Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China

 , ,
, , {kind=link}
{kind=link}
{kind=link}
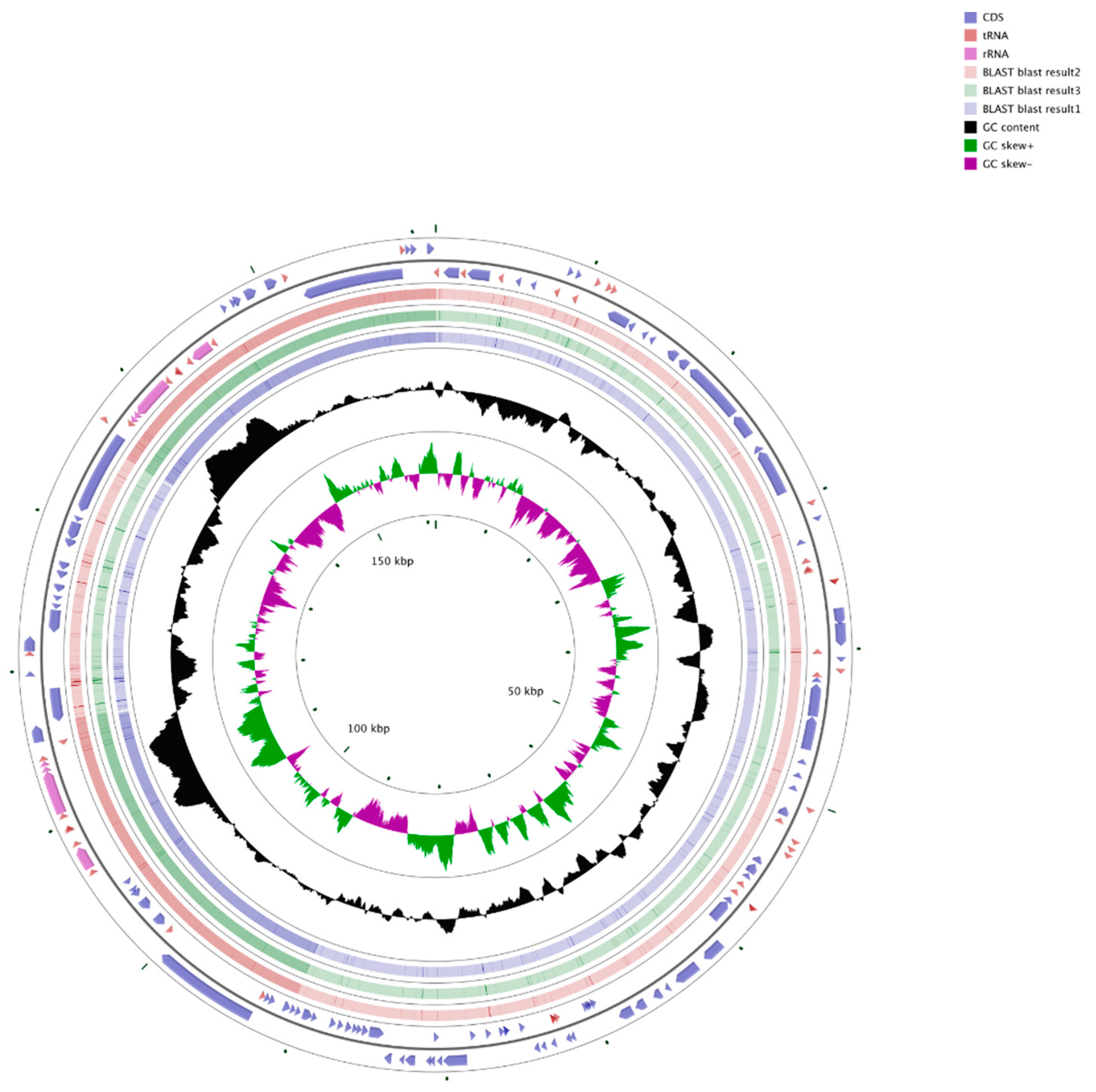{kind=link}
{kind=link}
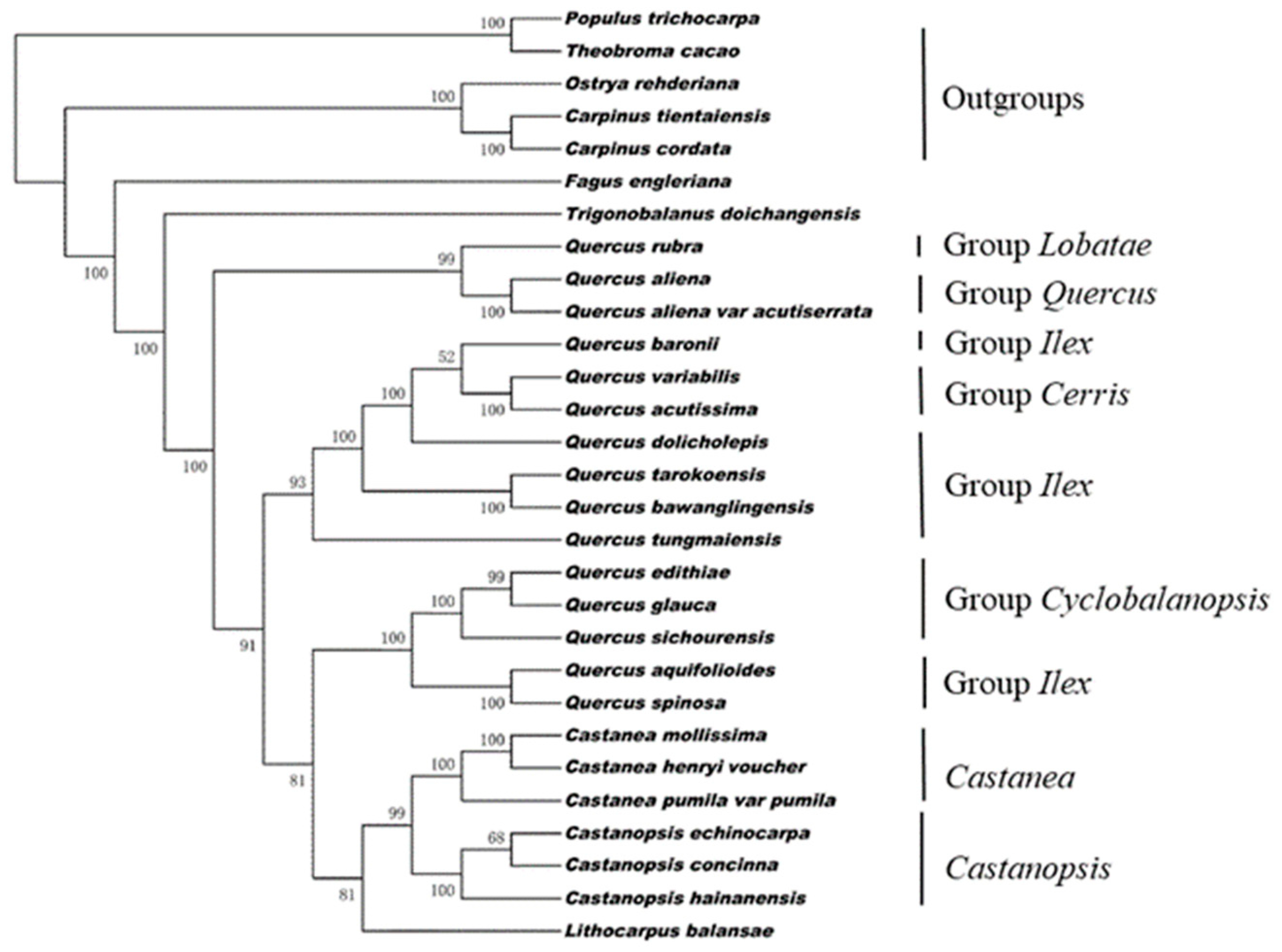{kind=link}
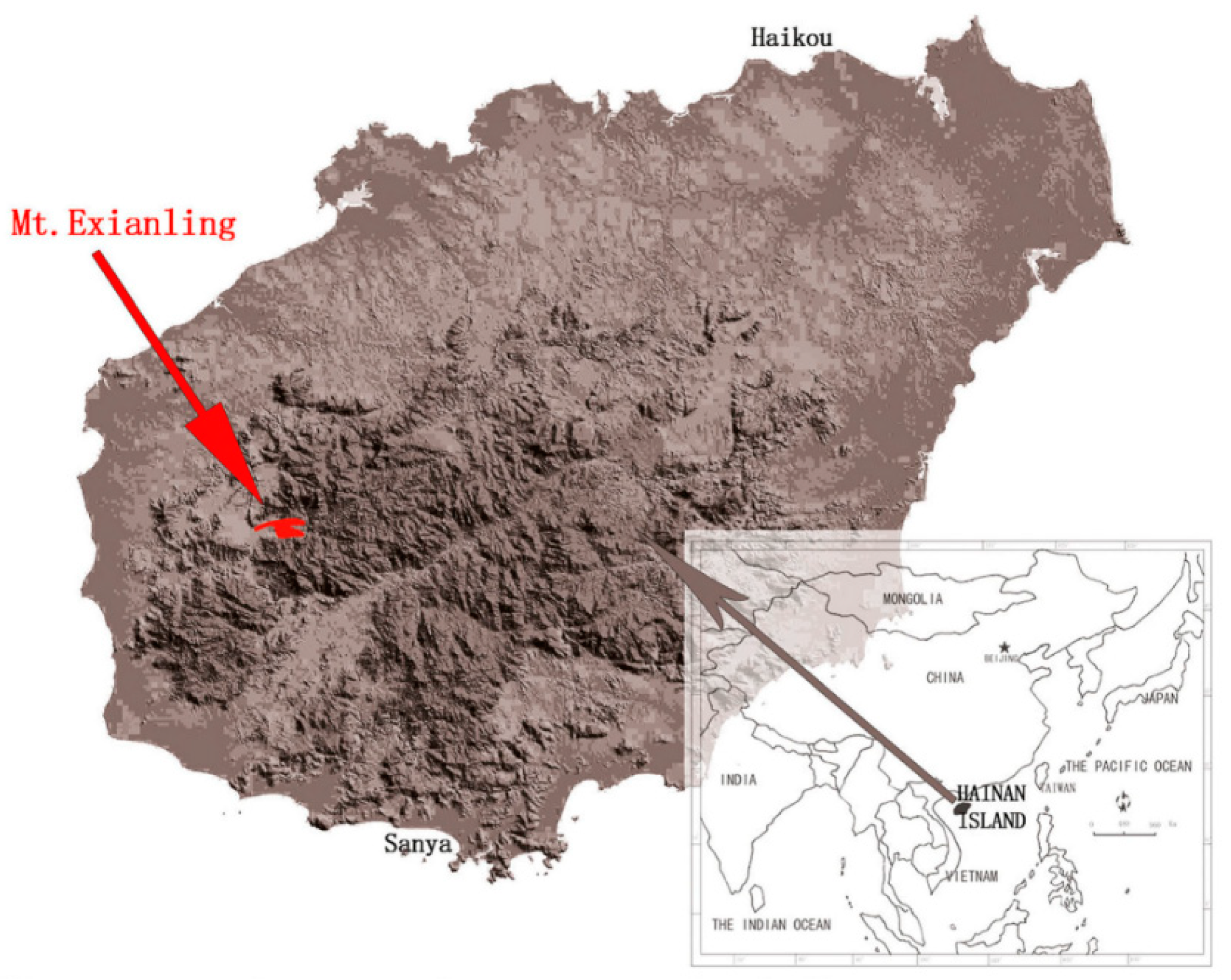{kind=link}
Abstract
Share and Cite
Liu, X.; Chang, E.-M.; Liu, J.-F.; Huang, Y.-N.; Wang, Y.; Yao, N.; Jiang, Z.-P. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China. Forests 2019, 10, 587. https://doi.org/10.3390/f10070587
Liu X, Chang E-M, Liu J-F, Huang Y-N, Wang Y, Yao N, Jiang Z-P. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China. Forests. 2019; 10(7):587. https://doi.org/10.3390/f10070587
Chicago/Turabian StyleLiu, Xue, Er-Mei Chang, Jian-Feng Liu, Yue-Ning Huang, Ya Wang, Ning Yao, and Ze-Ping Jiang. 2019. "Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China" Forests 10, no. 7: 587. https://doi.org/10.3390/f10070587
APA StyleLiu, X., Chang, E.-M., Liu, J.-F., Huang, Y.-N., Wang, Y., Yao, N., & Jiang, Z.-P. (2019). Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China. Forests, 10(7), 587. https://doi.org/10.3390/f10070587