Kinetics of Water Vapor Sorption in Wood Cell Walls: State of the Art and Research Needs




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of Experimental Methods for Determining Sorption Kinetics
2.1. Conditioning Chamber
2.2. Desiccator with Aqueous Solution
2.3. Quartz Helix Vacuum Balance
2.4. Automated Vacuum Balance
2.5. Automated Continuous-Flow Sorption Balance
3. Summary of Main Experimental Observations on Sorption Kinetics
3.1. Wood Species, Anatomy, and Specimen Geometry
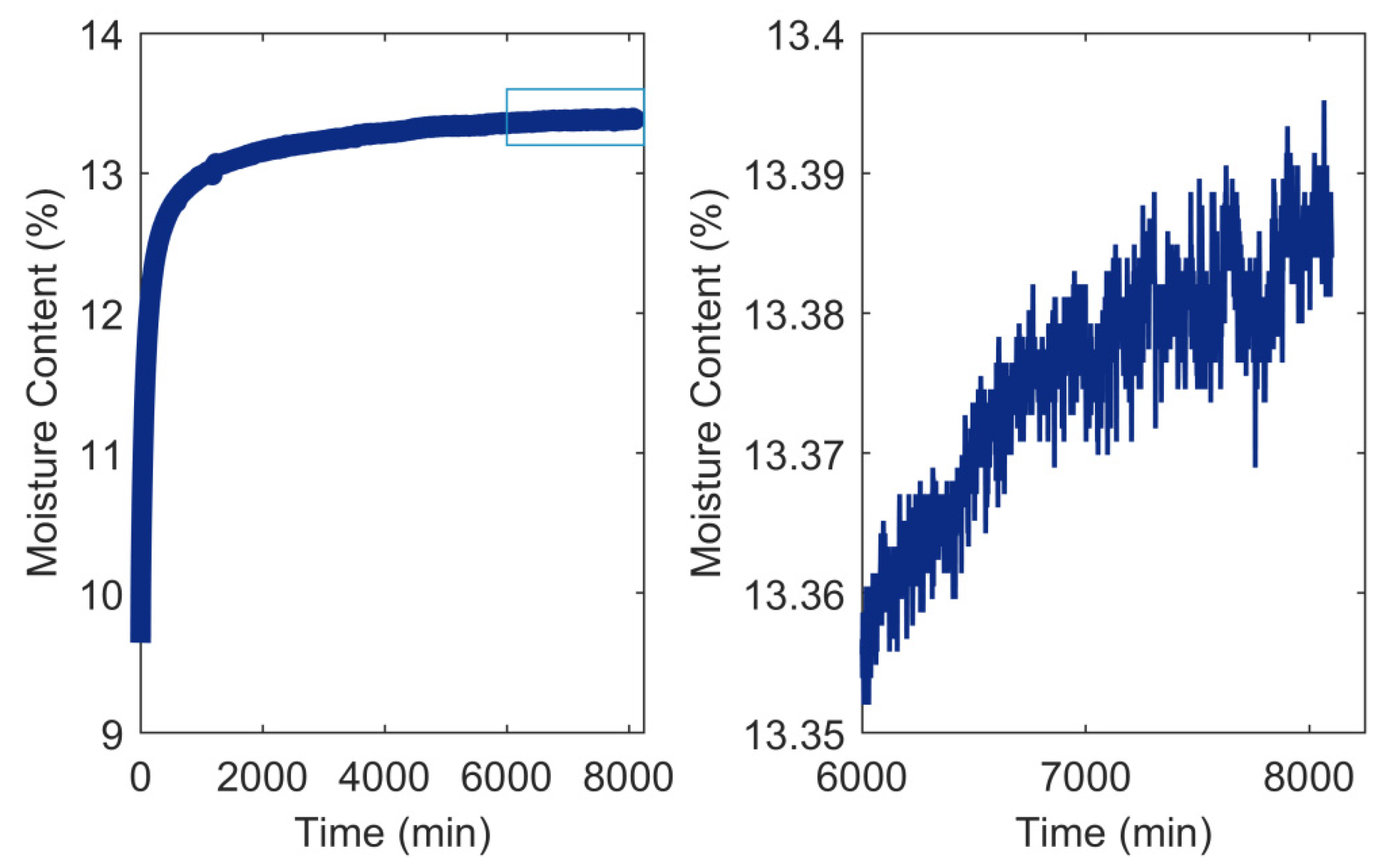
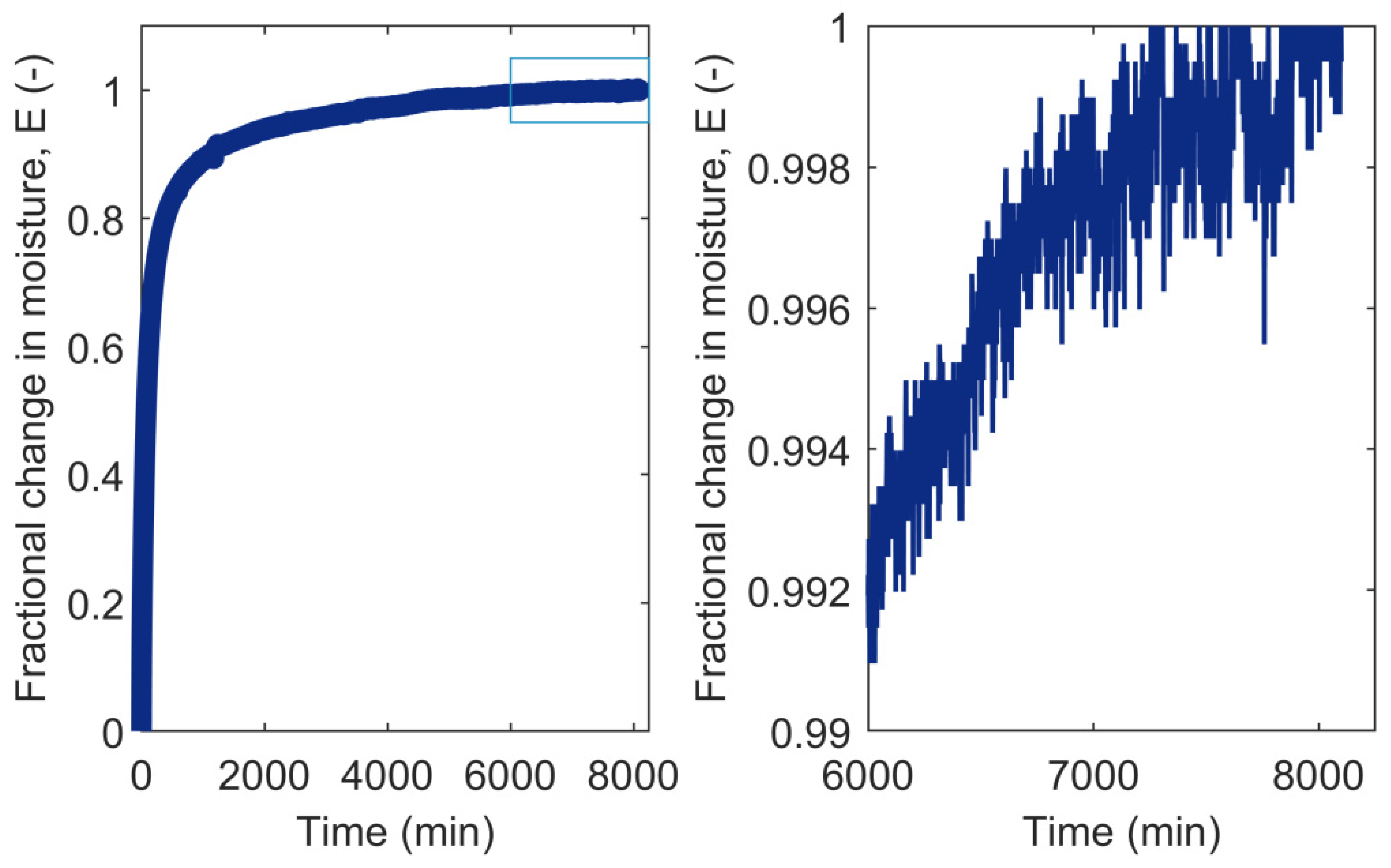
3.2. RH Level, RH Step Size, and Conditioning Time
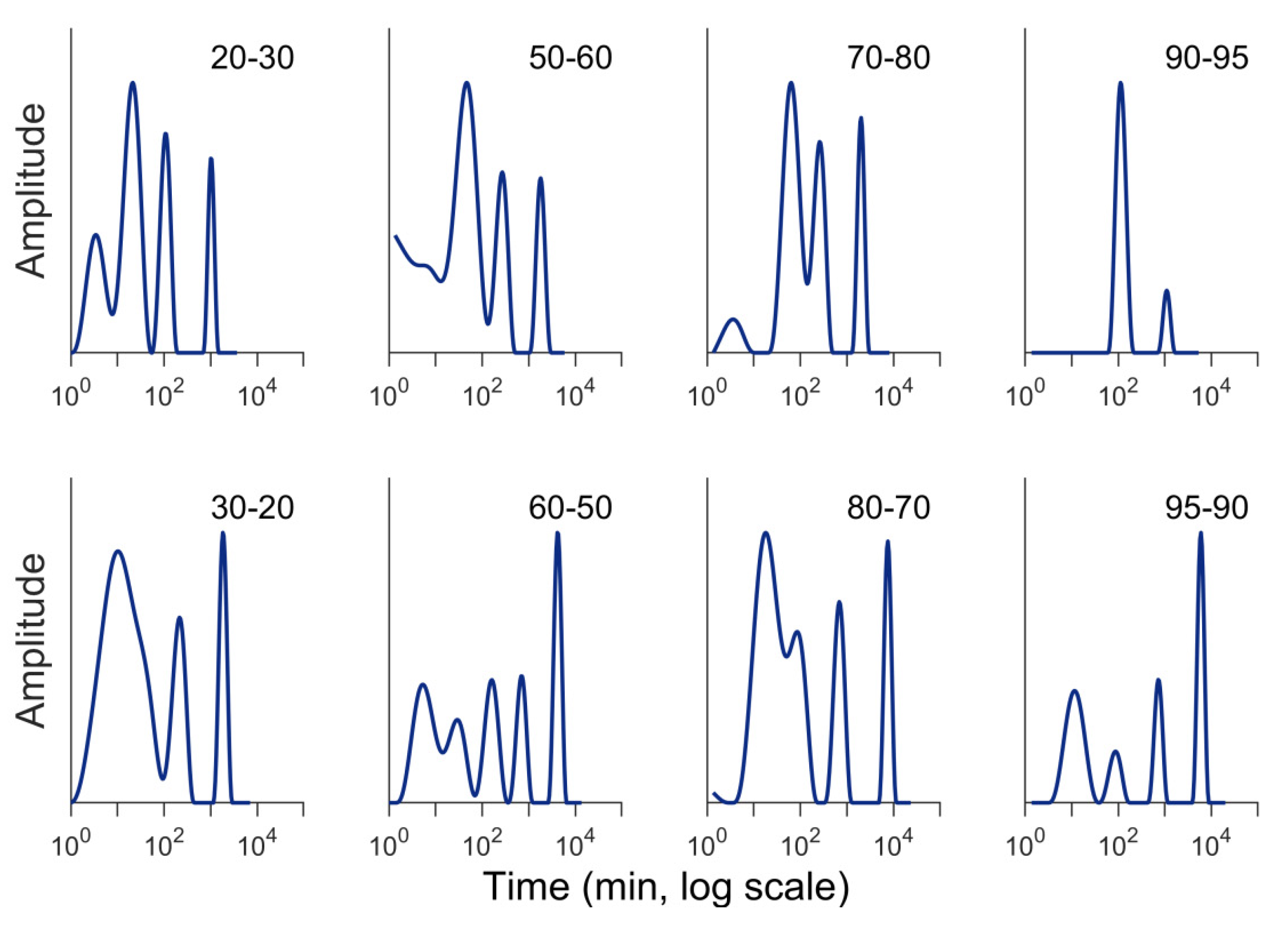
3.3. Absorption Versus Desorption
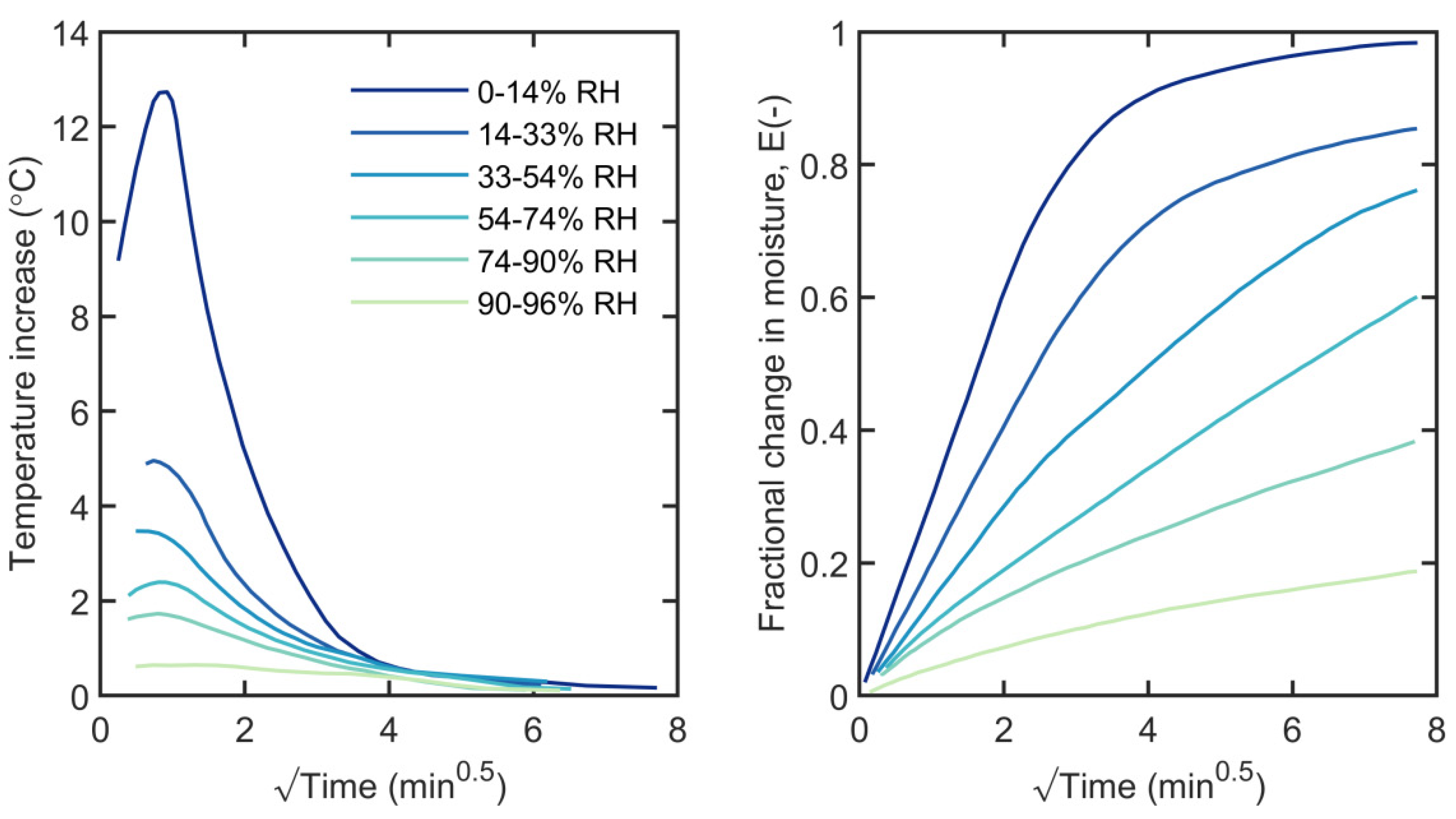
3.4. Temperature Changes During Sorption
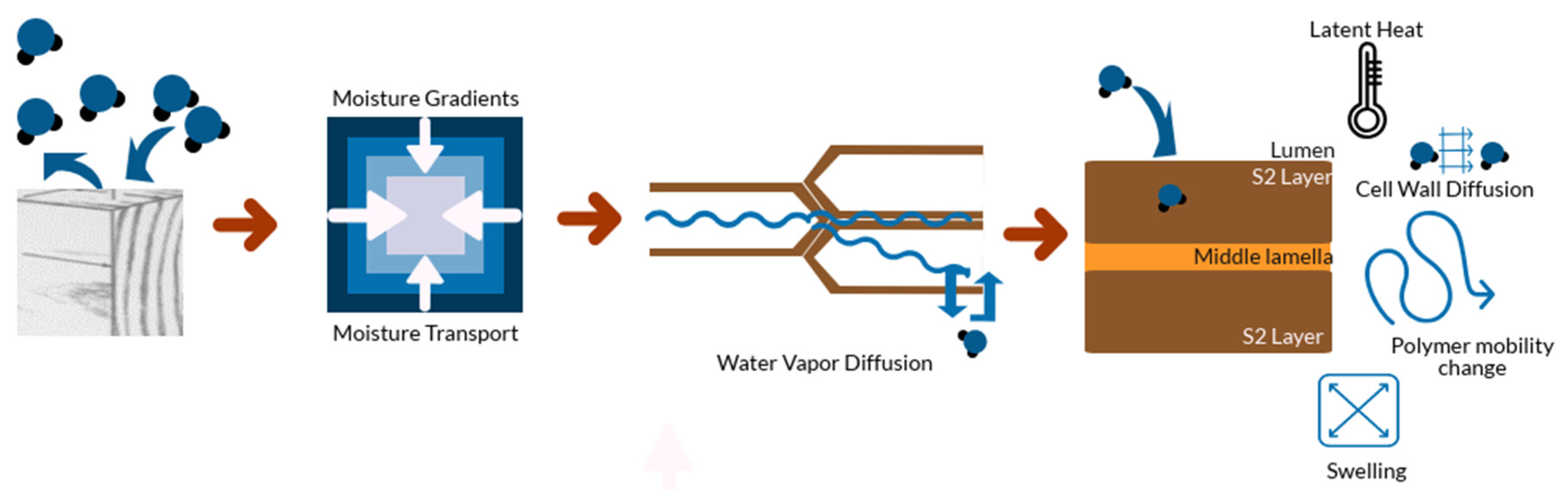
4. Physical Phenomena Involved in the Sorption Process
4.1. External Resistance to Vapor Transfer
4.2. Temperature Change
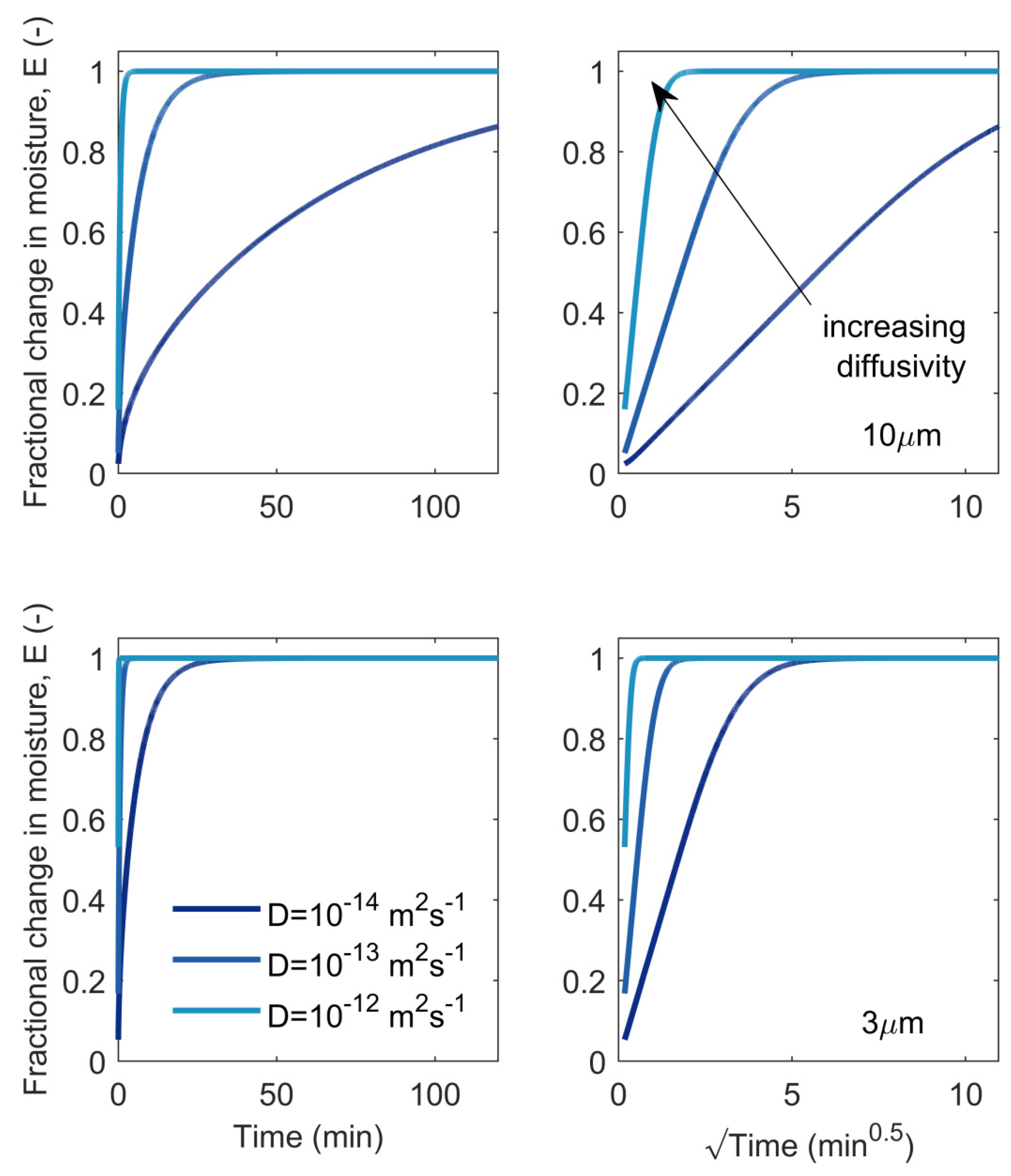
4.3. Diffusion of Bound Water within Cell Walls
4.4. Dimensional Changes and Associated Stresses
4.5. Polymer Mobility
5. Theoretical Models Describing Sorption Kinetics
5.1. Fickian Diffusion Model
5.2. Swelling Stress Models
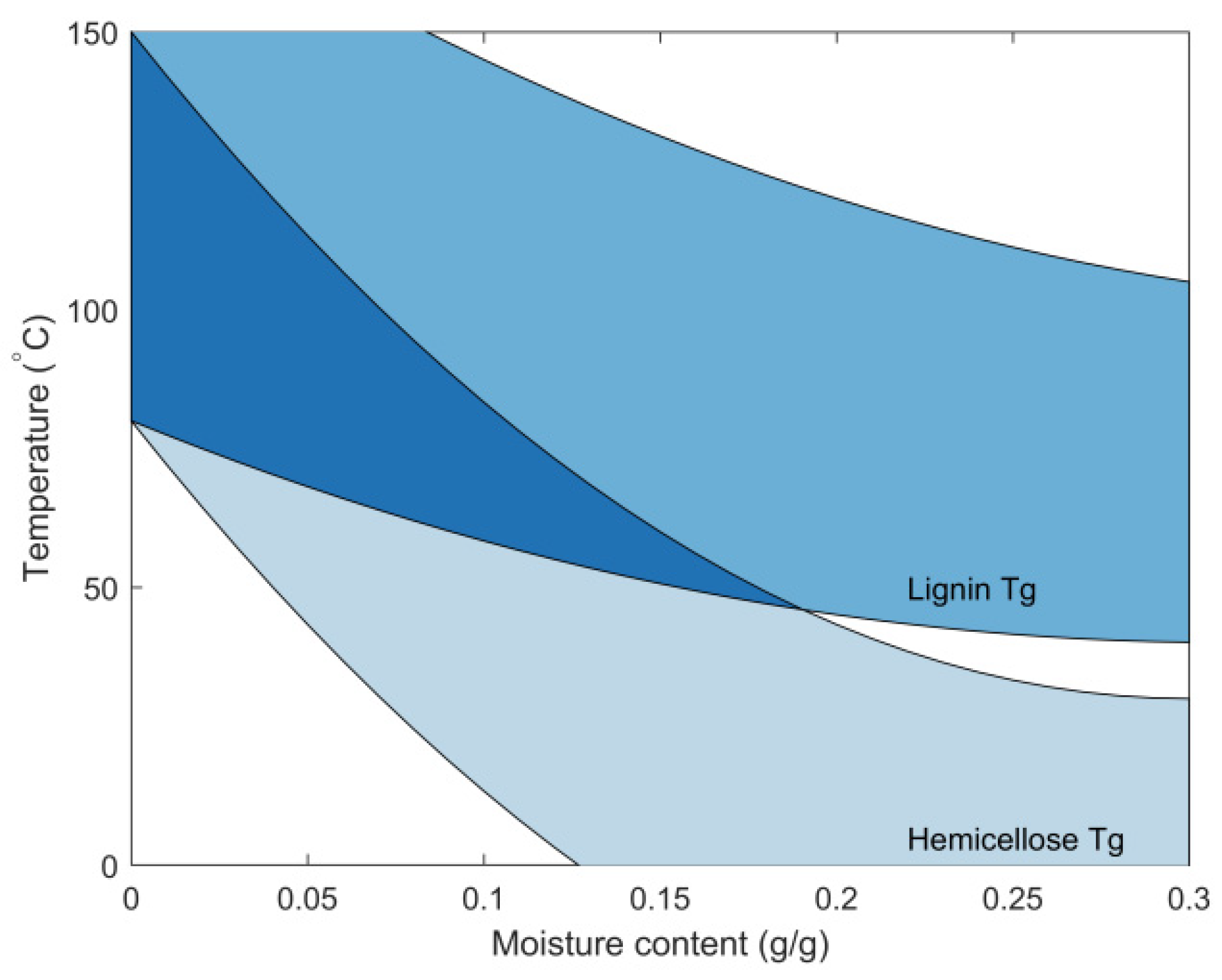
5.3. Thermally Limited Moisture Transport Model
5.4. Parallel Exponential Kinetics (PEK) Model
6. Conclusions and Perspective on Research Needs
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Green, M.C.; Karsh, J.E. The Case for Tall Wood Buildings—How Mass Timber Offers a Safe, Economical, and Environmental Friendly Alternative for Tall Building Structures; MGB Architecture + Design: Vancouver, BC, Canada, 2012; pp. 1–237. [Google Scholar]
- Iqbal, A. Recent developments in tall wood buildings. In Proceedings of the 1st International Conference on New Horizons in Green Civil Engineering, Victoria, BC, Canada, 25–27 April 2018; pp. 111–116. [Google Scholar]
- Jakes, J.E.; Arzola, X.; Bergman, R.; Ciesielski, P.; Hunt, C.G.; Rahbar, N.; Tshabalala, M.; Wiedenhoeft, A.C.; Zelinka, S.L. Not Just Lumber—Using Wood in the Sustainable Future of Materials, Chemicals, and Fuels. JOM 2016, 68, 2395–2404. [Google Scholar] [CrossRef]
- Thybring, E.E.; Kymäläinen, M.; Rautkari, L. Experimental techniques for characterising water in wood covering the range from dry to fully water-saturated. Wood Sci. Technol. 2018, 52, 297–329. [Google Scholar] [CrossRef]
- Fredriksson, M.; Thybring, E.E. Scanning or desorption isotherms? Characterising sorption hysteresis of wood. Cellulose 2018, 25, 4477–4485. [Google Scholar] [CrossRef]
- Hoffmeyer, P.; Engelund, E.T.; Thygesen, L.G. Equilibrium moisture content (EMC) in Norway spruce during the first and second desorptions. Holzforschung 2011, 65, 875–882. [Google Scholar] [CrossRef]
- Engelund, E.T.; Thygesen, L.G.; Svensson, S.; Hill, C.A.S. A critical discussion of the physics of wood-water interactions. Wood Sci. Technol. 2013, 47, 141–161. [Google Scholar] [CrossRef]
- Glass, S.V.; Boardman, C.R.; Thybring, E.E.; Zelinka, S.L. Quantifying and reducing errors in equilibrium moisture content measurements with dynamic vapor sorption (DVS) experiments. Wood Sci. Technol. 2018, 52, 909–927. [Google Scholar] [CrossRef]
- Stamm, A.J. Wood and Cellulose Science; The Ronald Press Company: New York, NY, USA, 1964. [Google Scholar]
- Skaar, C. Wood-Water Relations; Springer: New York, NY, USA, 1988. [Google Scholar]
- Siau, J.F. Wood: Influence of Moisture on Physical Properties; Department of Wood Science and Forest Products, Virginia Polytechnic Institute and State University: Blacksburg, VA, USA, 1995. [Google Scholar]
- Kiefer, S.; Robens, E. Some intriguing items in the history of volumetric and gravimetric adsorption measurements. J. Therm. Anal. Calorim. 2008, 94, 613–618. [Google Scholar] [CrossRef]
- Zeller, S.M. Humidity in Relation to Moisture Imbibition by Wood and to Spore Germination on Wood. Ann. Mo. Bot. Gard. 1920, 7, 51. [Google Scholar] [CrossRef]
- Glass, S.V.; Zelinka, S.L.; Johnson, J.A. Investigation of Historic Equilibrium Moisture Content Data from the Forest Products Laboratory; FPL-GTR-229; US Department of Agriculture, Forest Service, Forest Products Laboratory: Madison, WI, USA, 2014. [CrossRef]
- Hedlin, C.P. Sorption isotherms of twelve woods at subfreezing temperatures. For. Prod. J. 1968, 17, 43–48. [Google Scholar]
- Strømdahl, K. Water Sorption in Wood and Plant Fibres. Ph.D. Thesis, Technical University of Denmark, Lyngby, Denmark, 2000. [Google Scholar]
- Perre, P. Experimental device for the accurate determination of wood-water relations on micro-samples. Holzforschung 2007, 61, 419–429. [Google Scholar] [CrossRef]
- Zelinka, S.L.; Bourne, K.J.; Glass, S.V.; Boardman, C.R.; Lorenz, L.; Thybring, E.E. Apparatus for gravimetric measurement of moisture sorption isotherms for 1-100 g samples in parallel. Wood Fiber Sci. 2018, 50, 244–253. [Google Scholar] [CrossRef]
- Skaar, C.; Prichananda, C.; Davidson, R.W. Some aspects of moisture sorption dynamics in wood. Wood Sci. 1970, 2, 179–185. [Google Scholar]
- Comstock, G.L. Moisture diffusion coefficients in wood as calculated from adsorption, desorption, and steady state data. For. Prod. J. 1963, 13, 97–103. [Google Scholar]
- Choong, E.T.; Fogg, P.J. Moisture movement in six wood species. For. Prod. J. 1968, 18, 66–70. [Google Scholar]
- McNamara, W.S.; Hart, C.A. An analysis of interval and average diffusion coefficients for unsteady-state movement of moisture in wood. Wood Sci. 1971, 4, 37–45. [Google Scholar]
- Wadsö, L. Measurements of water vapour sorption in Wood Part 1. Instrumentation. Wood Sci. Technol. 1993, 27, 396–400. [Google Scholar] [CrossRef]
- Time, B. Hygroscopic Moisture Transport in Wood. Ph.D. Thesis, Norwegian University of Science and Technology, Trondheim, Norway, 1998. [Google Scholar]
- Håkansson, H. Retarded Sorption in Wood—Experimental Study, Analysis and Modelling. Ph.D. Thesis, Lund University, Lund, Sweden, 1998. [Google Scholar]
- Eitelberger, J.; Svensson, S. The sorption behavior of wood studied by means of an improved cup method. Transp. Porous Med. 2012, 92, 321–335. [Google Scholar] [CrossRef]
- Larian, M.; Lavine, I.; Mann, C.A.; Gauger, A.W. II—Sorption of water vapor by lignite, peat, and wood. Ind. Eng. Chem. 1930, 22, 1231–1234. [Google Scholar] [CrossRef]
- Lavine, I.; Gauger, A.W. Studies in the development of Dakota lignite. Ind. Eng. Chem. 1930, 22, 1226–1231. [Google Scholar] [CrossRef]
- Wadsö, L.; Svennberg, K.; Dueck, A. An experimentally simple method for measuring sorption isotherms. Dry. Technol. 2004, 22, 2427–2440. [Google Scholar] [CrossRef]
- Suchsland, O. Determination of sorption isotherms in minidesiccators. Wood Sci. 1980, 12, 214–217. [Google Scholar]
- Goulet, M. Phénomènes DE Second Ordre DE La Sorption D’Humidité Dans Le Bois AU Terme D’Un Conditionnement DE Trois Mois à Température Normale; Université Laval: Quebec City, QC, Canada, 1968; pp. 1–30. [Google Scholar]
- Greenspan, L. Humidity fixed points of binary saturated aqueous solutions. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1977, 81, 89. [Google Scholar] [CrossRef]
- McBain, J.W.; Bakr, A.M. A New Sorption Balance 1. J. Am. Chem. Soc. 1926, 48, 690–695. [Google Scholar] [CrossRef]
- Pidgeon, L.M.; Maass, O. The adsorption of water by wood. J. Am. Chem. Soc. 1930, 52, 1053–1069. [Google Scholar] [CrossRef]
- Pidgeon, L.M.; Maass, O. The penetration of water vapor into wood. Can. J. Res. 1930, 2, 318–326. [Google Scholar] [CrossRef]
- Stamm, A.J.; Woodruff, S.A. A convenient six-tube vapor sorption apparatus. Ind. Eng. Chem. 1941, 13, 0836–0838. [Google Scholar] [CrossRef]
- Kelsey, K.E. The sorption of water vapour by wood. Aust. J. Appl. Sci. 1957, 8, 42–54. [Google Scholar]
- Christensen, G.N.; Kelsey, K.E. The rate of sorption of water vapor by wood. Holz Roh Werkst 1959, 17, 178–188. [Google Scholar] [CrossRef]
- Christensen, G.N. The rate of sorption of water vapour by wood and pulp. Appita J. 1959, 13, 112–123. [Google Scholar]
- Christensen, G.N. Kinetics of sorption of water vapour by wood. Aust. J. Appl. Sci. 1960, 11, 295–304. [Google Scholar]
- Weichert, L. Investigations on sorption and swelling of spruce, beech and compressed beech wood at temperatures between 20 °C and 100 °C. Holz Roh Werkst 1963, 21, 290–300. [Google Scholar] [CrossRef]
- Spalt, H.A. The fundamentals of water sorption by wood. For. Prod. J. 1958, 8, 288–295. [Google Scholar]
- Kelly, M.W.; Hart, C.A. Water vapor sorption rates by wood cell walls. Wood Fiber Sci. 1970, 1, 270–282. [Google Scholar]
- Spalt, H.A. The sorption of water vapor by domestic and tropical woods. For. Prod. J. 1957, 7, 331–335. [Google Scholar]
- Sandstede, G.; Robens, E. Automatisierte Apparatur zur gravimetrischen Messung der Gassorption, insbesondere für die Bestimmung der spezifischen Oberfläche und der Porengröße. Chem. Ing. Tech. 1962, 34, 708–713. [Google Scholar] [CrossRef]
- Czanderna, A.; Vasofsky, R. Surface studies with the vacuum microbalance. Prog. Surf. Sci. 1979, 9, 45–82. [Google Scholar] [CrossRef]
- Rasmussen, M.D. Microcomputer-controlled gravimetric adsorption apparatus. Rev. Sci. Instrum. 1983, 54, 1558. [Google Scholar] [CrossRef]
- Buckton, G.; Beezer, A.E.; Newton, J.M. A vacuum microbalance technique for studies on the wettability of powders. J. Pharm. Pharmacol. 1986, 38, 713–720. [Google Scholar] [CrossRef]
- Astill, D.M.; Hall, P.L.; McConnell, J.D.C. An automated vacuum microbalance for measurement of adsorption isotherms. J. Phys. E Sci. Instrum. 1987, 20, 19–21. [Google Scholar] [CrossRef]
- Benham, M.J.; Ross, D.K. Experimental determination of absorption-desorption isotherms by computer-controlled gravimetric analysis. Z. Phys. Chem. 1989, 163, 25. [Google Scholar] [CrossRef]
- Jakieła, S.; Bratasz, Ł.; Kozłowski, R. Numerical modelling of moisture movement and related stress field in lime wood subjected to changing climate conditions. Wood Sci. Technol. 2008, 42, 21–37. [Google Scholar] [CrossRef]
- Bratasz, Ł.; Kozłowska, A.; Kozłowski, R. Analysis of water adsorption by wood using the Guggenheim-Anderson-de Boer equation. Holz Roh Werkst 2012, 70, 445–451. [Google Scholar] [CrossRef]
- Bergren, M.S. An automated controlled atmosphere microbalance for the measurement of moisture sorption. Int. J. Pharm. 1994, 103, 103–114. [Google Scholar] [CrossRef]
- Marshall, P.V.; Cook, P.A.; Williams, D.R. A new analytical technique for characterising the water vapour sorption properties of powders. In Proceedings of the International Symposium on Solid Oral Dosage Forms, Swedish Academy of Pharmaceutical Sciences, Stockholm, Sweden, 7–9 February 1994; p. 19A. [Google Scholar]
- Williams, D.R. The characterisation of powders by gravimetric water vapour sorption. Int. Labmate 1995, 20, 40–42. [Google Scholar]
- Murr, A.; Lackner, R. Analysis on the influence of grain size and grain layer thickness on the sorption kinetics of grained wood at low relative humidity with the use of water vapour sorption experiments. Wood Sci. Technol. 2018, 52, 753–776. [Google Scholar] [CrossRef]
- Thorell, A.; Wadsö, L. Determination of external mass transfer coefficients in dynamic sorption (DVS) measurements. Dry. Technol. 2018, 36, 332–340. [Google Scholar] [CrossRef]
- Christensen, G.N. The rate of sorption of water vapor by thin materials. In Volume Four: Principles and Methods of Measuring Moisture in Liquids and Solids, 1st ed.; Winn, P.N., Ed.; Reinhold Publishing Corporation: New York, NY, USA, 1965; pp. 279–293. [Google Scholar]
- Wadsö, L. Measurements of water-vapor sorption in wood. Part 2. Results. Wood Sci. Technol. 1993, 28, 59–65. [Google Scholar] [CrossRef]
- Christensen, G.N.; Hergt, H.F.A. Effect of previous history on kinetics of sorption by wood cell walls. J. Polym. Sci. Part A1 Polym. Chem. 1969, 7, 2427–2430. [Google Scholar] [CrossRef]
- Kelsey, K.E.; Clarke, L.N. The heat of sorption of water by wood. Aust. J. Appl. Sci. 1956, 7, 160–175. [Google Scholar]
- Hearmon, R.F.S.; Burcham, J.N. Specific heat and heat of wetting of wood. Nature 1955, 176, 978. [Google Scholar] [CrossRef]
- Kühlmann, G. Untersuchung der thermischen Eigenschaften von Holz und Spanplatten in Abhängigkeit von Feuchtigkeit und Temperatur im hygroskopischen Bereich. Holz Roh Werkst 1962, 20, 259–270. [Google Scholar] [CrossRef]
- Lee, L.-K.; Ruthven, D.M. Analysis of thermal effects in adsorption rate measurements. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1979, 75, 2406–2422. [Google Scholar] [CrossRef]
- Wang, P.; Meldon, J.H.; Sung, N. Heat effects in sorption of organic vapors in rubbery polymers. J. Appl. Polym. Sci. 1996, 59, 937–944. [Google Scholar] [CrossRef]
- Chihara, K.; Suzuki, M.; Kawazoe, K. Effect of heat generation on measurement of adsorption rate by gravimetric method. Chem. Eng. Sci. 1976, 31, 505–507. [Google Scholar] [CrossRef]
- Papadopoulos, A.N.; Hill, C.A.S.; Gkaraveli, A. Determination of surface area and pore volume of holocellulose and chemically modified wood flour using the nitrogen adsorption technique. Holz Roh Werkst 2003, 61, 453–456. [Google Scholar] [CrossRef]
- Rafsanjani, A.; Stiefel, M.; Jefimovs, K.; Mokso, R.; Derome, D.; Carmeliet, J. Hygroscopic swelling and shrinkage of latewood cell wall micropillars reveal ultrastructural anisotropy. J. R. Soc. Interface 2014, 11. [Google Scholar] [CrossRef]
- Murata, K.; Masuda, M. Microscopic observation of transverse swelling of latewood tracheid: Effect of macroscopic/mesoscopic structure. J. Wood. Sci. 2006, 52, 283–289. [Google Scholar] [CrossRef]
- Patera, A.; Van den Bulcke, J.; Boone, M.N.; Derome, D.; Carmeliet, J. Swelling interactions of earlywood and latewood across a growth ring: Global and local deformations. Wood Sci. Technol. 2018, 52, 91–114. [Google Scholar] [CrossRef]
- Seifert, J. Sorption and swelling of wood and wood base materials.2. Swelling behavior of wood and wood base materials. Holz Roh Werkst 1972, 30, 294–303. [Google Scholar] [CrossRef]
- Hartley, I.D.; Avramidis, S. Static dimensional changes of Sitka spruce and Western hemlock influenced by sorption conditions. J. Inst. Wood Sci. 1996, 14, 83–88. [Google Scholar]
- Ma, Q.; Rudolph, V. Dimensional change behavior of Carribean pine using an environmental scanning electron microscope. Dry. Technol. 2006, 24, 1397–1403. [Google Scholar] [CrossRef]
- Salmén, L.; Burgert, I. Cell wall features with regard to mechanical performance. A review COST Action E35 2004-2008: Wood machining—Micromechanics and fracture. Holzforschung 2009, 63, 121–129. [Google Scholar] [CrossRef]
- Salmén, L.; Bergström, E. Cellulose structural arrangement in relation to spectral changes in tensile loading FTIR. Cellulose 2009, 16, 975–982. [Google Scholar] [CrossRef]
- Matthews, J.F.; Skopec, C.E.; Mason, P.E.; Zuccato, P.; Torget, R.W.; Sugiyama, J.; Himmel, M.E.; Brady, J.W. Computer simulation studies of microcrystalline cellulose Iβ. Carbohydr. Res. 2006, 341, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Thybring, E.E.; Thygesen, L.G.; Burgert, I. Hydroxyl accessibility in wood cell walls as affected by drying and re-wetting procedures. Cellulose 2017, 24, 2375–2384. [Google Scholar] [CrossRef]
- Christensen, G.N.; Kelsey, K.E. The sorption of water vapor by the constituents of wood. Holz Roh Werkst 1959, 17, 189–203. [Google Scholar] [CrossRef]
- Fernandes, A.N.; Thomas, L.H.; Altaner, C.M.; Callow, P.; Forsyth, V.T.; Apperley, D.C.; Kennedy, C.J.; Jarvis, M.C. Nanostructure of cellulose microfibrils in spruce wood. Proc. Natl. Acad. Sci. USA 2011, 108, E1195–E1203. [Google Scholar] [CrossRef]
- Blomberg, J.; Persson, B. Swelling pressure of semi-isostatically densified wood under different mechanical restraints. Wood Sci. Technol. 2007, 41, 401–415. [Google Scholar] [CrossRef]
- Mazzanti, P.; Colmars, J.; Gril, J.; Hunt, D.; Uzielli, L. A hygro-mechanical analysis of poplar wood along the tangential direction by restrained swelling test. Wood Sci. Technol. 2014, 48, 673–687. [Google Scholar] [CrossRef]
- Tarkow, H.; Turner, H.D. The swelling pressure of wood. For. Prod. J. 1958, 8, 193–197. [Google Scholar]
- Plaza, N.Z.; Pingali, S.V.; Qian, S.; Heller, W.T.; Jakes, J.E. Informing the improvement of forest products durability using small angle neutron scattering. Cellulose 2016, 23, 1593–1607. [Google Scholar] [CrossRef]
- Chen, W.; Lickfield, G.C.; Yang, C.Q. Molecular modeling of cellulose in amorphous state. Part I: Model building and plastic deformation study. Polymer 2004, 45, 1063–1071. [Google Scholar] [CrossRef]
- Bonfield, P.W.; Mundy, J.; Robson, D.J.; Dinwoodie, J.M. The modelling of time-dependant deformation in wood using chemical kinetics. Wood Sci. Technol. 1996, 30, 105–115. [Google Scholar] [CrossRef]
- Yoon, J.; Cai, S.; Suo, Z.; Hayward, R.C. Poroelastic swelling kinetics of thin hydrogel layers: Comparison of theory and experiment. Soft Matter 2010, 6, 6004. [Google Scholar] [CrossRef]
- Petropoulos, J.H.; Sanopoulou, M.; Papadokostaki, K.G. Physically insightful modeling of non-Fickian kinetic regimes encountered in fundamental studies of isothermal sorption of swelling agents in polymeric media. Eur. Polym. J. 2011, 47, 2053–2062. [Google Scholar] [CrossRef]
- Crank, J. A theoretical investigation of the influence of molecular relaxation and internal stress on diffusion in polymers. J. Polym. Sci. 1953, 11, 151–168. [Google Scholar] [CrossRef]
- Neumann, S.; Marom, G. Prediction of moisture diffusion parameters in composite materials under stress. J. Compos. Mater. 1987, 21, 68–80. [Google Scholar] [CrossRef]
- Neumann, S.; Marom, G. Free-volume dependent moisture diffusion under stress in composite materials. J. Mater. Sci. 1986, 21, 26–30. [Google Scholar] [CrossRef]
- Roy, S.; Vengadassalam, K.; Wang, Y.; Park, S.; Liechti, K.M. Characterization and modeling of strain assisted diffusion in an epoxy adhesive layer. Int. J. Solids Struct. 2006, 43, 27–52. [Google Scholar] [CrossRef]
- Yaniv, G.; Ishai, O. Coupling between stresses and moisture diffusion in polymeric adhesives. Polym. Eng. Sci. 1987, 27, 731–739. [Google Scholar] [CrossRef]
- Hui, C.Y.; Wu, K.C.; Lasky, R.C.; Kramer, E.J. Case II diffusion in polymers.1. Transient swelling. J. Appl. Phys. 1987, 61, 5129–5136. [Google Scholar] [CrossRef]
- Long, F.A.; Richman, D. Concentration gradients for diffusion of vapors in glassy polymers and their relation to time dependent diffusion phenomena. J. Am. Chem. Soc. 1960, 82, 513–519. [Google Scholar] [CrossRef]
- Long, F.A.; Watt, I. Concentration gradients during sorption of vapor into polymers in the glassy state. J. Polym. Sci. 1956, 21, 554–557. [Google Scholar] [CrossRef]
- Newns, A.C. The sorption and desorption kinetics of water in a regenerated cellulose. Trans. Faraday Soc. 1956, 52, 1533–1545. [Google Scholar] [CrossRef]
- Crank, J.; Park, G.S. Diffusion in high polymers: Some anomalies and their significance. Trans. Faraday Soc. 1951, 47, 1072–1084. [Google Scholar] [CrossRef]
- Bagley, E.; Long, F.A. Two-stage sorption and desorption of organic vapors in cellulose acetate. J. Am. Chem. Soc. 1955, 77, 2172–2178. [Google Scholar] [CrossRef]
- Sadoh, T.; Christensen, G.N. Rate of sorption of water vapour by hemicellulose. Aust. J. Appl. Sci. 1964, 15, 297–308. [Google Scholar]
- Irvine, G.M. The glass transitions of lignin and hemicellulose and their measurement by differential thermal-analysis. Tappi J. 1984, 67, 118–121. [Google Scholar]
- Kelley, S.S.; Rials, T.G.; Glasser, W.G. Relaxation behavior of the amorphous components of wood. J. Mater. Sci. Lett. 1987, 22, 617–624. [Google Scholar] [CrossRef]
- Olsson, A.M.; Salmén, L. The softening behavior of hemicelluloses related to moisture. ACS Symp. Ser. 2004, 864, 184–197. [Google Scholar] [CrossRef]
- Paes, S.S.; Sun, S.M.; MacNaughtan, W.; Ibbett, R.; Ganster, J.; Foster, T.J.; Mitchell, J.R. The glass transition and crystallization of ball milled cellulose. Cellulose 2010, 17, 693–709. [Google Scholar] [CrossRef]
- Szcześniak, L.; Rachocki, A.; Tritt-Goc, J. Glass transition temperature and thermal decomposition of cellulose powder. Cellulose 2008, 15, 445–451. [Google Scholar] [CrossRef]
- Zelinka, S.L.; Glass, S.V.; Jakes, J.E.; Stone, D.S. A solution thermodynamics definition of the fiber saturation point and the derivation of a wood–water phase (state) diagram. Wood Sci. Technol. 2016, 50, 443–462. [Google Scholar] [CrossRef]
- Duda, J.L.; Vrentas, J.S.; Ju, S.T.; Liu, H.T. Prediction of diffusion coefficients for polymer-solvent systems. AIChE J. 1982, 28, 279–285. [Google Scholar] [CrossRef]
- Matsuoka, S.; Bair, H.E.; Bearder, S.S.; Kern, H.E.; Ryan, J.T. Analysis of non-linear stress relaxation in polymeric glasses. Polym. Eng. Sci. 1978, 18, 1073–1080. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Frisch, H.L.; Stern, S.A. Diffusion of small molecules in polymers. Crit. Rev. Solid State Mater. Sci. 1983, 11, 123–187. [Google Scholar] [CrossRef]
- Crank, J. Some methods of deducing the diffusion coefficient and its concentration dependence from sorption experiments. Trans. Faraday Soc. 1955, 51, 1632. [Google Scholar] [CrossRef]
- Stamm, A.J. Diffusion of water into uncoated cellophane.2. From steady-state diffusion measurements. J. Phys. Chem. 1956, 60, 83–86. [Google Scholar] [CrossRef]
- Hansen, N.M.L.; Blomfeldt, T.O.J.; Hedenqvist, M.S.; Plackett, D.V. Properties of plasticized composite films prepared from nanofibrillated cellulose and birch wood xylan. Cellulose 2012, 19, 2015–2031. [Google Scholar] [CrossRef]
- Heikkilä, M.I.; Willför, S.M.; Mikkonen, K.S.; Tenkanen, M. Films from Glyoxal-Crosslinked Spruce Galactoglucomannans Plasticized with Sorbitol. Int. J. Polym. Sci. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Stamm, A.J. Bound water diffusion into wood in the fiber direction. For. Prod. J. 1959, 9, 27–32. [Google Scholar]
- Taguchi, A.; Murata, K.; Nakano, T. Observation of cell shapes in wood cross-sections during water adsorption by confocal laser-scanning microscopy (CLSM). Holzforschung 2010, 64, 627–631. [Google Scholar] [CrossRef]
- Wadsö, L. A test of different methods to evaluate the diffusivity from unsteady-state sorption measurements. Dry. Technol. 1994, 12, 1863–1876. [Google Scholar] [CrossRef]
- Sanopoulou, M.; Roussis, P.P.; Petropoulos, J.H. A detailed study of the viscoelastic nature of vapor sorption and transport in a cellulosic polymer. I. Origin and physical implications of deviations from Fickian sorption kinetics. J. Polym. Sci. Part B Polym. Phys. 1995, 33, 993–1005. [Google Scholar] [CrossRef]
- Joshi, S.; Astarita, G. Diffusion-relaxation coupling in polymers which show two-stage sorption phenomena. Polymer 1979, 20, 455–458. [Google Scholar] [CrossRef]
- Berens, A.R.; Hopfenberg, H.B. Diffusion and relaxation in glassy polymer powders: 2. Separation of diffusion and relaxation parameters. Polymer 1978, 19, 489–496. [Google Scholar] [CrossRef]
- Camera-Roda, G.; Sarti, G.C. Mass transport with relaxation in polymers. AIChE J. 1990, 36, 851–860. [Google Scholar] [CrossRef]
- Meng, Y.; Xia, Y.; Young, T.M.; Cai, Z.; Wang, S. Viscoelasticity of wood cell walls with different moisture content as measured by nanoindentation. RSC Adv. 2015, 5, 47538–47547. [Google Scholar] [CrossRef]
- Kojima, Y.; Yamamoto, H. Effect of moisture content on the longitudinal tensile creep behavior of wood. J. Wood Sci. 2005, 51, 462–467. [Google Scholar] [CrossRef]
- Olsson, A.M.; Salmén, L.; Eder, M.; Burgert, I. Mechano-sorptive creep in wood fibres. Wood Sci. Technol. 2007, 41, 59–67. [Google Scholar] [CrossRef]
- Willems, W. Thermally limited wood moisture changes: Relevance for dynamic vapour sorption experiments. Wood Sci. Technol. 2017, 51, 751–770. [Google Scholar] [CrossRef]
- Kohler, R.; Dück, R.; Ausperger, B.; Alex, R. A numeric model for the kinetics of water vapor sorption on cellulosic reinforcement fibers. Compos. Interface 2003, 10, 255–276. [Google Scholar] [CrossRef]
- Hill, C.A.S.; Norton, A.; Newman, G. Analysis of the water vapour sorption behaviour of Sitka spruce [Picea sitchensis (Bongard) Carr.] based on the parallel exponential kinetics model. Holzforschung 2010, 64, 469–473. [Google Scholar] [CrossRef]
- Kohler, R.; Alex, R.; Brielmann, R.; Ausperger, B. A New Kinetic Model for Water Sorption Isotherms of Cellulosic Materials. Macromol. Symp. 2006, 244, 89–96. [Google Scholar] [CrossRef]
- Kachrimanis, K.; Noisternig, M.F.; Griesser, U.J.; Malamataris, S. Dynamic moisture sorption and desorption of standard and silicified microcrystalline cellulose. Eur. J. Pharm. Biopharm. 2006, 64, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Okubayashi, S.; Griesser, U.; Bechtold, T. A kinetic study of moisture sorption and desorption on lyocell fibers. Carbohydr. Polym. 2004, 58, 293–299. [Google Scholar] [CrossRef]
- Okubayashi, S.; Griesser, U.J.; Bechtold, T. Moisture sorption/desorption behavior of various manmade cellulosic fibers. J. Appl. Polym. Sci. 2005, 97, 1621–1625. [Google Scholar] [CrossRef]
- Zaihan, J.; Hill, C.A.S.; Curling, S.; Hashim, W.S.; Hamdan, H. The kinetics of water vapour sorption: Analysis using parallel exponential kinetics model on six Malaysian hardwoods. J. Trop. For. Sci. 2010, 22, 107–117. [Google Scholar]
- Belbekhouche, S.; Bras, J.; Siqueira, G.; Chappey, C.; Lebrun, L.; Khelifi, B.; Marais, S.; Dufresne, A. Water sorption behavior and gas barrier properties of cellulose whiskers and microfibrils films. Carbohydr. Polym. 2011, 83, 1740–1748. [Google Scholar] [CrossRef]
- Cordin, M.; Griesser, U.J.; Bechtold, T. Analysis of moisture sorption in lyocell-polypropylene composites. Cellulose 2017, 24, 1837–1847. [Google Scholar] [CrossRef]
- Guo, X.; Wu, Y.; Xie, X. Water vapor sorption properties of cellulose nanocrystals and nanofibers using dynamic vapor sorption apparatus. Sci. Rep. 2017, 7, 14207. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.J.; Hill, C.A.S.; Xiao, Z.F.; Jalaludin, Z.; Militz, H.; Mai, C. Water vapor sorption kinetics of wood modified with glutaraldehyde. J. Appl. Polym. Sci. 2010, 117, 1674–1682. [Google Scholar] [CrossRef]
- Hill, C.A.S.; Norton, A.; Newman, G. The water vapor sorption behavior of flax fibers-analysis using the parallel exponential kinetics model and determination of the activation energies of sorption. J. Appl. Polym. Sci. 2010, 116, 2166–2173. [Google Scholar] [CrossRef]
- Lundahl, M.J.; Cunha, A.G.; Rojo, E.; Papageorgiou, A.C.; Rautkari, L.; Arboleda, J.C.; Rojas, O.J. Strength and water interactions of cellulose I filaments wet-spun from cellulose nanofibril hydrogels. Sci. Rep. 2016, 6, 30695. [Google Scholar] [CrossRef] [PubMed]
- Mutungi, C.; Schuldt, S.; Onyango, C.; Schneider, Y.; Jaros, D.; Rohm, H. Dynamic moisture sorption characteristics of enzyme-resistant recrystallized cassava starch. Biomacromolecules 2011, 12, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Thybring, E.E.; Boardman, C.R.; Glass, S.V.; Zelinka, S.L. The parallel exponential kinetics model is unfit to characterize moisture sorption kinetics in cellulosic materials. Cellulose 2019, 26, 723–735. [Google Scholar] [CrossRef]
- Glass, S.V.; Boardman, C.R.; Zelinka, S.L. Short hold times in dynamic vapor sorption measurements mischaracterize the equilibrium moisture content of wood. Wood Sci. Technol. 2017, 51, 243–260. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thybring, E.E.; Glass, S.V.; Zelinka, S.L. Kinetics of Water Vapor Sorption in Wood Cell Walls: State of the Art and Research Needs. Forests 2019, 10, 704. https://doi.org/10.3390/f10080704
Thybring EE, Glass SV, Zelinka SL. Kinetics of Water Vapor Sorption in Wood Cell Walls: State of the Art and Research Needs. Forests. 2019; 10(8):704. https://doi.org/10.3390/f10080704
Chicago/Turabian StyleThybring, Emil Engelund, Samuel V. Glass, and Samuel L. Zelinka. 2019. "Kinetics of Water Vapor Sorption in Wood Cell Walls: State of the Art and Research Needs" Forests 10, no. 8: 704. https://doi.org/10.3390/f10080704
APA StyleThybring, E. E., Glass, S. V., & Zelinka, S. L. (2019). Kinetics of Water Vapor Sorption in Wood Cell Walls: State of the Art and Research Needs. Forests, 10(8), 704. https://doi.org/10.3390/f10080704