On the Experimental Assessment of the Molecular-Scale Interactions between Wood and Water


Abstract
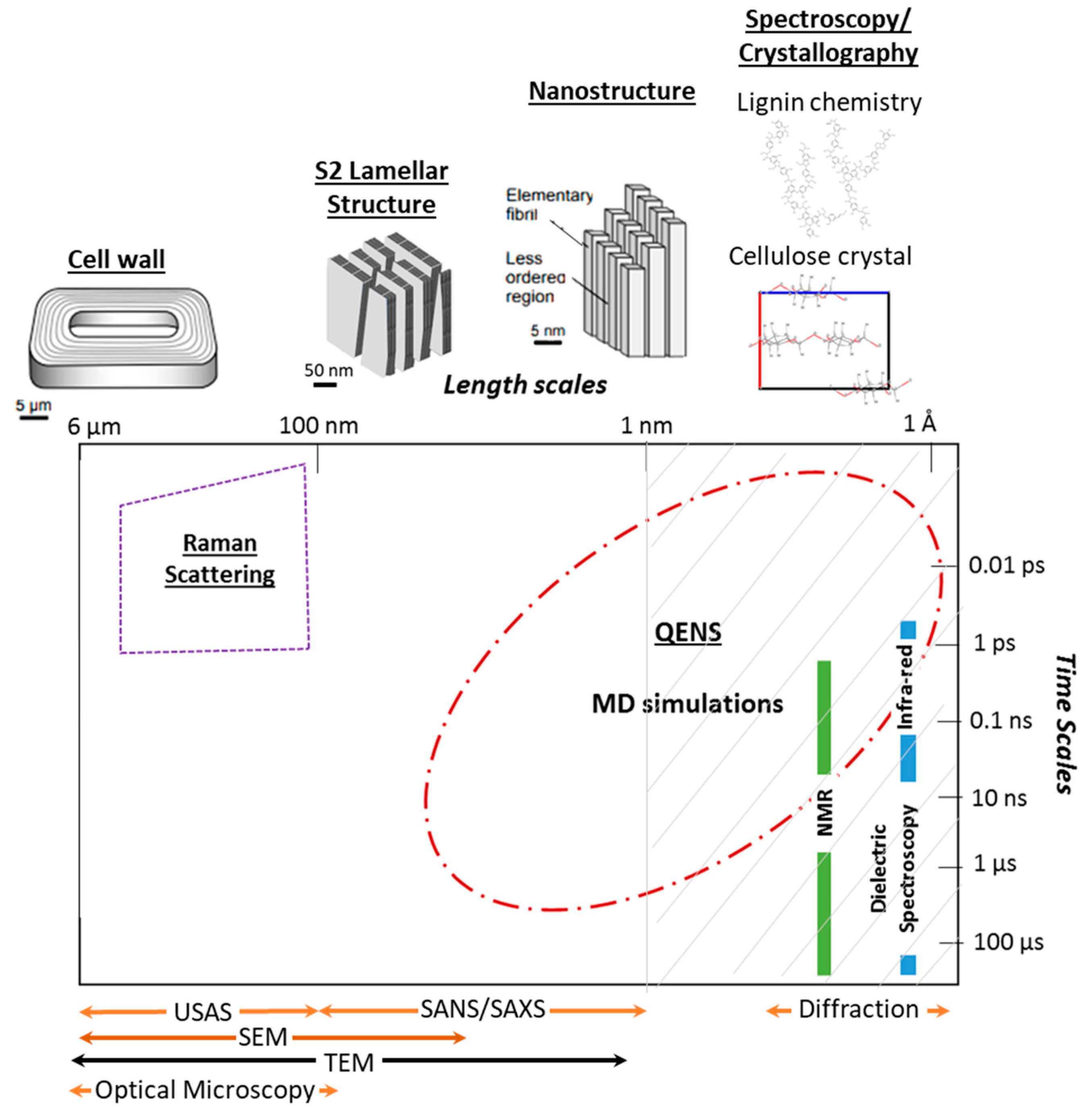
1. Introduction
2. Relevant Terminology
Unmodified Wood
- Cellulose accounts for 40%–45% of the cell wall in softwoods and 38%–49% in hardwoods [29]. In unmodified wood, cellulose is found in the cellulose microfibrils, which are bundles of cellulose elementary fibrils that are formed by an ordered arrangement of 18 to 24 chains. Only non-crystalline cellulose is accessible to water and at the elementary fibril level only surface chains are thought to be accessible [30]. Cellulose is a linear polysaccharide whose primary unit is the cellobiose. Each primary unit consists of two glucose units linked by a glycosidic bond [29]. Cellulose in higher order plants can be naturally found in two crystalline forms, namely and [31]. The main differences between the two allomorphs are hydrogen bonding, crystal stacking and number of cellulose chains per crystal. In the triclinic cellulose there is only one cellulose chain per crystal and the crystals are stacked longitudinally by glucose units that vary slightly in conformation. This allomorph is generally found as the dominant crystalline phase in bacterial cellulose and algae [32,33]. In the monoclinic cellulose there are two alternating cellulose chains held together by intramolecular hydrogen bonds in each crystal and its crystals are stacked paralleled to each other by forming inter-molecular hydrogen bonds. While spectroscopy studies have suggested that both forms of cellulose can coexist in the native plants [32,34], the dominant crystalline form in wood is [33,35]. The monoclinic structure found in wood is typically described in terms of the a, b, c, and lattice parameters, which can be solved by tracking the main diffraction peaks, namely the (200), the (10), the (110), and (004) [8,32,36]. The longitudinal axis of the cellulose fibers is parallel to the c-axis. The cellulose chains are hydrogen-bonded into planar sheets along the b-axis, and these sheets are stacked via Van der Waal interactions along the a-axis. Most evidence supports that cellulose has both hydrophilic and hydrophobic surfaces [6], with the (110), (010), and (10) planes being the most hydrophilic ones [37]. Although the degree of cellulose crystallinity can vary across wood species and measuring methods, values between 50 to 65% are typical for both softwoods and hardwoods [38]. The remaining cellulose is typically considered amorphous or para-crystalline, which is an intermediate phase between and , and is more laterally disordered and mobile than crystalline cellulose [6,39].
- Hemicelluloses can be found in the middle lamella that holds together wood cell walls as well as inside the matrix of the cell walls [40]. It usually accounts for 15%–25% of the wood’s dry weight [29]. Hemicelluloses are polysaccharides with a lower degree of polymerization than cellulose. Usually, they have an equatorial β-1,4-linked glycosyl residue backbone [41] and despite its backbone structure similarities to cellulose it lacks crystallinity. The specific composition of the hemicelluloses can vary across wood types, species, and even wood cell walls [42]. The main hemicellulose in hardwoods is xylan and in softwoods it is galactoglucomannan [43]. In softwoods, xylan is typically found in the primary wall [41], while the major hemicellulosic components found in the secondary cell are partially acetylated galactoglucomannans and arabinoglucuronoxylan [44]. Different hemicelluloses have varying degrees of affinity between cellulose and lignin and hence they act as a bridge between the two components [45]. For instance, glucomannans have a higher affinity to cellulose than xylans [46], whereas xylans associate mostly with lignin [46] despite their ability to interact with cellulose hydrophobic surfaces [47]. Hemicelluloses are much more accessible to water than cellulose. In the hemicelluloses, it is assumed that most of all of its hydroxyl groups are accessible because of its lack of crystallinity [48] and the estimated concentration of available hydroxyl groups ranges from 8.6 to 18.8 mmol/g in glucomannan, whereas in xylan is 14.4 mmol/g [30].
- Lignin is a three-dimensional aromatic polymer network that usually accounts for 26%–34% of the wood dry weight in softwoods and 23%–30% in hardwoods [29]. It is made of phenylpropane units that are linked by ether and carbon-carbon bonds [29,49]. In the cell wall lignin is typically formed after polysaccharide synthesis and thus its supramolecular structure is determined by the space available when lignin is deposited, meaning that middle lamella lignin differs from secondary cell wall lignin [50]. In the middle lamella, lignin is thought to form 3D globules made of folded chain oligolignols, whereas in the secondary cell wall it forms tubular structures that surround the hemicellulose coated microfibrils. Recent evidence, based on molecular dynamics simulations, have also suggested that the aromatic rings of lignin align themselves parallel to the cellulose fibrils; though this order seems limited to the first monolayer of lignin [51]. Due to its lack of crystallinity, it is expected that most of its hydroxyl groups will be available to water [48]. Nonetheless, lignin is the least hydrophilic wood polymer and its concentration of accessible hydroxyl groups is comparable to the theoretical accessibility of cellulose [30].
- Water in wood can exist as bound water and free water. The total amount of water in wood is called the moisture content and it is defined as the mass of water over the mass of oven dried wood [1]. The equilibrium moisture content varies according to the environmental conditions, such as relative humidity (RH) and temperature. In wood science, bound water typically refers to water that is bound within the wood cell wall by intermolecular attractions and free water refers to water that is found within empty cavities inside the wood structure, such as lumina and pits [1]. Different groups have proposed that water within wood can be found in two or three states [52]. While, it has been shown that the freezable bound water found via differential calorimetry measurements is caused by sample preparation [53], some scattering techniques have been able to discern between populations of water molecules that have different types of local environment. Hence, in this paper, all bound water will be referred to as absorbed water. It is known that changes in the local environment experienced by water molecules affect their properties and state [54]. In the literature, water that interacts strongly with the wood polymers has been referred to as slow water, strongly-bonded water, and, in some instances, non-freezing water. Here, it will be referred to as strongly-bound water. Water that interacts loosely with the wood polymers and/or is more likely interacting with other water molecules has been referred to as fast water, weakly-bonded water, and/or bulk-like water. Here, the term loosely-bound water will be used.
- Extractives correspond to non-structural components that can be typically removed with solvents. These components are typically classified into three subgroups: aliphatic compounds, terpenes, terpenoids, and phenolic compounds [55]. Although, the total content and composition of these components can vary greatly between species and even within different parts of a tree, the majority of the extractives are typically found in the heartwood [44]. In general, extractives account for 2% to 10% of the wood’s dry mass, although some durable species can have higher values [55]. It is widely accepted that extractives play a role in increasing the durability of wood, particularly, in terms of decay and termite resistance [56]. Moreover, studies on the bulk swelling of wood in organic liquids have shown that extractives can lower the rate of maximum swelling [55]. However, in situ studies focusing on the effect of the extractives on the molecular-scale interactions between wood and water have been difficult likely due to the variability in content and composition of the extractives. NMR experiments have shown that wood with higher content of wood extractives have different bound water T2 values than other temperate species [57], yet it is still unclear whether the extractives play a role in the dynamics of the bound water or the mobility of the wood polymers.
3. Moisture-Dependent Behavior of the Wood Polymers
4. State of Water Absorbed in Wood and Wood Polymers
5. Needs for Future Research
Funding
Conflicts of Interest
References
- Glass, S.V.; Zelinka, S.L. Moisture relations and physical properties of wood. In Wood Handbook: Wood as an Engineering Material; U.S. Dept. of Agriculture, Forest Service, Forest Products Laboratory: Madison, WI, USA, 2010; pp. 4.1–4.19. [Google Scholar]
- Stamm, A.J. SHRINKING and SWELLING of WOOD. Ind. Eng. Chem. 1935, 27, 401–406. [Google Scholar] [CrossRef]
- Rafsanjani, A.; Stiefel, M.; Jefimovs, K.; Mokso, R.; Derome, D.; Carmeliet, J. Hygroscopic Swelling and Shrinkage of Latewood Cell Wall Micropillars Reveal Ultrastructural Anisotropy. J. R. Soc. Interface 2014, 11, 20140126. [Google Scholar] [CrossRef] [PubMed]
- Derome, D.; Griffa, M.; Koebel, M.; Carmeliet, J. Hysteretic Swelling of Wood at Cellular Scale Probed by Phase-Contrast X-Ray Tomography. J. Struct. Biol. 2011, 173, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Jakob, H.F.; Tschegg, S.E.; Fratzl, P. Hydration Dependence of the Wood-Cell Wall Structure in Picea Abies. A Small-Angle X-Ray Scattering Study. Macromolecules 1996, 29, 8435–8440. [Google Scholar] [CrossRef]
- Fernandes, A.N.; Thomas, L.H.; Altaner, C.M.; Callow, P.; Forsyth, V.T.; Apperley, D.C.; Kennedy, C.J.; Jarvis, M.C. Nanostructure of Cellulose Microfibrils in Spruce Wood. Proc. Natl. Acad. Sci 2011, 108, E1195–E1203. [Google Scholar] [CrossRef]
- Plaza, N.Z.; Pingali, S.V.; Qian, S.; Heller, W.T.; Jakes, J.E. Informing the Improvement of Forest Products Durability Using Small Angle Neutron Scattering. Cellulose 2016, 23, 1593–1607. [Google Scholar] [CrossRef]
- Zabler, S.; Paris, O.; Burgert, I.; Fratzl, P. Moisture Changes in the Plant Cell Wall Force Cellulose Crystallites to Deform. J. Struct. Biol 2010, 171, 133–141. [Google Scholar] [CrossRef]
- Kuttich, B.; Grefe, A.K.; Kröling, H.; Schabel, S.; Stühn, B. Molecular Mobility in Cellulose and Paper. RSC Adv. 2016, 6, 32389–32399. [Google Scholar] [CrossRef]
- Petridis, L.; O’Neill, H.M.; Johnsen, M.; Fan, B.; Schulz, R.; Mamontov, E.; Maranas, J.; Langan, P.; Smith, J.C.; Neill, H.M.O.; et al. Hydration Control of the Mechanical and Dynamical Properties of Cellulose. Biomacromolecules 2014, 15, 4152–4159. [Google Scholar] [CrossRef]
- Bellissent-Funel, M.C. Hydration Processes in Biology: Theoretical and Experimental Approaches; NATO Science Series, Series A, Life sciences; v. 305; IOS Press: Amsterdam, The Netherlands, 1999; ISBN 9789051994391. [Google Scholar]
- Cousins, W.J. Young’s Modulus of Hemicellulose as Related to Moisture Content. Wood Sci. Technol. 1978, 12, 161–167. [Google Scholar] [CrossRef]
- Lenth, C.A.; Kamke, F.A. Moisture Dependent Softening Behavior of Wood. Wood Fiber Sci. 2001, 33, 492–507. [Google Scholar]
- Zelinka, S.L.; Gleber, S.C.; Vogt, S.; López, G.M.R.; Jakes, J.E. Threshold for Ion Movements in Wood Cell Walls below Fiber Saturation Observed by X-Ray Fluorescence Microscopy (XFM). Holzforschung 2015, 69, 441–448. [Google Scholar] [CrossRef]
- Lindh, E.L.; Bergenstråhle-Wohlert, M.; Terenzi, C.; Salmén, L.; Furó, I. Non-Exchanging Hydroxyl Groups on the Surface of Cellulose Fibrils: The Role of Interaction with Water. Carbohydr. Res. 2016, 434, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, C.; Prakobna, K.; Berglund, L.A.; Furó, I. Nanostructural Effects on Polymer and Water Dynamics in Cellulose Biocomposites: 2H and 13C NMR Relaxometry. Biomacromolecules 2015, 16, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Terenzi, C.; Furó, I.; Berglund, L.A.; Wohlert, J. Hydration-Dependent Dynamical Modes in Xyloglucan from Molecular Dynamics Simulation of 13 C NMR Relaxation Times and Their Distributions. Biomacromolecules 2018, 19, 2567–2579. [Google Scholar] [CrossRef] [PubMed]
- Kulasinski, K.; Guyer, R.; Derome, D.; Carmeliet, J. Water Diffusion in Amorphous Hydrophilic Systems: A Stop and Go Process. Langmuir 2015, 31, 10843–10849. [Google Scholar] [CrossRef]
- Kulasinski, K.; Guyer, R.; Keten, S.; Derome, D.; Carmeliet, J. Impact of Moisture Adsorption on Structure and Physical Properties of Amorphous Biopolymers. Macromolecules 2015, 48. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Z.; Du, X.; Chen, L. Contribution of Different State of Adsorbed Water to the Sub-T g Dynamics of Cellulose. Carbohydr. Polym. 2019, 210, 322–331. [Google Scholar] [CrossRef]
- Kulasinski, K.; Salmén, L.; Derome, D.; Carmeliet, J. Moisture Adsorption of Glucomannan and Xylan Hemicelluloses. Cellulose 2016, 23, 1629–1637. [Google Scholar] [CrossRef]
- Einfeldt, J.; Meißner, D.; Kwasniewski, A. Polymerdynamics of Cellulose and Other Polysaccharides in Solid State-Secondary Dielectric Relaxation Processes. Prog. Polym. Sci. 2001, 26, 1419–1472. [Google Scholar] [CrossRef]
- Davison, B.H.; Pu, Y.; Vural, D.; Parks, J.M.; Sokolov, A.P.; Petridis, L.; Mamontov, E.; Ragauskas, A.J.; Smith, J.C.; Pingali, S.V.; et al. Impact of Hydration and Temperature History on the Structure and Dynamics of Lignin. Green Chem. 2018, 20, 1602–1611. [Google Scholar]
- O’Neill, H.; Pingali, S.V.; Petridis, L.; He, J.; Mamontov, E.; Hong, L.; Urban, V.; Evans, B.; Langan, P.; Smith, J.C.; et al. Dynamics of Water Bound to Crystalline Cellulose. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qing, Y.; Wu, Y.; Wu, Q. Molecular Association of Adsorbed Water with Lignocellulosic Materials Examined by Micro-FTIR Spectroscopy. Int. J. Biol. Macromol. 2016, 83, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Plaza, N.Z. Neutron Scattering Studies of Nano-Scale Wood-Water Interactions. Ph.D. Thesis, University of Wisconsin–Madison, Madison, WI, USA, August 2017. [Google Scholar]
- Martínez-Sanz, M.; Gidley, M.J.; Gilbert, E.P. Application of X-Ray and Neutron Small Angle Scattering Techniques to Study the Hierarchical Structure of Plant Cell Walls: A Review. Carbohydr. Polym. 2015, 125, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Wiedenhoeft, A.C.; Miller, R.B. Structure and Function of Wood. In Handbook of Wood Chemistry and Wood Composites; Rowell, R.M., Ed.; CRC Press: Boca Raton, FL, USA, 2005; pp. 9–32. [Google Scholar]
- Rowell, R.; Pettersen, R.; Han, J.S.; Rowell, J.S.; Tshabalala, M. Cell Wall Chemistry. In Handbook of Wood Chemistry and Wood Composites; CRC Press: Boca Raton, FL, USA, 2005; pp. 35–74. [Google Scholar]
- Engelund, E.T.; Thygesen, L.G.; Svensson, S.; Hill, C.A.S. A Critical Discussion of the Physics of Wood-Water Interactions. Wood Sci. Technol. 2013, 47, 141–161. [Google Scholar] [CrossRef]
- Wada, M.; Nishiyama, Y.; Chanzy, H.; FForsyth, T.; Langan, P. The Structure of Celluloses. Int. Cent. Diffr. Data 2008, 51, 138–144. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Sugiyama, J.; Chanzy, H.; Langan, P. Crystal Structure and Hydrogen Bonding System in Cellulose Iα from Synchrotron X-Ray and Neutron Fiber Diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef]
- Sugiyama, J.; Vuong, R.; Chanzy, H. Electron Diffraction Study on the Two Crystalline Phases Occurring in Native Cellulose from an Algal Cell Wall. Macromolecules 1991, 24, 4168–4175. [Google Scholar] [CrossRef]
- Driemeier, C.; Francisco, L.H. X-Ray Diffraction from Faulted Cellulose I Constructed with Mixed Iα–Iβ Stacking. Cellulose 2014, 21, 3161–3169. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Johnson, G.P.; French, A.D.; Forsyth, V.T.; Langan, P. Neutron Crystallography, Molecular Dynamics, and Quantum Mechanics Studies of the Nature of Hydrogen Bonding in Cellulose Iβ. Biomacromolecules 2008, 9, 3133–3140. [Google Scholar] [CrossRef]
- Abe, K.; Yamamoto, H. Change in Mechanical Interaction between Cellulose Microfibril and Matrix Substance in Wood Cell Wall Induced by Hygrothermal Treatment. J. Wood Sci. 2006, 52, 107–110. [Google Scholar] [CrossRef]
- Zhao, Z.; Crespi, V.H.; Kubicki, J.D.; Cosgrove, D.J.; Zhong, L. Molecular Dynamics Simulation Study of Xyloglucan Adsorption on Cellulose Surfaces: Effects of Surface Hydrophobicity and Side-Chain Variation. Cellulose 2014, 21, 1025–1039. [Google Scholar] [CrossRef]
- Agarwal, U.P.; Reiner, R.R.; Ralph, S.A. Estimation of Cellulose Crystallinity of Lignocelluloses Using Near-IR FT-Raman Spectroscopy and Comparison of the Raman and Segal-WAXS Methods. J. Agric. Food Chem. 2013, 61, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.T.; Wickholm, K.; Iversen, T. A CP/MAS 13C NMR Investigation of Molecular Ordering in Celluloses. Carbohydr. Res. 1997, 302, 19–25. [Google Scholar] [CrossRef]
- Wiedenhoeft, A.C. Structure and Function of Wood. In Wood handbook: Wood as an Engineering Material; U.S. Dept. of Agriculture, Forest Service, Forest Products Laboratory: Madison, WI, USA, 2010; pp. 9–33. [Google Scholar]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Sun, X.F.; Tomkinson, J. Hemicelluloses and Their Derivatives. ACS Sym. 2003, 846, 2–22. [Google Scholar]
- Terrett, O.M.; Dupree, P. Covalent Interactions between Lignin and Hemicelluloses in Plant Secondary Cell Walls. Curr. Opin. Biotechnol. 2019, 56, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Rowell, R.; Pettersen, R.; Tshabalala, M. Cell Wall Chemistry. In Handbook of Wood Chemistry and Wood Composites, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 33–72. ISBN 9781439853801. [Google Scholar]
- Hansen, C.M.; Björkman, A. The Ultrastructure of Wood from a Solubility Parameter Point of View. Holzforschung 1998, 52, 335–344. [Google Scholar] [CrossRef]
- Åkerholm, M.; Salmén, L. Interactions between Wood Polymers Studied by Dynamic FT-IR Spectroscopy. Polymer (Guildf) 2001, 42, 963–969. [Google Scholar] [CrossRef]
- Busse-Wicher, M.; Gomes, T.C.F.; Tryfona, T.; Nikolovski, N.; Stott, K.; Grantham, N.J.; Bolam, D.N.; Skaf, M.S.; Dupree, P. The Pattern of Xylan Acetylation Suggests Xylan May Interact with Cellulose Microfibrils as a Twofold Helical Screw in the Secondary Plant Cell Wall of Arabidopsis Thaliana. Plant J. 2014, 79, 492–506. [Google Scholar] [CrossRef]
- Rowell, R.M.; Service, F. Moisture Sorption Properties of Acetylated Lignocellulosic Fibers. In Proceedings of the 10th Cellulose Conference; Schuerch, C., Ed.; John Wiley & Sons, Inc.: Syracuse, NY, USA, 1989; pp. 343–355. [Google Scholar]
- Duval, A.; Lawoko, M. A Review on Lignin-Based Polymeric, Micro- and Nano- Structured Materials. React. Funct. Polym. 2014, 85, 78–96. [Google Scholar] [CrossRef]
- Terashima, N.; Yoshida, M.; Hafrén, J.; Fukushima, K.; Westermark, U. Proposed Supramolecular Structure of Lignin in Softwood Tracheid Compound Middle Lamella Regions. Holzforschung 2012, 66, 907–915. [Google Scholar] [CrossRef]
- Besombes, S.; Mazeau, K. The Cellulose/Lignin Assembly Assessed by Molecular Modeling. Part 2: Seeking for Evidence of Organization of Lignin Molecules at the Interface with Cellulose. Plant Physiol. Biochem. 2005, 43, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; McDonald, P.J.; Gardiner, B.A. A Study of Water Exchange in Wood by Means of 2D NMR Relaxation Correlation and Exchange. Holzforschung 2010, 64, 259–266. [Google Scholar] [CrossRef]
- Zelinka, S.L.; Lambrecht, M.J.; Glass, S.V.; Wiedenhoeft, A.C.; Yelle, D.J. Examination of Water Phase Transitions in Loblolly Pine and Cell Wall Components by Differential Scanning Calorimetry. Thermochimica Acta 2012, 533, 39–45. [Google Scholar] [CrossRef]
- Etzler, F.M. A comparison of the properties of vicinal water in silica, clay, wood, cellulose and other polymeric materials. In Water Relationships in Foods; Springer: Boston, MA, USA, 1991; pp. 805–822. [Google Scholar]
- Mantanis, G.I.; Young, R.A.; Rowell, R.M. Swelling of Wood Part III. Effect of Temperature and Extractives on Rate and Maximum Swelling. Holzforschung 1995, 49, 239–248. [Google Scholar] [CrossRef]
- Kirker, G.T.; Blodgett, A.B.; Arango, R.A.; Lebow, P.K.; Clausen, C.A. The Role of Extractives in Naturally Durable Wood Species. Int. Biodeterior. Biodegrad. 2013, 82, 53–58. [Google Scholar] [CrossRef]
- Almeida, G.; Gagné, S.; Hernández, R.E. A NMR Study of Water Distribution in Hardwoods at Several Equilibrium Moisture Contents. Wood Sci. Technol. 2007, 41, 293–307. [Google Scholar] [CrossRef]
- Sugino, H.; Sugimoto, H.; Miki, T.; Kanayama, K. Fine structure changes of wood during moisture adsorption and desorption process analyzed by X-ray diffraction measurement. J. Jpn. Wood Res. Soc. 2007, 53, 82–89. [Google Scholar] [CrossRef][Green Version]
- Agarwal, U.P.; Ralph, S.A.; Baez, C.; Reiner, R.S.; Verrill, S.P. Effect of Sample Moisture Content on XRD-Estimated Cellulose Crystallinity Index and Crystallite Size. Cellulose 2017, 24, 1971–1984. [Google Scholar] [CrossRef]
- Kljun, A.; Benians, T.A.S.; Goubet, F.; Meulewaeter, F.; Knox, J.P.; Blackburn, R.S. Comparative Analysis of Crystallinity Changes in Cellulose I Polymers Using ATR-FTIR, X-Ray Diffraction, and Carbohydrate-Binding Module Probes. Biomacromolecules 2011, 12, 4121–4126. [Google Scholar] [CrossRef] [PubMed]
- Ahvenainen, P.; Kontro, I.; Svedström, K. Comparison of Sample Crystallinity Determination Methods by X-Ray Diffraction for Challenging Cellulose I Materials. Cellulose 2016, 23, 1073–1086. [Google Scholar] [CrossRef]
- Heiner, A.P.; Kuutti, L.; Teleman, O. Comparison of the Interface between Water and Four Surfaces of Native Crystalline Cellulose by Molecular Dynamics Simulations. Carbohydr. Res. 1998, 306, 205–220. [Google Scholar] [CrossRef]
- Einfeldt, J.; Meißner, D.; Kwasniewski, A. Molecular Interpretation of the Main Relaxations Found in Dielectric Spectra of Cellulose–Experimental Arguments. Cellulose 2004, 11, 137–150. [Google Scholar] [CrossRef]
- Kaminski, K.; Kaminska, E.; Ngai, K.L.; Paluch, M.; Wlodarczyk, P.; Kasprzycka, A.; Szeja, W. Identifying the Origins of Two Secondary Relaxations in Polysaccharides. J. Phys. Chem. B 2009, 113, 10088–10096. [Google Scholar] [CrossRef] [PubMed]
- Thybring, E.E.; Kymäläinen, M.; Rautkari, L. Experimental Techniques for Characterising Water in Wood Covering the Range from Dry to Fully Water-Saturated. Wood Sci. Technol. 2018, 52, 297–329. [Google Scholar] [CrossRef]
- Chen, S.H. Quasi-Elastic and Inelastic Neutron Scattering and Molecular Dynamics of Water at Supercooled Temperature. In Hydrogen-Bonded Liquids; Dore, J.C., Teixeira, J., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1989; pp. 289–332. [Google Scholar]
- Gao, X.; Zhuang, S.; Jin, J.; Cao, P. Bound Water Content and Pore Size Distribution in Swollen Cell Walls Determined by NMR Technology. Bioresources 2015, 10, 8208–8224. [Google Scholar] [CrossRef]


{kind=link}
| Material Studied | Experimental Techniques Used | Findings Findings |
|---|---|---|
| Spruce Earlywood sections | Synchrotron X-ray Diffraction (XRD) | Measured compression of the cellulose crystalline lattice with increasing hydration [8]. |
| Various wood species | Cu Kα XRD | Showed moisture-induced changes in the XRD diffraction profiles [6,36,58,59], including increased crystallinity [58,59]. |
| Microcrystalline cellulose from cotton | 2H Magic-angle spinning nuclear magnetic resonance (MAS NMR) and Fourier-transform Infrared (FTIR) Spectroscopy MD simulations were also used in analysis of the data. | Provided evidence that only two hydroxyl sites on the surface of the cellulose were accessible to water molecules [15]. |
| Hemicelluloses films from rye arabinoxylan and Konjac glucomannan | FTIR | Accessibility of hydroxyl sites to D2O vapor exchange [21]. |
| Microcrystalline cellulose from cotton | 13C Cross-polarization Magic-angle Spinning (CP MAS) NMR | Tracked T1 values quantify molecular motions [15]. |
| Nanocomposites made from cellulose nanofibrils (CNF) and tamarind xyloglucan (XG) | 13C CP MAS NMR | Quantified increased molecular mobility of CNF and XG via tracking the T1 values and relaxation rates [16]. |
| Bacterial cellulose [10] and lignin from vanilla beans [23] | Quasielastic neutron scattering (QENS) combined with MD simulations | Tracked increase in mobility in terms of the mean square displacement of the polymers as a function of hydration level. |
| Loblolly pine latewood | QENS | Showed that overall mobility increased but no average mean square displacement (MSD) was calculated [26]. |
| Various polysaccharides including cellulose | Dielectric relaxation spectroscopy (DRS), and low-field NMR | DRS showed that increasing the moisture content leads to increased activation energy and cooperativity between local chains [22]. For cellulose, all relaxations (β, βwet and γ) exhibited a moisture-dependence [20]. |
| Material Studied | Experimental Techniques Used | Findings |
|---|---|---|
| Spruce | 2D T1-T2 1H Nuclear Magnetic Resonance (NMR) | Found two different spin relaxation times, attributed to less mobile water capable of swelling wood, and mobile water in voids [52]. |
| Nanocomposites made out of cellulose nanofibrils (CNFs) and tamarind xyloglucan (XG) | NMR 2H relaxometry | Found that water is less mobile in XG than CNFs and their water distribution is also different [16]. |
| Cellulose extracted from corn | Low-field NMR and dielectric relaxation spectroscopy (DRS) | Found evidence for three populations of water: tightly-bound, which was found incorporated in all relaxation units, freezable loosely-bound, and non-freezable loosely-bound, which acted as a plasticizer [20]. |
| Gingko biloba | Micro Fourier-transform infrared spectroscopy (FTIR) | Found three water populations: strongly bonded, moderately bonded, and weakly bonded to hydrogen. The relative content of the water populations changed with humidity [25]. |
| Bacterial cellulose | Quasielastic neutron scattering (QENS) and Molecular Dynamics (MD) simulations | Found two water populations and described the dynamics of one using a jump diffusion model [24]. |
| Loblolly pine latewood | QENS | Found two water populations: tightly-bound and loosely-bound water. The diffusive behavior of both populations was quantified using a jump diffusion model [26]. |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaza, N.Z. On the Experimental Assessment of the Molecular-Scale Interactions between Wood and Water. Forests 2019, 10, 616. https://doi.org/10.3390/f10080616
Plaza NZ. On the Experimental Assessment of the Molecular-Scale Interactions between Wood and Water. Forests. 2019; 10(8):616. https://doi.org/10.3390/f10080616
Chicago/Turabian StylePlaza, Nayomi Z. 2019. "On the Experimental Assessment of the Molecular-Scale Interactions between Wood and Water" Forests 10, no. 8: 616. https://doi.org/10.3390/f10080616
APA StylePlaza, N. Z. (2019). On the Experimental Assessment of the Molecular-Scale Interactions between Wood and Water. Forests, 10(8), 616. https://doi.org/10.3390/f10080616