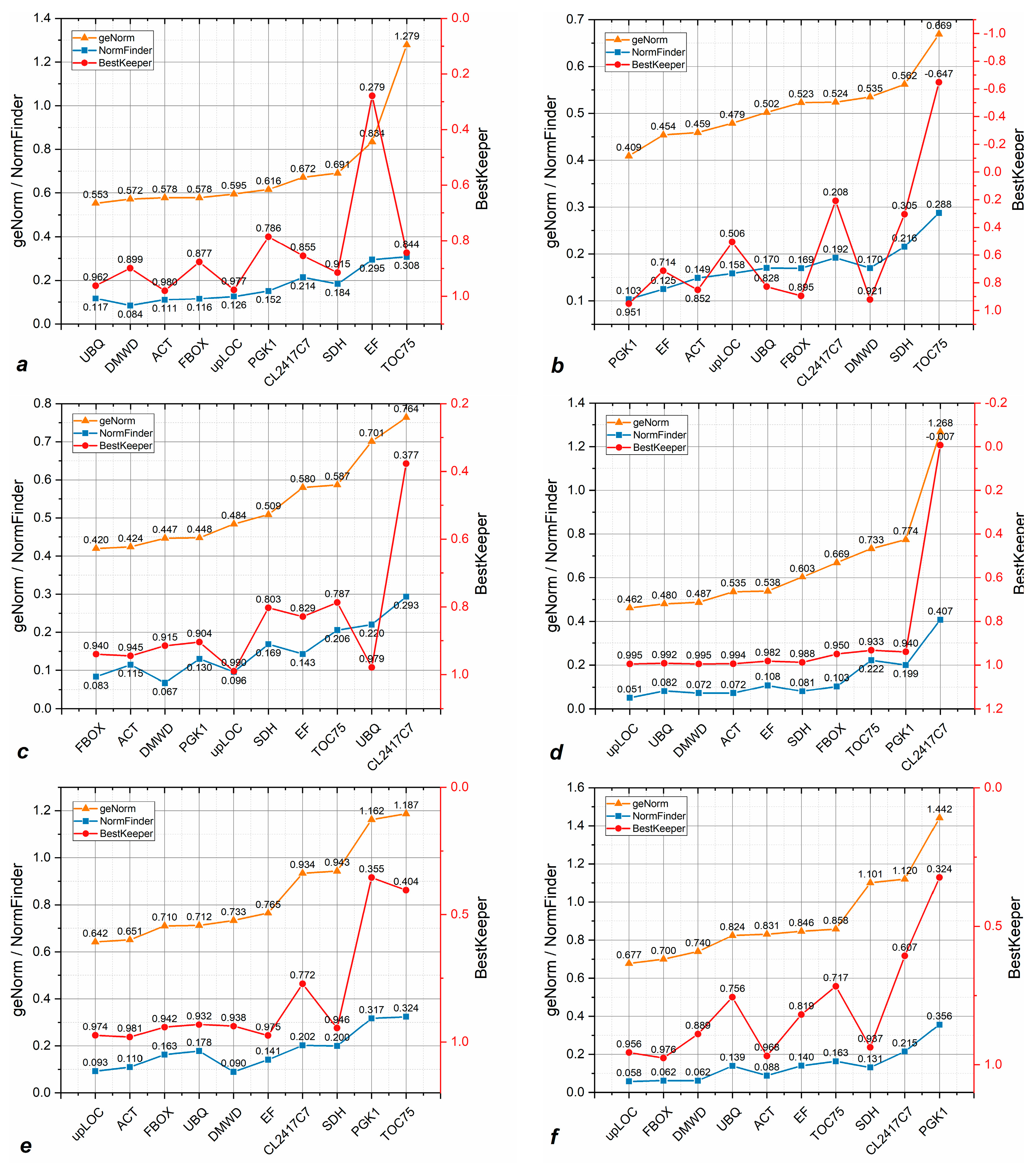
3.3. Candidate Reference Genes Show Variable Stability Across Sample Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mills, J.S.; White, R. Natural resins of art and archaeology their sources, chemistry, and identification. Stud. Conserv. 1977, 22, 12–31. [Google Scholar]
- Yang, N.; Liu, L.; Tao, W.; Duan, J.; Tian, L. Diterpenoids from Pinus massoniana resin and their cytotoxicity against A431 and A549 cells. Phytochemistry 2010, 71, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Editorial Board of Flora of China, Chinese Academy of Science. Flora of China, 1st ed.; Science Press: Beijing, China, 1978; Volume 7, pp. 263, 266. (In Chinese) [Google Scholar]
- Luo, S.; Zou, H.; Liang, S. Study on the introgressive hybridization between Pinus hwangshanensis and P. massoniana. Sci. Silvae Sin. 2001, 37, 118–122. (In Chinese) [Google Scholar]
- Zhai, D.; He, Z.; Feng, J.; Zheng, Y. Study on introgression between Pinus hwangshanensis and Pinus massoniana by using inter-simple sequence repeat marker (ISSR). For. Sci. Technol. 2012, 37, 4–6. (In Chinese) [Google Scholar]
- Li, S.; Chen, Y.; Gao, H.; Yin, T. Potential chromosomal introgression barriers revealed by linkage analysis in a hybrid of Pinus massoniana and P. hwangshanensis. BMC Plant Biol. 2010, 10. [Google Scholar] [CrossRef]
- Mo, J.; Xu, J.; Cao, Y.; Yang, L.; Yin, T.; Hua, H.; Zhao, H.; Guo, Z.; Yang, J.; Shi, J. Pinus massoniana introgression hybrids display differential expression of reproductive genes. Forests 2019, 10, 230. [Google Scholar] [CrossRef]
- Weis, J.H.; Tan, S.S.; Martin, B.K.; Wittwer, C.T. Detection of rare mRNAs via quantitative RT-PCR. Trends Genet. 1992, 8, 263–264. [Google Scholar] [CrossRef]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR–a perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. BioTechniques 2005, 39, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Karge, W.H.; Schaefer, E.J.; Ordovas, J.M. Quantification of mRNA by polymerase chain reaction (PCR) using an internal standard and a nonradioactive detection method. Methods Mol. Biol. 1998, 110, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xie, W.; Wang, S.; Wu, Q.; Yang, N.; Yang, X.; Pan, H.; Zhou, X.; Bai, L.; Xu, B. Reference gene selection for qRT-PCR analysis in the sweetpotato whitefly, Bemisia tabaci (Hemiptera: Aleyrodidae). PLoS ONE 2013, 8, e53006. [Google Scholar] [CrossRef] [PubMed]
- Robledo, D.; Hernández-Urcera, J.; Cal, R.M.; Pardo, B.G.; Sánchez, L.; Martínez, P.; Viñas, A. Analysis of qPCR reference gene stability determination methods and a practical approach for efficiency calculation on a turbot (Scophthalmus maximus) gonad dataset. BMC Genom. 2014, 15, 648. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Ma, J.; Guo, Q.; Li, X.; Wang, H.; Lu, M. Selection of reference genes for quantitative real-time PCR in bamboo (Phyllostachys edulis). PLoS ONE 2013, 8, e56573. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–11. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Dodd, A.; Lai, D.; Mcnabb, W.C.; Love, D.R. Validation of zebrafish (Danio rerio) reference genes for quantitative real-time RT-PCR normalization. Acta Biochim. Biophys. Sin. 2007, 39, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Ponton, F.; Chapuis, M.; Pernice, M.; Sword, G.A.; Simpson, S.J. Evaluation of potential reference genes for reverse transcription-qPCR studies of physiological responses in Drosophila melanogaster. J. Insect Physiol. 2011, 57, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Plusquin, M.; DeGheselle, O.; Cuypers, A.; Geerdens, E.; Van Roten, A.; Artois, T.; Smeets, K. Reference genes for qPCR assays in toxic metal and salinity stress in two flatworm model organisms. Ecotoxicology 2012, 21, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Zhou, L.; Lim, Q.; Zou, R.; Stephanopoulos, G.; Too, H. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol. Biol. 2011, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, J.; Huang, S.; Guo, T.; Deng, L.; Hua, W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 2014, 538, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Behringer, D.; Zimmermann, H.; Ziegenhagen, B.; Liepelt, S. Differential gene expression reveals candidate genes for drought stress response in Abies alba (Pinaceae). PLoS ONE 2015, 10, e124564. [Google Scholar] [CrossRef]
- de Vega-Bartol, J.J.; Santos, R.R.; Simões, M.; Miguel, C.M. Normalizing gene expression by quantitative PCR during somatic embryogenesis in two representative conifer species: Pinus pinaster and Picea abies. Plant Cell Rep. 2013, 32, 715–729. [Google Scholar] [CrossRef]
- Ren, R.; Huang, F.; Gao, R.; Dong, X.; Peng, J.; Cao, F.; Li, M. Selection and validation of suitable reference genes for RT-qPCR analysis in dove tree (Davidia involucrata Baill.). Trees 2019, 1–13. [Google Scholar] [CrossRef]
- Chao, W.S.; Doğramaci, M.; Foley, M.E.; Horvath, D.P.; Anderson, J.V. Selection and validation of endogenous reference genes for qRT-PCR analysis in leafy spurge (Euphorbia esula). PLoS ONE 2012, 7, e42839. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Z.; Hu, Y.; Tan, J.; Jia, J.; Xu, H.; Chen, X. Reference genes selection for quantitative gene expression studies in Pinus massoniana L. Trees 2016, 30, 685–696. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, Q.; Dong, H.; Zhou, Z.; Hao, Y.; Chen, X.; Xu, L. Selection of reference genes for real-time quantitative PCR in Pinus massoniana post nematode inoculation. PLoS ONE 2016, 11, e147224. [Google Scholar] [CrossRef] [PubMed]
- Gibson, U.E.; Heid, C.A.; Williams, P.M. A novel method for real time quantitative RT-PCR. Genome Res. 1996, 6, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, H.; Shan, T.; Pang, S. Selection of reference genes for real-time RT-PCR normalization in brown alga Undaria pinnatifida. J. Appl. Phycol. 2019, 31, 787–793. [Google Scholar] [CrossRef]
- Wang, B.; Du, H.; Yao, Z.; Ren, C.; Ma, L.; Wang, J.; Zhang, H.; Ma, H. Validation of reference genes for accurate normalization of gene expression with quantitative real-time PCR in Haloxylon ammodendron under different abiotic stresses. Physiol. Mol. Biol. Plants 2018, 24, 455–463. [Google Scholar] [CrossRef]
- Gunning, P.W.; Ghoshdastider, U.; Whitaker, S.; Popp, D.; Robinson, R.C. The evolution of compositionally and functionally distinct actin filaments. J. Cell Sci. 2015, 128, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, Y.; Ban, L.; Wang, Z.; Feng, G.; Li, J.; Gao, H. Selection of reliable reference genes for quantitative real-time RT-PCR in alfalfa. Genes Genet. Syst. 2015, 90, 175–180. [Google Scholar] [CrossRef]
- Chong, G.; Kuo, F.; Tsai, S.; Lin, C. Validation of reference genes for cryopreservation studies with the gorgonian coral endosymbiont Symbiodinium. Sci. Rep. 2017, 7, 39396. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, J.; Ji, A.; Zhu, Y.; Zhang, X.; Hu, Y.; Song, J.; Chen, S. Genome-wide selection of superior reference genes for expression studies in Ganoderma lucidum. Gene 2015, 574, 352–358. [Google Scholar] [CrossRef]
- Duan, Z.; Lamendola, D.E.; Yusuf, R.Z.; Penson, R.T.; Preffer, F.I.; Seiden, M.V. Overexpression of human phosphoglycerate kinase 1 (PGK1) induces a multidrug resistance phenotype. Anticancer Res. 2002, 22, 1933–1941. [Google Scholar]
- Wang, J.; Ying, G.; Wang, J.; Jung, Y.; Lu, J.; Zhu, J.; Pienta, K.J.; Taichman, R.S. Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor microenvironment in prostate cancer progression. Cancer Res. 2010, 70, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, V.R.; Whistler, T.; Janna’R, M.; Unger, E.R.; Rajeevan, M.S. Identification of phosphoglycerate kinase 1 (PGK1) as a reference gene for quantitative gene expression measurements in human blood RNA. BMC Res. Notes 2011, 4, 324. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Bächner, D.; Coerwinkel, M.; Wormskamp, N.; Hameister, H.; Wieringa, B. Structural organization and developmental expression pattern of the mouse WD-repeat gene DMR-N9 immediately upstream of the myotonic dystrophy locus. Hum. Mol. Genet. 1995, 4, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.J.; McCurrach, M.; Rundle, S.A.; Harley, H.G.; Crow, S.R.; Sohn, R.; Thirion, J.; Hamshere, M.G.; Buckler, A.J.; Harper, P.S. Genomic organization and transcriptional units at the myotonic dystrophy locus. Genomics 1993, 18, 673–679. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Sample Code | Sample Detail | Environmental Information |
|---|---|---|---|
| P. massoniana | MCN | cone | 116.04 E, 29.50 N, 78 m (a.s.l. 1) |
| MYS | young stem | ||
| MMN | mature needle | ||
| MYN | young needle | VETC 2 | |
| Z pine | ZCN | cone | 115.98 E, 29.54 N, 730 m (a.s.l. 1) |
| ZYS | young stem | ||
| ZMN | mature needle | ||
| ZYN | young needle | VETC 2 |
| Gene Code | Gene Name | Primer Sequence (5′–3′) | Product Size (bp) | Accession Number |
|---|---|---|---|---|
| upLOC | uncharacterized protein LOC103705956 | F: CACCTTCCGCTTCTTCTA | 95 | MN172175 |
| R: TTGAATGACTCTTCTTGATGG | ||||
| SDH | succinate dehydrogenase | F: AGACCTTGATGTTAAGAATGC | 127 | MN172176 |
| R: TTCCTGTCCAGCCTTATC | ||||
| ACT | actin | F: ATTCTCCTCACCGAAGCACC | 188 | MN172177 |
| R: ATGGGAACTGTGTGGCTGAC | ||||
| EF | elongation factor | F: TTGGGACTGTGCCTGTTGGT | 206 | MN172178 |
| R: AGATGCCACGTAACCACGCT | ||||
| TOC75 | protein TOC75-3 | F: TGAAGGAGGAATAATTGTTGAG | 128 | MN172179 |
| R: CACCTGGCTGAATTGAAG | ||||
| DMWD | dystrophia myotonica WD repeat-containing protein | F: GGACTTGGTGATGGATGA | 133 | MN172180 |
| R: TGGAGCAGATTGAAGAAGA | ||||
| FBOX | F-box family protein | F: TTGCTTCCTTGTAACATCTG | 81 | MN172181 |
| R: CACCTCTTAATATCCAACTCTAC | ||||
| PGK1 | phosphoglycerate kinase 1 | F: TGCCAAGGTTATTCTTACAAG | 136 | MN172182 |
| R: CGGTCCAATACAATCATCTG | ||||
| UBQ | ubiquitin | F: CTTATGACGATGTTGTGGAG | 122 | MN172183 |
| R: TCTGAATCTGATGGCTTGA | ||||
| CL2417C7 | unknown | F: TCAATCTAAGCCTTCTAGCA | 87 | MN172184 |
| R: TCCGTCATACCAACATCTT |
| Sample | Recommended | NOT Recommended |
|---|---|---|
| MCN + ZCN | ACT | TOC75, EF |
| MYS + ZYS | PGK1 | TOC75 |
| MMN + ZMN | upLOC, ACT, DMWD | CL2417C7 |
| MYN + ZYN | upLOC | CL2417C7 |
| M_ALL | upLOC, ACT | TOC75 |
| Z_ALL | upLOC | PGK1 |
| MZ_ALL | upLOC, ACT | PGK1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, J.; Xu, J.; Jin, W.; Yang, L.; Yin, T.; Shi, J. Identification of Reference Genes for Quantitative Gene Expression Studies in Pinus massoniana and Its Introgression Hybrid. Forests 2019, 10, 787. https://doi.org/10.3390/f10090787
Mo J, Xu J, Jin W, Yang L, Yin T, Shi J. Identification of Reference Genes for Quantitative Gene Expression Studies in Pinus massoniana and Its Introgression Hybrid. Forests. 2019; 10(9):787. https://doi.org/10.3390/f10090787
Chicago/Turabian StyleMo, Jiaxing, Jin Xu, Wenjing Jin, Liwei Yang, Tongming Yin, and Jisen Shi. 2019. "Identification of Reference Genes for Quantitative Gene Expression Studies in Pinus massoniana and Its Introgression Hybrid" Forests 10, no. 9: 787. https://doi.org/10.3390/f10090787
APA StyleMo, J., Xu, J., Jin, W., Yang, L., Yin, T., & Shi, J. (2019). Identification of Reference Genes for Quantitative Gene Expression Studies in Pinus massoniana and Its Introgression Hybrid. Forests, 10(9), 787. https://doi.org/10.3390/f10090787