In this Section, we present two new heuristic algorithms for the MaxRTC problem.
4.1. FastTree
The first heuristic algorithm has a bottom-up greedy approach, which is faster than the other previously known algorithms employing a simple data structure.
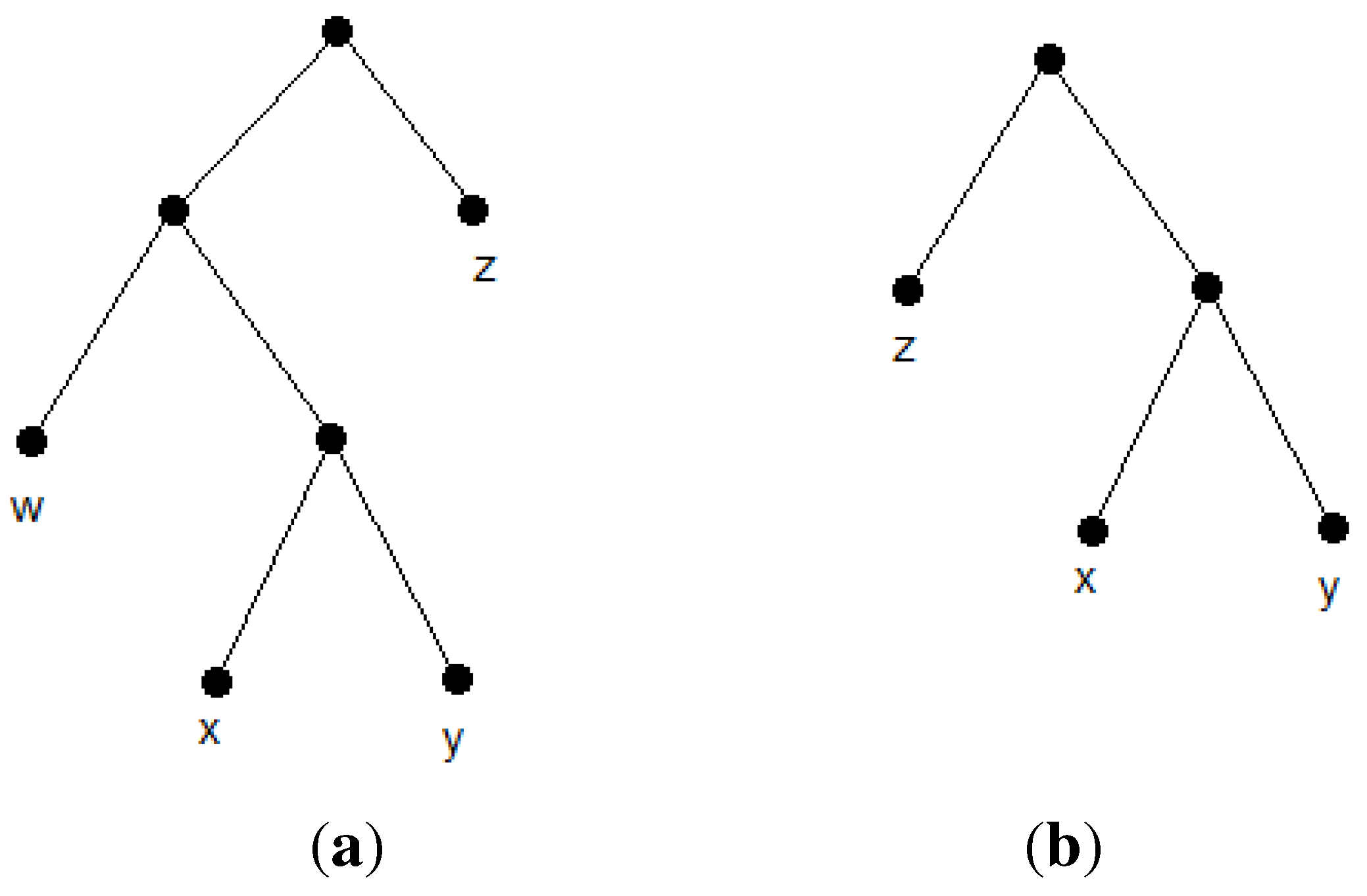
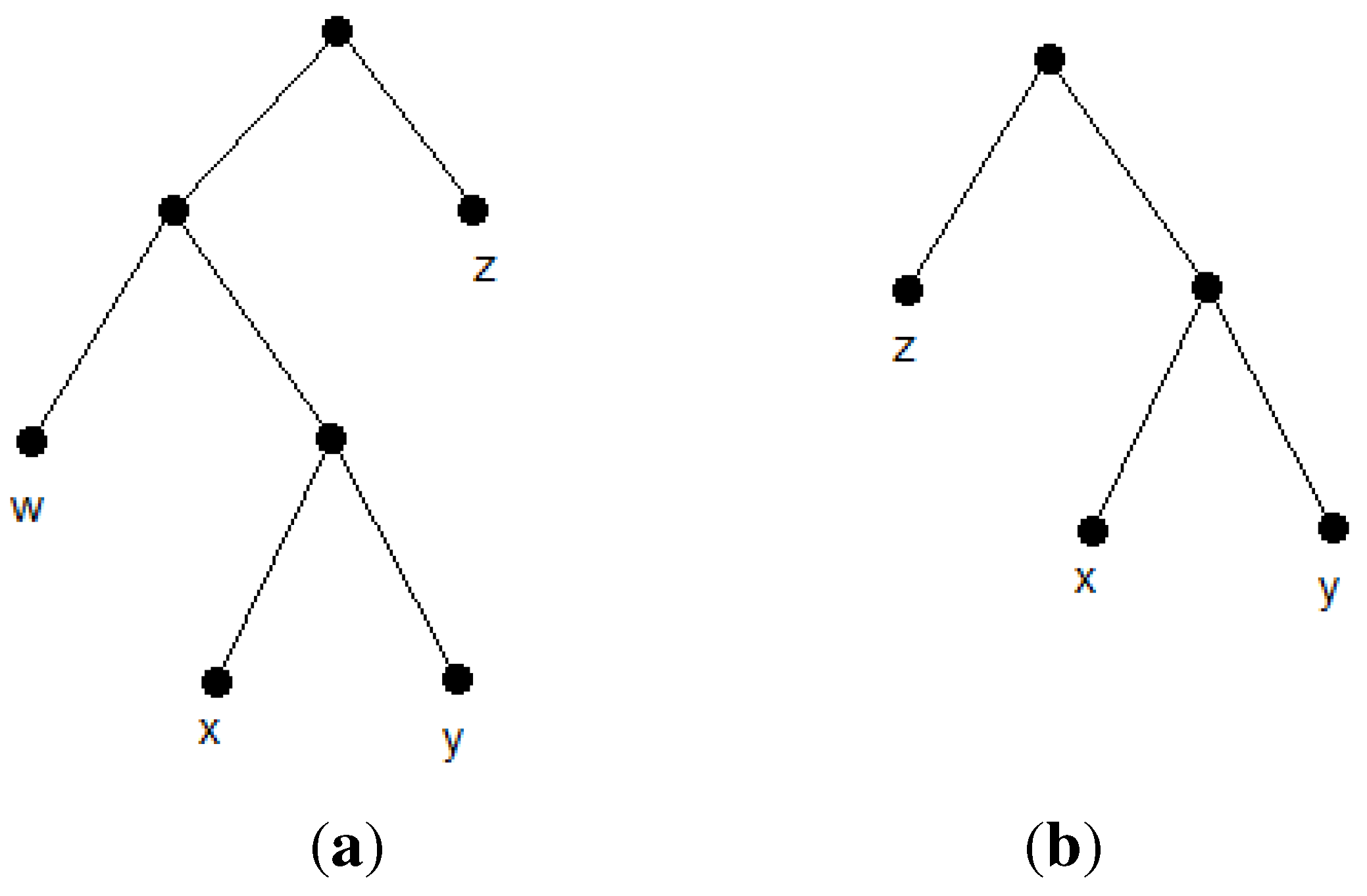
Let
R(T) denote the set of all triplets consistent with a given tree,
T.
R(T) is called the
reflective triplet set of
T. It forms a minimally dense triplet set and represents
T uniquely [
17]. Now, we define the
closeness of the pair,
{i,j}. The closeness of the pair,
{i,j},
, is defined as the number of triplets of the form,
ij|k, in a triplet set. Clearly, for any arbitrary tree,
T, the closeness of a cherry species equals
, which is maximum in
R(T). The reason is that every cherry species has a triplet with every other species. Now, suppose we contract every cherry species of the form, {
i,j}, to their parents,
, and then update
R(T) as follows. For each contracted cherry species, {
i,j}, we remove triplets of the form,
ij|
k, from
R(T) and replace
i and
j with
within the remaining triplets. The updated set,
, would be the reflective triplet set for the new tree,
. Observe that, for cherries of the form,
, in
,
and
would equal n-3 in
R(T). Similarly, for cherries of the form,
, in
,
,
,
and
would equal n-4 in
R(T). This forms the main idea of the first heuristic algorithm. We first compute the closeness of pairs of species by visiting triplets. Furthermore, sorting the pairs according to their closeness gives us the reconstruction order of the tree. This routine outputs the unique tree,
T, for any given reflective triplet set,
R(T). Yet, we have to consider that the input triplet set is not always a reflective triplet set. Consequently, the reconstruction order produced by sorting may not be the right order. However, if the loss of triplets admits a uniform distribution, it will not affect the reconstruction order. An approximate solution for this problem is refining the closeness. This can be done by reducing the closeness of the pairs, {
i,k} and {
j,k}, for any visited triplet of the form,
ij|
k. Thus, if the pair, {
i,j}, is actually cherries, then the probability of choosing the pairs, {
i,k} or {
j,k}, before choosing the pair, {
i,j}, due to triplet loss, will be reduced. We call this algorithm FastTree. See Algorithm 1 for the whole algorithm.
| Algorithm 1 FastTree |
- 1:
Initialize a forest, F, consisting of n one-node trees labeled by species. - 2:
for each triplet of the form ij|k do - 3:
: = +1 - 4:
: = −1 - 5:
: = −1 - 6:
end for - 7:
Create a list, L, of pairs of species. - 8:
Sort L according to the refined closeness of pairs with a linear-time sorting algorithm. - 9:
while |L|>0 do - 10:
Remove the pair, {i,j}, with maximum, . - 11:
if i and j are not in the same tree then - 12:
Add a new node and connect it to roots of trees containing i and j. - 13:
end if - 14:
end while - 15:
if F has more than one tree then - 16:
Merge trees in any order, until there would be only one tree. - 17:
end if - 18:
return the tree in F
|
Theorem 1. FastTree runs in time.
Proof. Initializing a forest in Step 1 takes
time. Steps 2–6 take
time. We know that the closeness is an integer value between 0 and
. Thus, we can employ a linear-time sorting algorithm [
18]. There are
possible pairs; therefore, Step 8 takes
time. Similarly, the while loop in Step 9 takes
time. Each removal in Step 10 can be done in
time. By employing optimal data structures, which are used for disjoint-set unions [
18], the amortized time complexity of Steps 11 and 12 will be
, where
is the inverse of the function,
, and
A is the well-known fast-growing
Ackermann function. Furthermore, Step 16 takes
time. Hence, the running time of FastTree would be
. ☐
Since
,
is less than four for any practical input size,
n. In comparison to the fast version of Aho
et al.’s algorithm, FastTree employs a simpler data structure and, in comparison to Aho
et al.’s original algorithm, it has lower time complexity. Yet, the most important advantage of FastTree to Aho
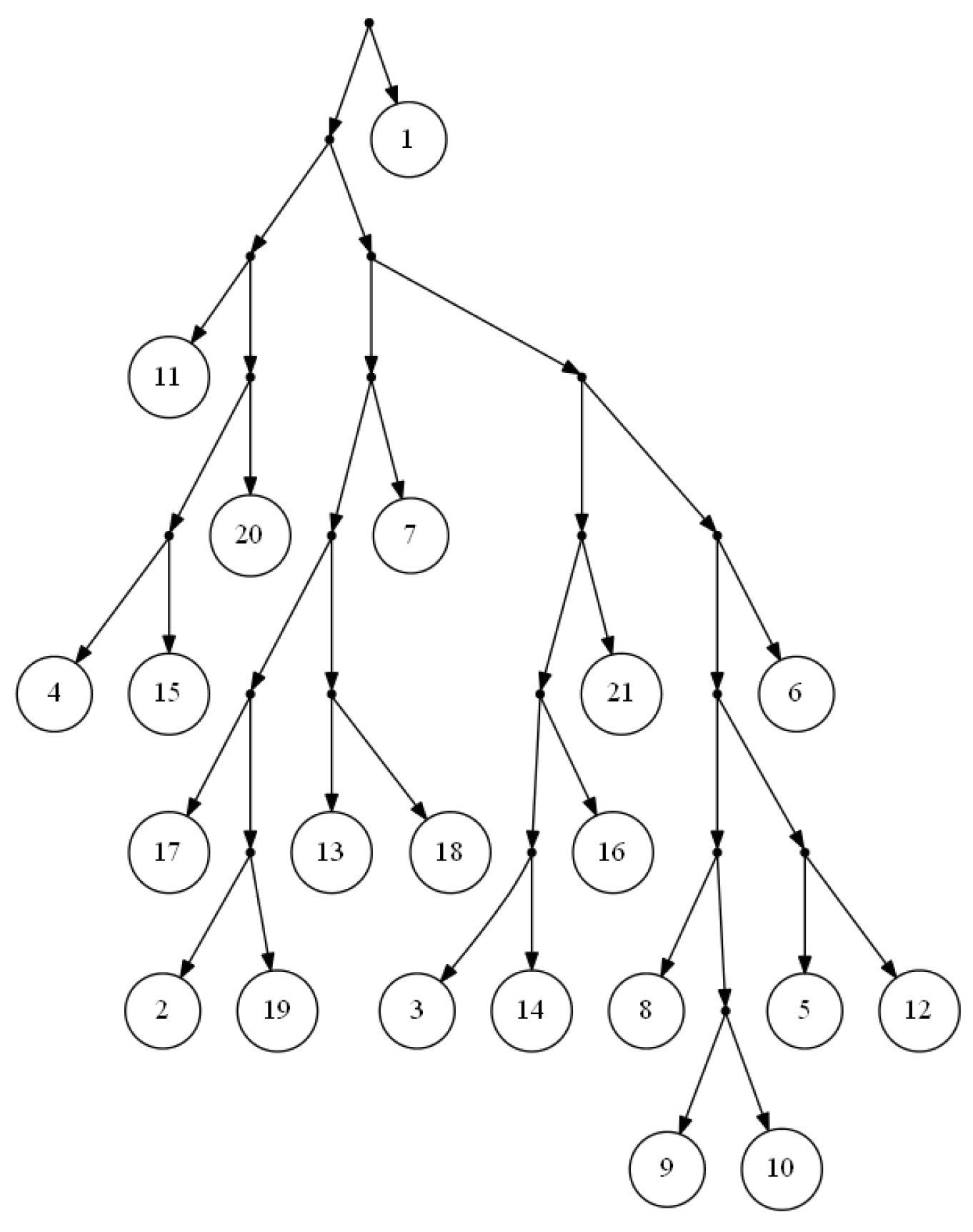
et al.’s algorithm is that it will not stick if there is not a consistent tree with the input triplets, and it will output a proper tree in such a way that the clusters are very similar to that of the real network. The tree in
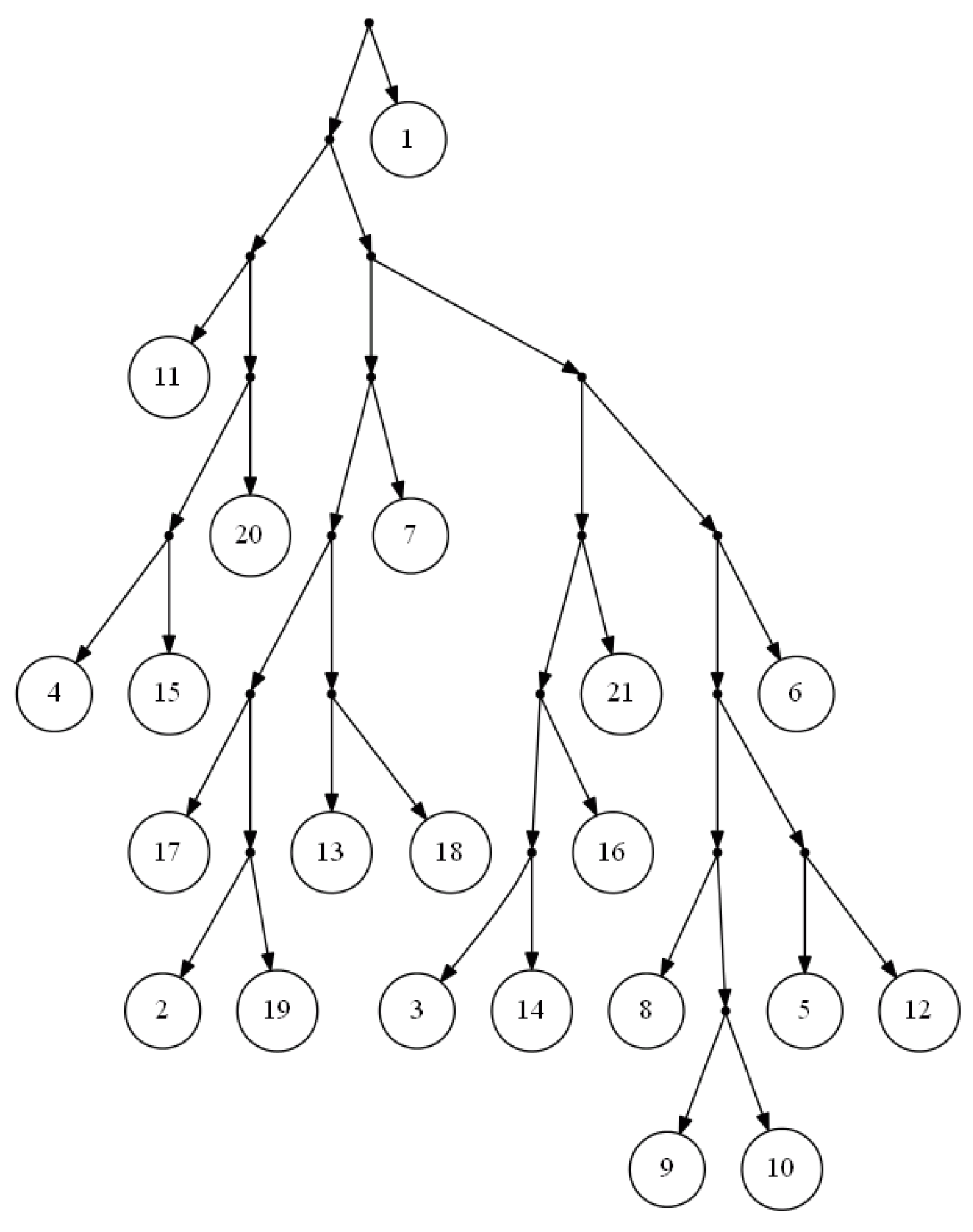
Figure 2 is the output of FastTree on a dense set of triplets based on the yeast,
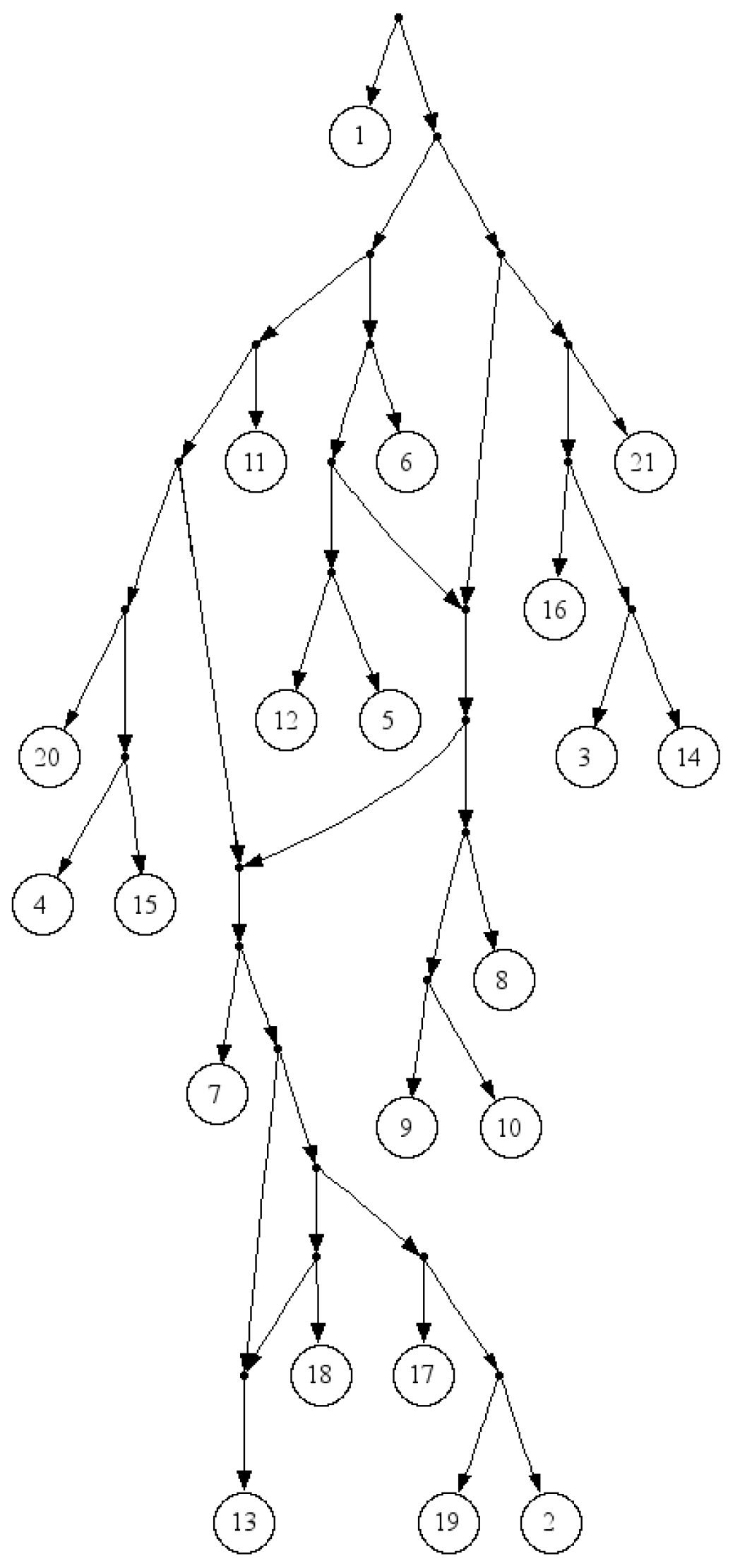
Cryptococcus gattii, data. There is no consistent tree with the whole triplet set; however, Van Iersel
et al. [
19] presented a level-2 network consistent with the set (see
Figure 3). This set is available online [
20]. In comparison to BPMR and BPMF, FastTree runs much faster for large sets of triplets and species. However, for highly sparse triplet sets, the output of FastTree may satisfy considerably less triplets than the tree constructed by BPMF or BPMR.
Figure 2.
Output of FastTree for a dense triplet set of the yeast, Cryptococcus gattii, data.
Figure 2.
Output of FastTree for a dense triplet set of the yeast, Cryptococcus gattii, data.
4.2. BPMTR
Before explaining the second heuristic algorithm, we need to review BPMF [
11] and BPMR [
16]. BPMF utilizes a bottom-up approach similar to hierarchical clustering. Initially, there are
n trees, each containing a single node representing one of
n given species. In each iteration, the algorithm computes a function, called
, for each combination of two trees. Furthermore, two trees with the maximum
are merged into a single tree by adding a new node as the common parent of the selected trees. Wu [
11] introduced six alternatives for computing the
using combinations of
w,
p and
t. (see
Table 1). However, in each run, one of the six alternatives must be used. In the function,
,
w is the number of triplets satisfied by merging
and
, which is the number of triplets of the form
ij|
k, in which
i is in
,
j is in
and
k is neither in
nor in
. The value of
p is the number of triplets that are in conflict with merging
and
. It is the number of triplets of the form,
ij|
k, in which
i is in
,
k is in
and
j is neither in
nor in
. The value of
t is the total number of triplets of the form,
ij|
k, in which
i is in
and
j is
. Wu compared the BPMF with
One-Leaf-Split and
Min-Cut-Split and showed that BPMF works better on randomly generated triplet sets. He also pointed out that none of the six alternatives of
is significantly better than the other.
Figure 3.
A Level-2 network for a dense triplet set of the yeast, Cryptococcus gattii, data.
Figure 3.
A Level-2 network for a dense triplet set of the yeast, Cryptococcus gattii, data.
Table 1.
The six alternatives of e_score.
Table 1.
The six alternatives of e_score.
| If-Penalty | | Ratio Type | |
|---|
| False | w | w/(w + p) | w/t |
| True | w − p | (w − p)/(w + p) | (w − p)/t |
Maemura
et al. [
16] introduced a modified version of BPMF, called BPMR, that outperforms the results of BPMF. BPMR works very similarly in comparison to BPMF, except for a reconstruction step used in BPMR. Suppose
and
are two trees having the maximum,
, at some iteration and are selected to merge into a new tree. By merging
and
, some triplets will be satisfied, but some other triplets will be in conflict. Without loss of generality, suppose
has two subtrees, namely the left subtree and the right subtree. In addition, suppose a triplet,
ij|
k, in which
i is in the left subtree of
,
k is in the right subtree of
and
j is in
. Observe that by merging
and
, the mentioned triplet becomes inconsistent. However, swapping
with the right subtree of the
satisfies this triplet, while some other triplets become inconsistent. It is possible that the resulting tree of this swap satisfies more triplets than the primary tree. This is the main idea behind the BPMR. In BPMR, in addition to the regular merging of
and
,
is swapped with the left and the right subtree of
, and also,
is swapped with the left and the right tree of
. Finally, among these five topologies, we choose the one that satisfies the most triplets.
Suppose the left subtree of the also has two subtrees. Swapping with one of these subtrees would probably satisfy new triplets, while some old ones would become inconsistent. There are examples in which this swap results in a tree that satisfies more triplets. This forms our second heuristic idea that swapping of with every subtree of should be checked. should also be swapped with every subtree of . At every iteration of BPMF after choosing two trees maximizing the , the algorithm tests every possible swapping of these two trees with subtrees of each other and, then, chooses the tree with the maximum consistency of the triplets. We call this algorithm BPMTR (Best Pair Merge with Total Reconstruction). See Algorithm 2 for details of the BPMTR.
| Algorithm 2 BPMTR |
- 1:
Initialize a set, T, consisting of n one-node trees labeled by species. - 2:
while |T|>1 do - 3:
Find and remove two trees, , , with maximum . - 4:
Create a new tree, , by adding a common parent to and - 5:
: = - 6:
for each subtree of do - 7:
Let be the tree constructed by swapping with - 8:
if the number of consistent triplets with was larger than the number of triplets consistent with then - 9:
: = - 10:
end if - 11:
end for - 12:
for each subtree of do - 13:
Let be the tree constructed by swapping with - 14:
if the number of consistent triplets with was larger than the number of triplets consistent with then - 15:
: = - 16:
end if - 17:
end for - 18:
Add to T. - 19:
end while - 20:
return the tree in T
|
Theorem 2. BPMTR runs in time.
Proof. Step 1 takes
time. In Step 2, initially,
T contains
n clusters, but in each iteration, two clusters merge into a cluster. Hence, the while loop in Step 2 takes
time. In Step 3,
is computed for every subset of
T of size two. By applying Bender and Farach-Colton’s preprocessing algorithm [
21], which runs in
time for a tree with
n nodes, every LCAquery can be answered in
time. Therefore, the consistency of a triplet with a cluster can be checked in
time. Since there are
m triplets, Step 3 takes
time. In Steps 5, 9 and 15,
is a pointer that stores the best topology found so far during each iteration of the while loop in
time. The complexity analysis of the loops in Steps 6–11 and 12–17 are similar, and it is enough to consider one. Every rooted binary tree with
n leaves has
internal nodes, so the total number of swaps in Step 7 for any two clusters will be at most
. In Step 8, computing the number of consistent triplets with
takes no more than
time. Steps 4, 7 and 18 are implementable in
time. Accordingly, the running time of Steps 2–19 would be:
Step 20 takes time. Hence, the time complexity of BPMTR is . ☐
We tested BPMTR over randomly generated triplet sets with
n = 15, 20 species and
m = 500, 1,000 triplets. We experimented hundreds of times for each combination of
n and
m. The results in
Table 2 indicate that BPMTR outperforms BPMR. However, in these hundreds of tests, there were a few examples of BPMR performing better than BPMTR. For
n = 30 and
m = 1,000, in 62 triplet sets out of a hundred randomly generated triplet sets, BPMTR satisfied more triplets. In 34 triplet sets, BPMR and BPMTR had the same results, and in four triplet sets, BPMR satisfied more triplets.
Table 2.
Performance results of Best Pair Merge with Total Reconstruction (BPMTR) in comparison to Best Pair Merge with Reconstruction (BPMR).
Table 2.
Performance results of Best Pair Merge with Total Reconstruction (BPMTR) in comparison to Best Pair Merge with Reconstruction (BPMR).
| No. of species and triplets | % better results | % worse results |
|---|
| n = 20, m = 500 | %29 | %0.0 |
| n = 20, m = 1000 | %37 | %1 |
| n = 30, m = 500 | %61 | %3 |
| n = 30, m = 1000 | %62 | %4 |


{kind=link}
{kind=link}
{kind=link}