Algorithm for the Analysis of Tryptophan Fluorescence Spectra and Their Correlation with Protein Structural Parameters

Abstract
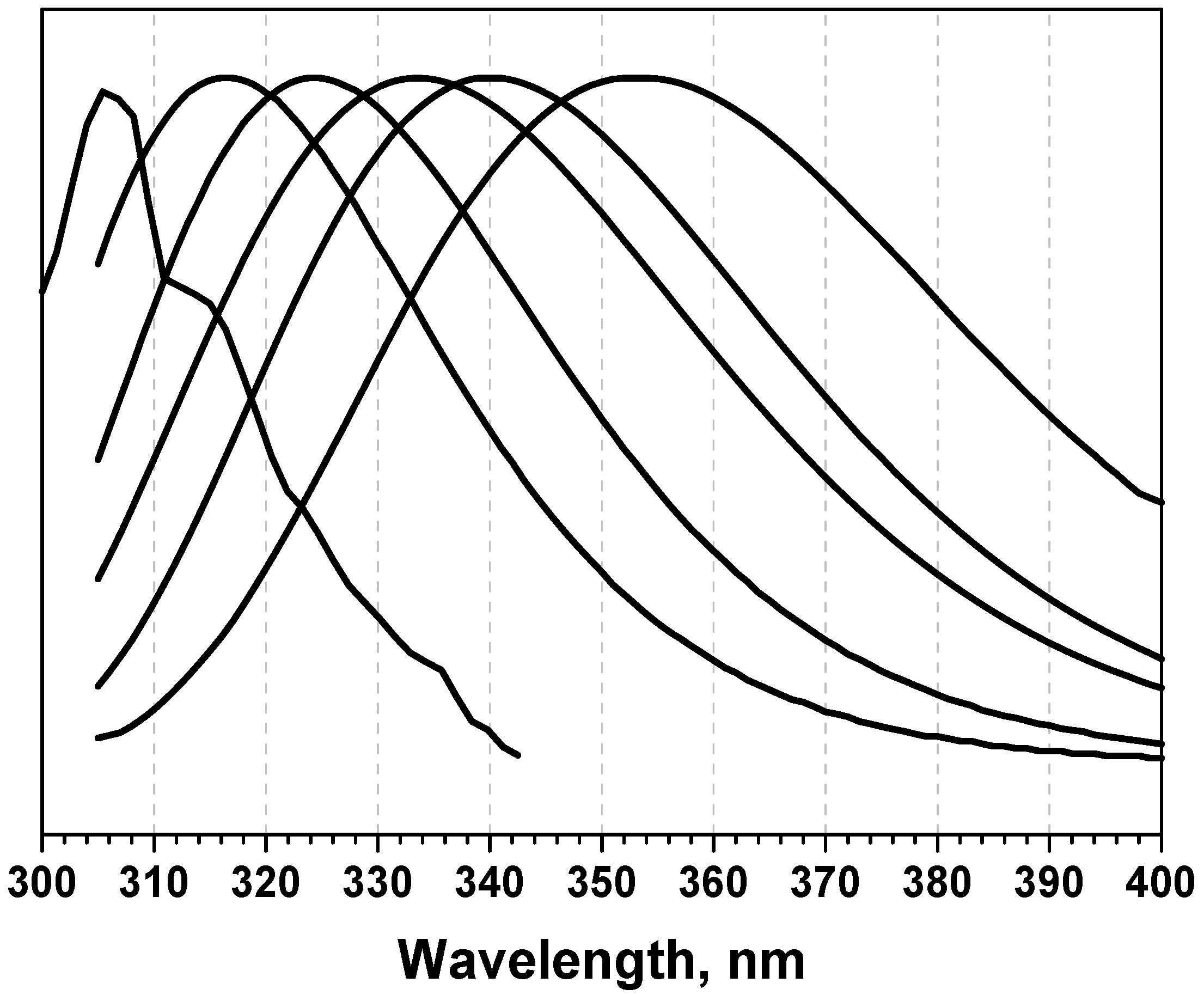
:
1. Methods for the Analysis of Protein Fluorescence Spectra
1.1. Protein fluorescence
1.2. Complex nature of protein fluorescence spectra
1.3. Decomposition algorithms
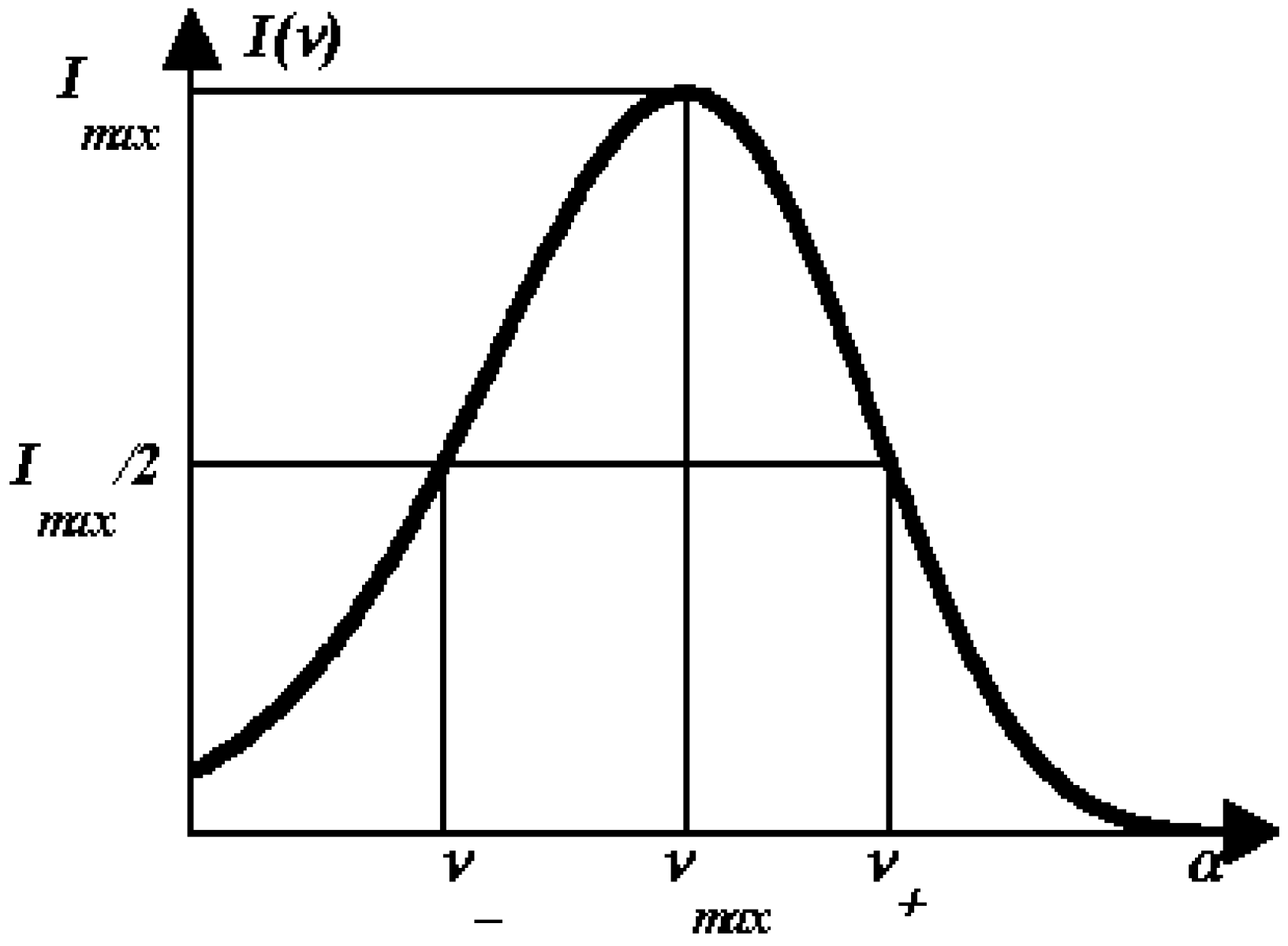
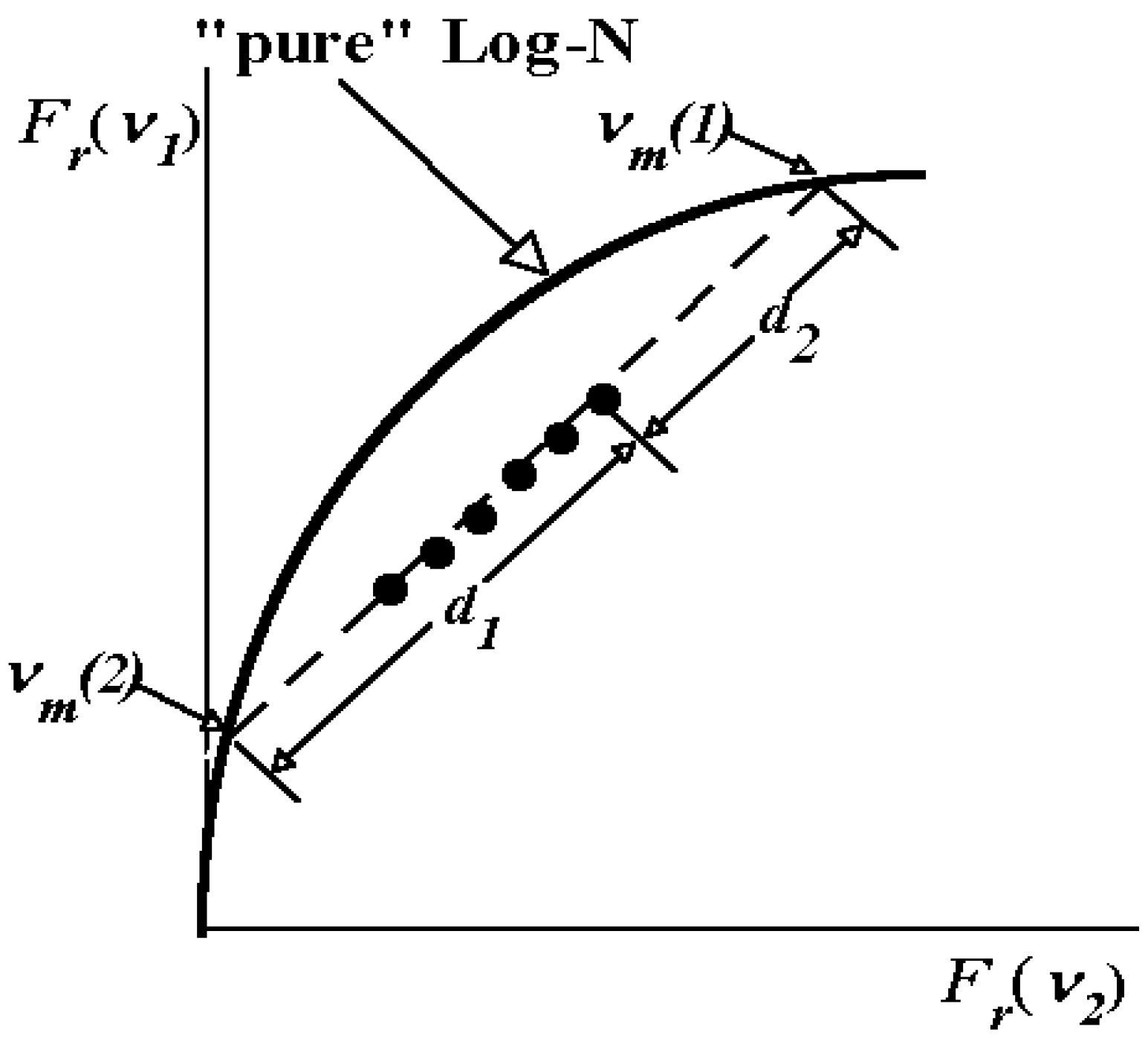
1.4. Log-normal function for describing spectral curves
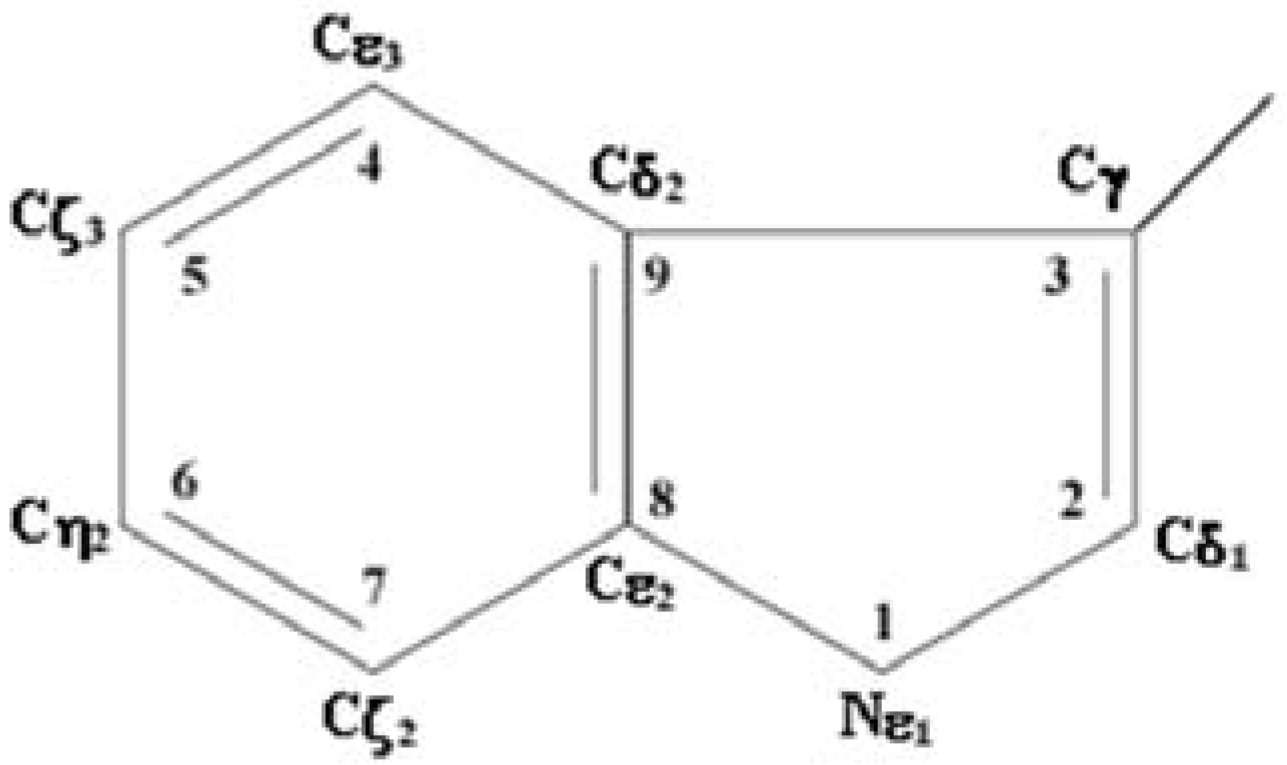
1.5. SIMS and PHREQ decomposition algorithms
- 1)
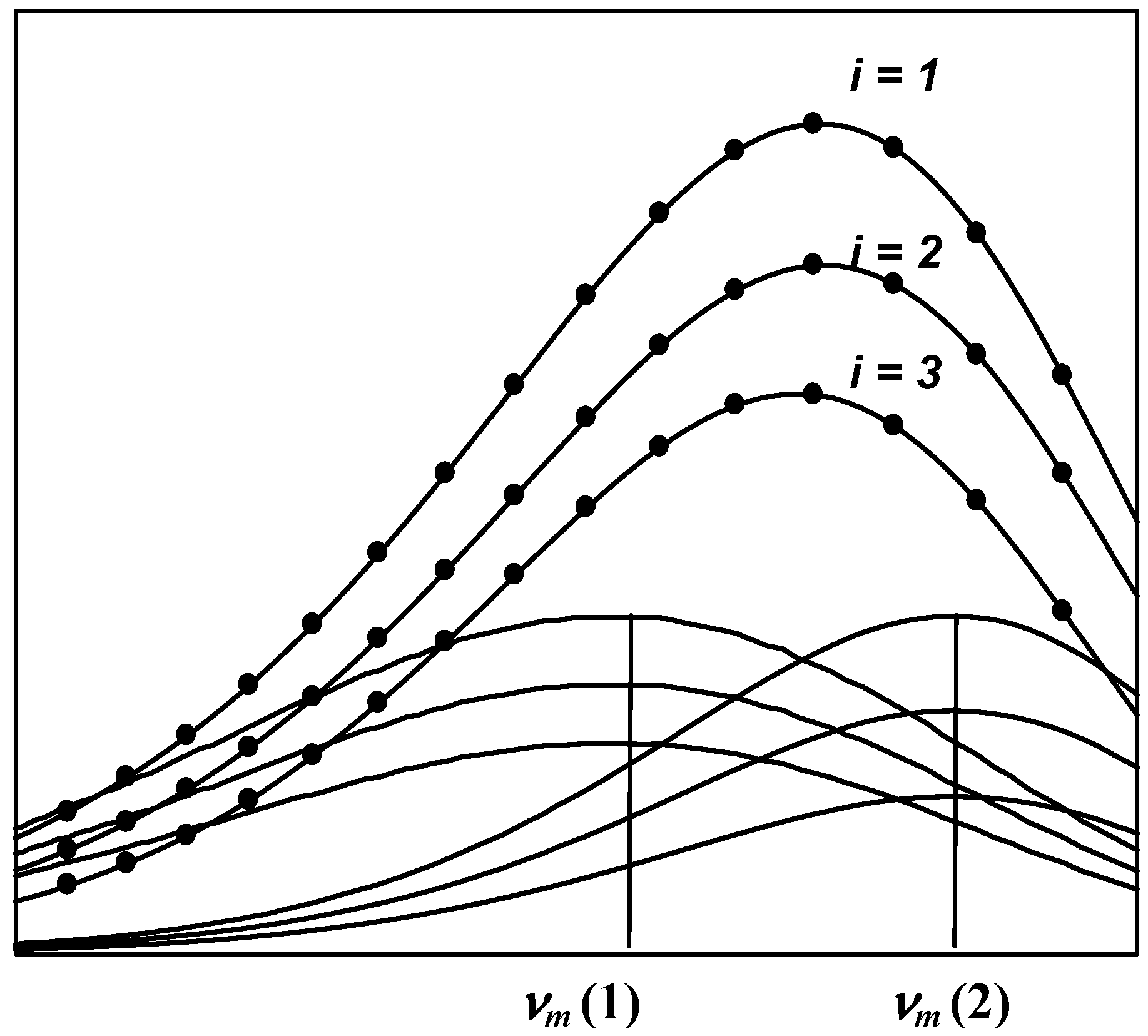
- The spectrum of an elementary component on the frequency (wave number) scale is described by a bi-parametric (maximal amplitude and position of the maximum) log-normal function (Equation 2 and 3).
- 2)
- The shape and position of the tryptophan emission spectra remain unchanged by quenching of the fluorescence by small water-soluble quenchers [34,35]. Thus, a series of spectra measured at various quencher concentrations represents a sum of spectral components whose position and shape are constant at all quencher concentrations, while the relative spectral contributions (intensities) are changed. This allows the analysis of all spectra measured at different concentrations of quencher in a global mode.
- 3)
- The change in amplitudes of the individual components induced by quenching obeys the Stern-Volmer law (Equation 1).
- 4)
- The number of experimental points under analysis greatly exceeds the number of parameters sought. This approach attenuates the effect of occasional noise [49].
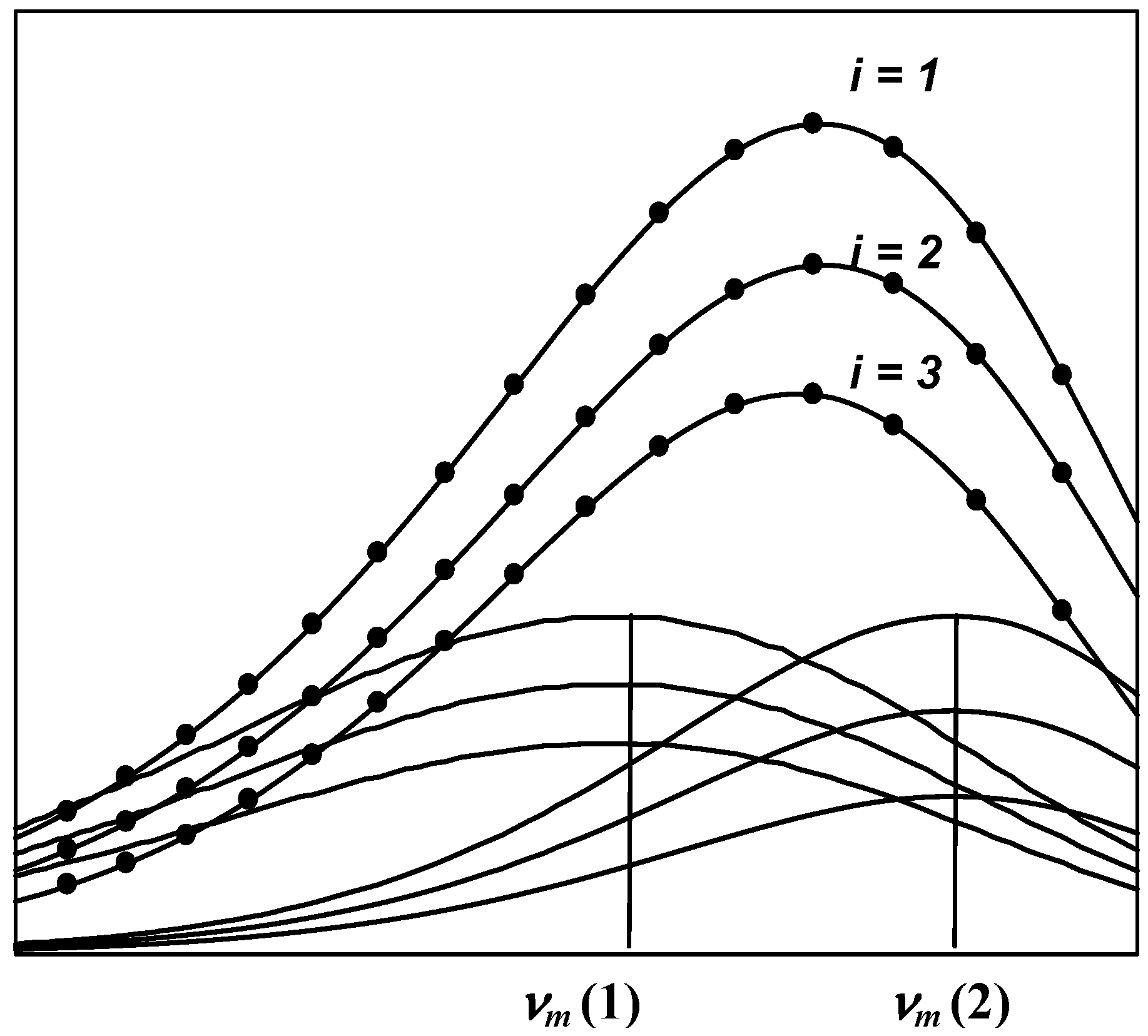
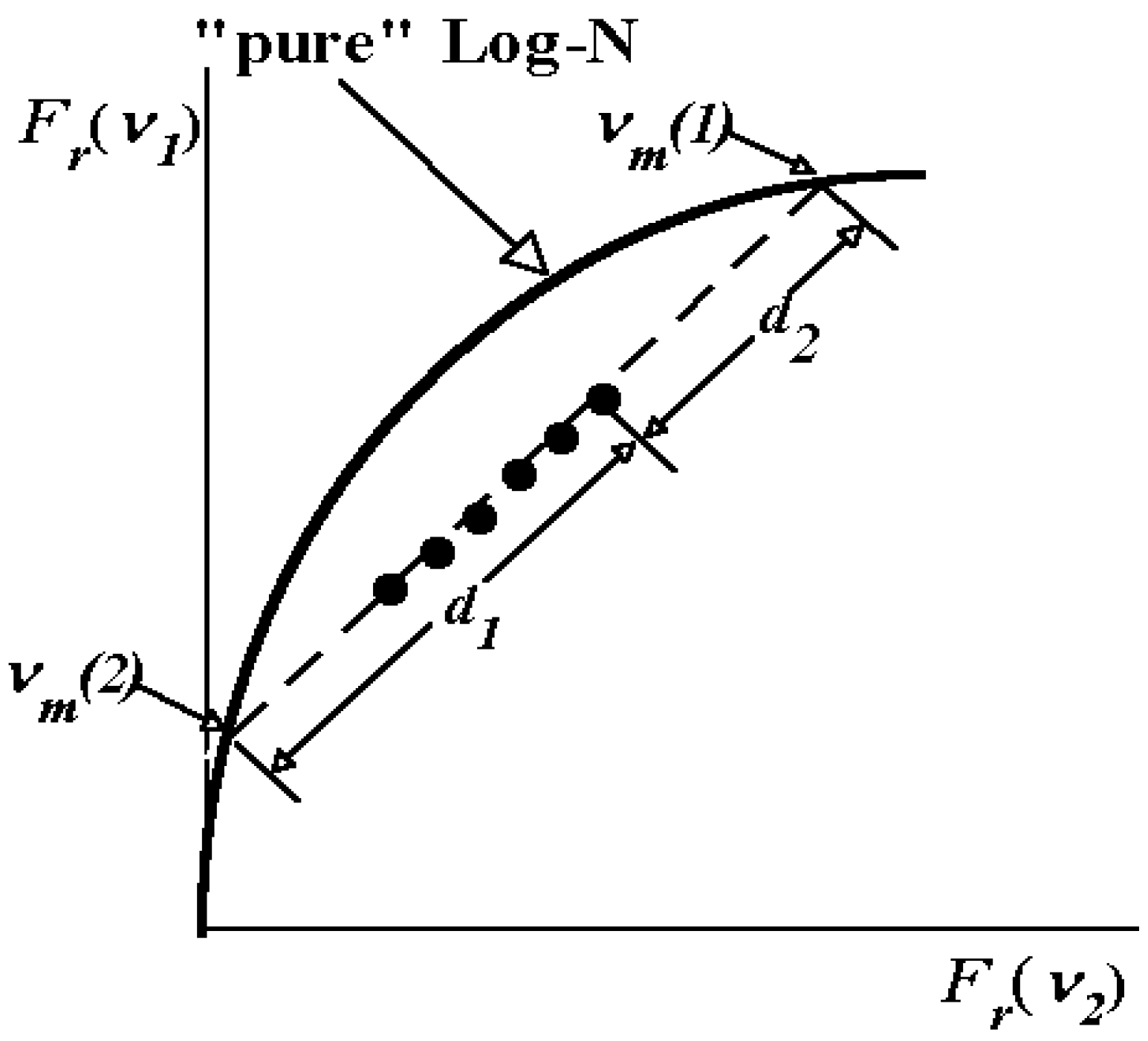
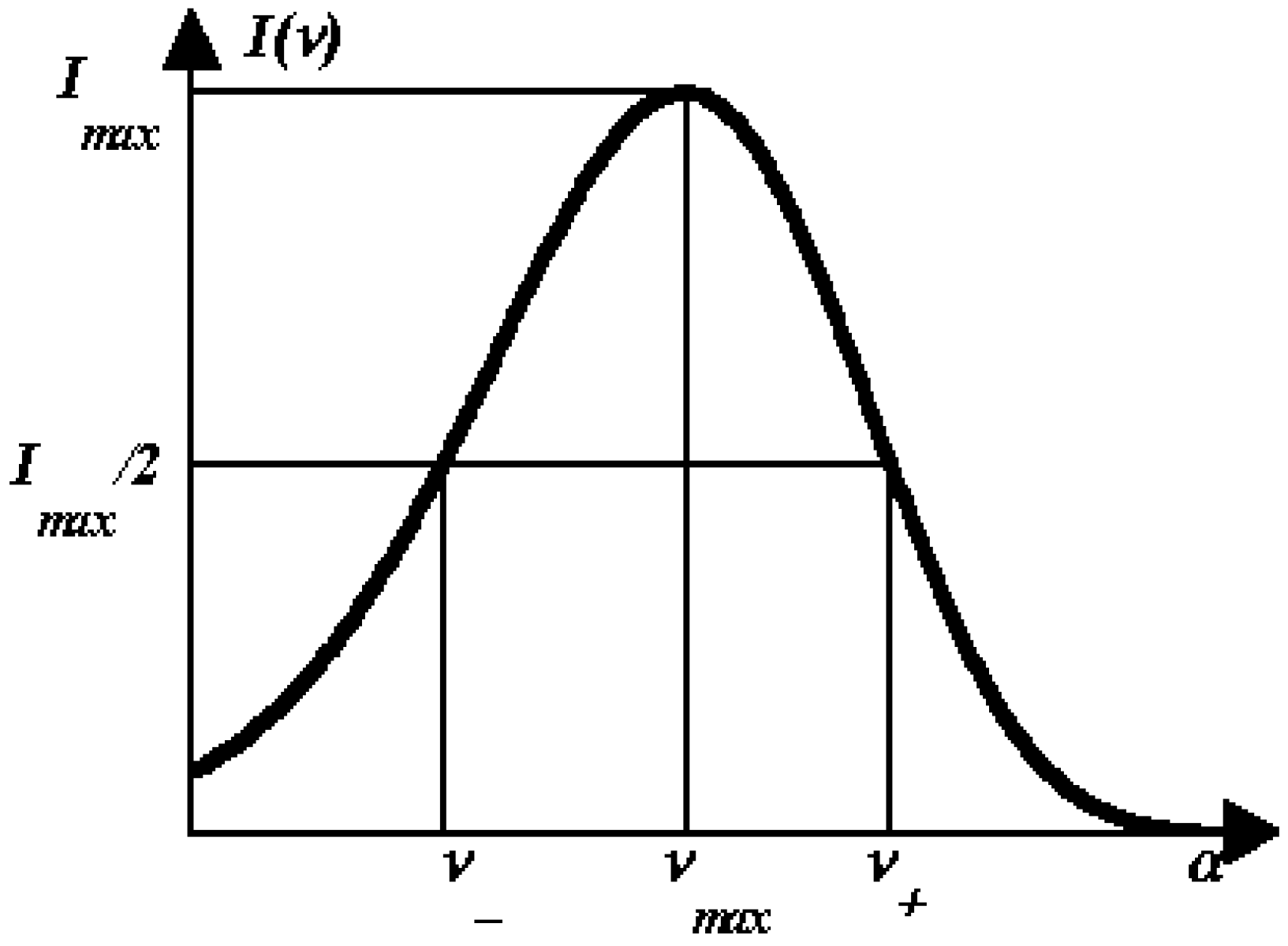
1.6. Accuracy of decomposition algorithms
2. Algorithm for the Analysis of Structural Properties of Environment of Tryptophan Residues from Atomic Structures of Proteins
3. Correlation between Spectral and Structural Properties of Tryptophan Residues
3.1. Statistical classification approaches
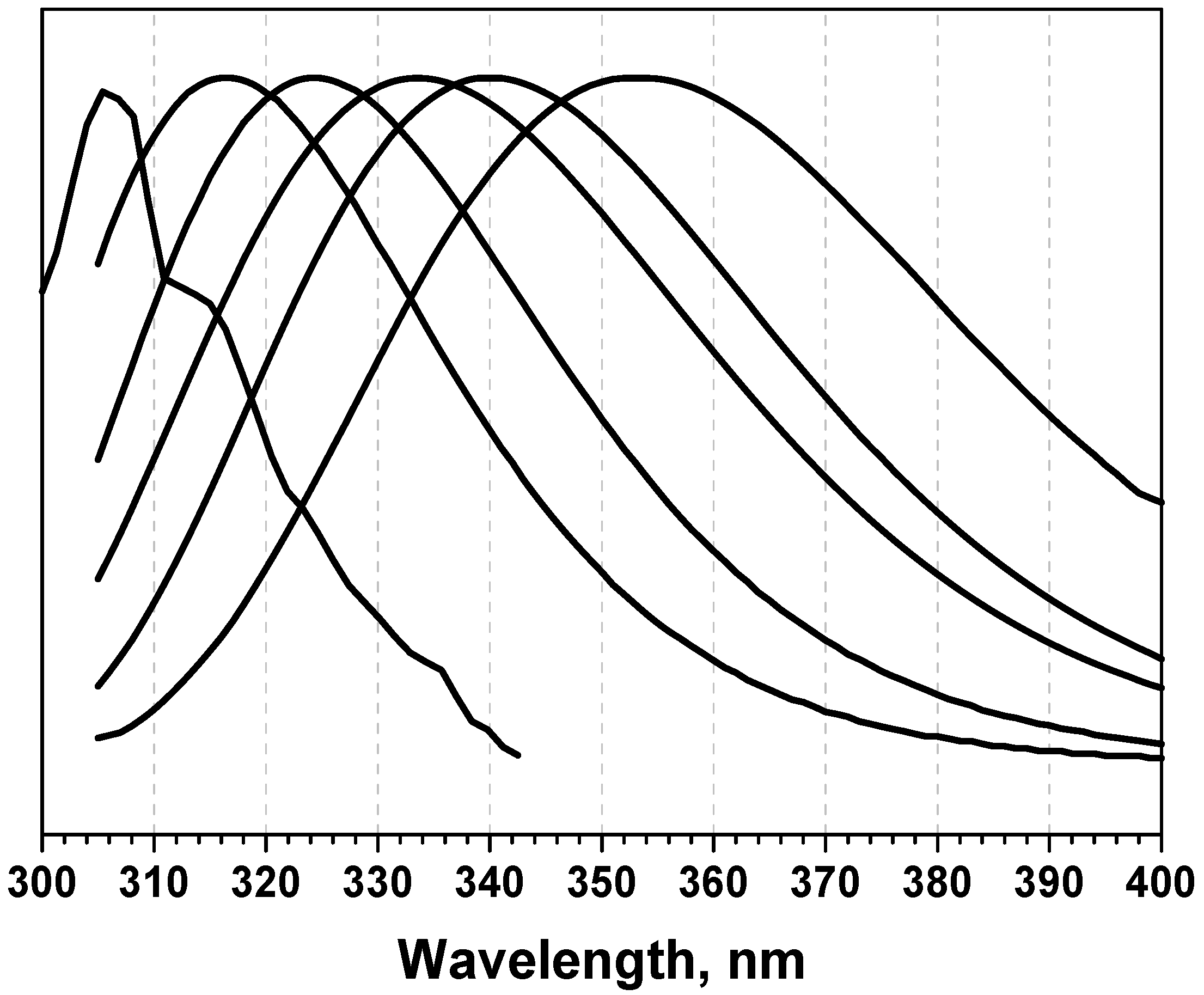
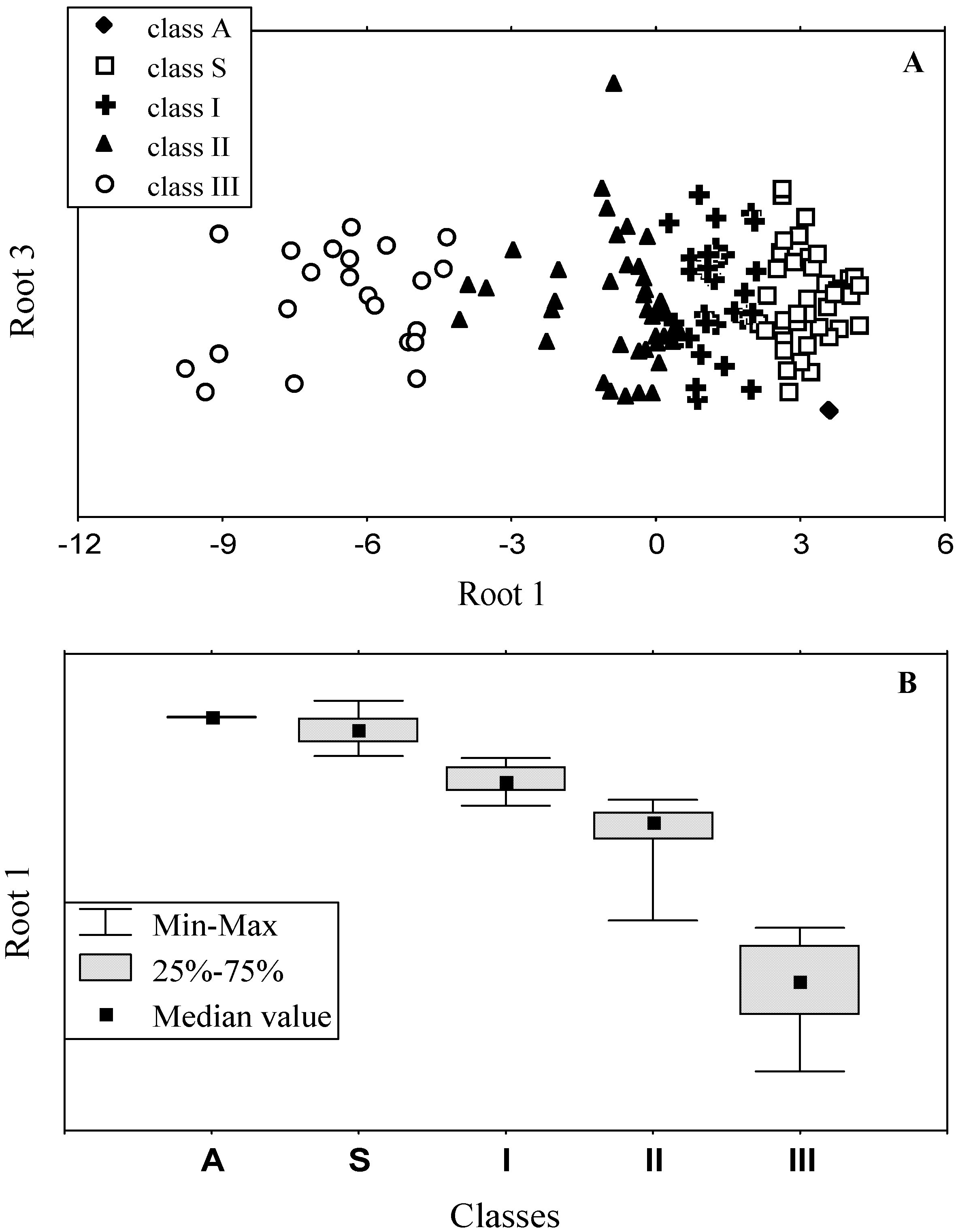
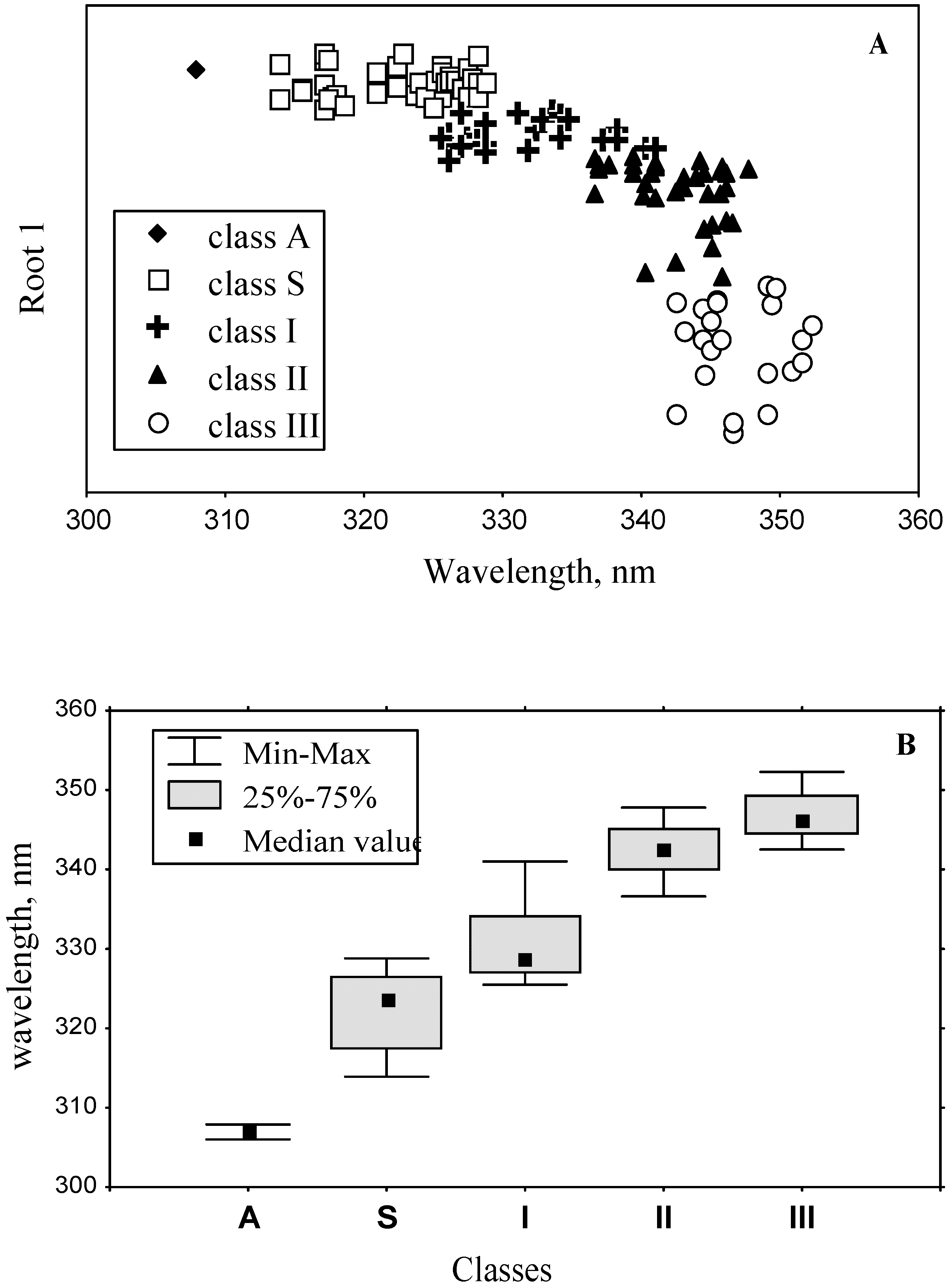
3.2. Classification of tryptophan residues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
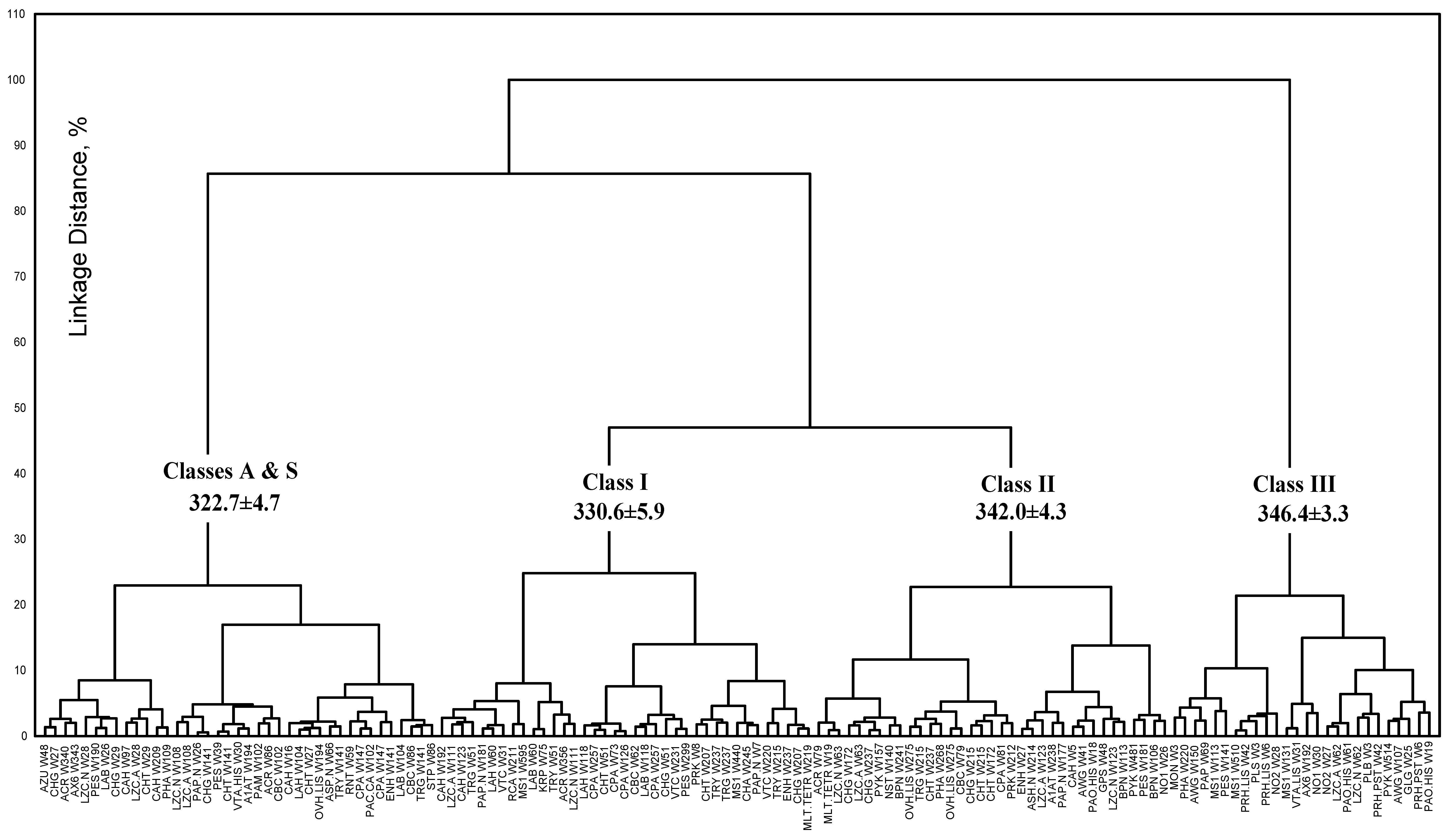
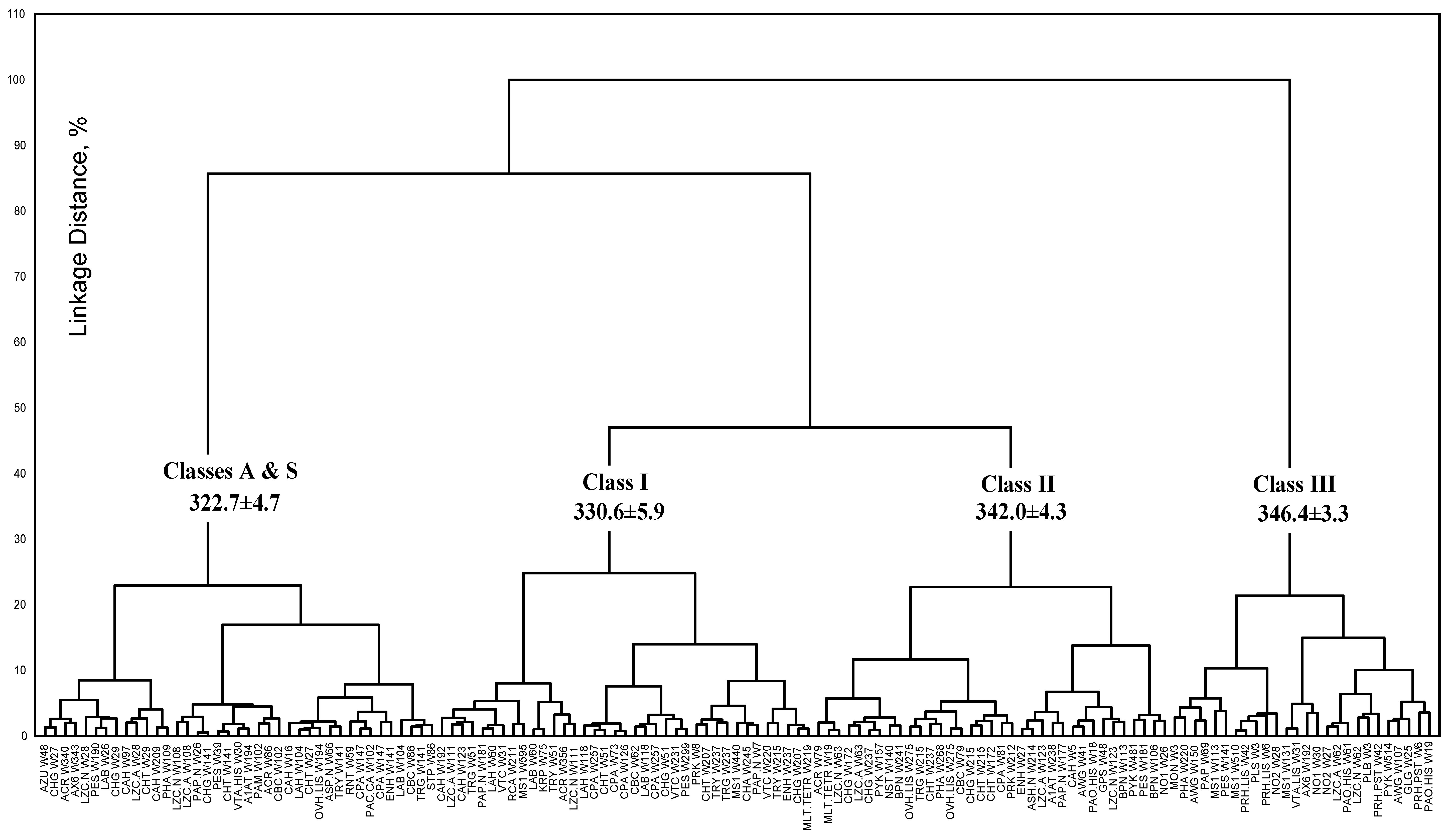{kind=link}
| Spectral and structural parameters * | Class A | Class S | Class I | Class II | Class III | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| The wavelengths of the most probable spectral positions (nm) revealed from an analysis of the fluorescence spectra of 160 proteins | 308 | 321–325 | 330–333 | 341–344 | 346–350 | ||||||
| Acc (averaged value of the relative solvent accessibility of the nine atoms of indole ring of the tryptophan fluorophore.) | 1.9 | 0.8 ± 1.4 | 6.0 ± 3.6 | 14.8 ± 7.5 | 55.3 ± 15.9 | ||||||
| Acc1-7 (averaged value of the relative solvent accessibility of 1 and 7 atoms of the tryptophan fluorophore) | 0.0 | 1.0 ± 2.2 | 11.2 ± 8.5 | 26.7 ± 19.1 | 71.1 ± 19.5 | ||||||
| Den (packing density: the number of neighbor atoms at a distance < 7.5 Å from the indole ring) | 138.3 | 148.3 ± 8.5 | 129.3 ± 9.1 | 109.3 ± 12.6 | 62.7 ± 18.8 | ||||||
| A (relative polarity of environment: portion of the atoms of the polar groups amongst all the atoms around the tryptophan residue at a distance <7.5 Å) | 23.5 | 34.5 ± 5.8 | 39.3 ± 5.5 | 45.1 ± 7.4 | 65.5 ± 13.9 | ||||||
| B (B-factor: crystallographic B-factors of the atoms of the polar groups normalized to the mean B-factor value of all the Cα atoms in the crystal structure) | 0.61 | 0.89 ± 0.17 | 1.11 ± 0.20 | 1.23 ± 0.32 | 1.54 ± 0.55 | ||||||
| R (“Dynamic accessibility” [R = Acc.B], a dynamic characteristic of the tryptophan microenvironment) | 0.9 | 0.7 ± 1.2 | 6.7 ± 4.0 | 18.2 ± 10.3 | 85.2 ± 30.9 |
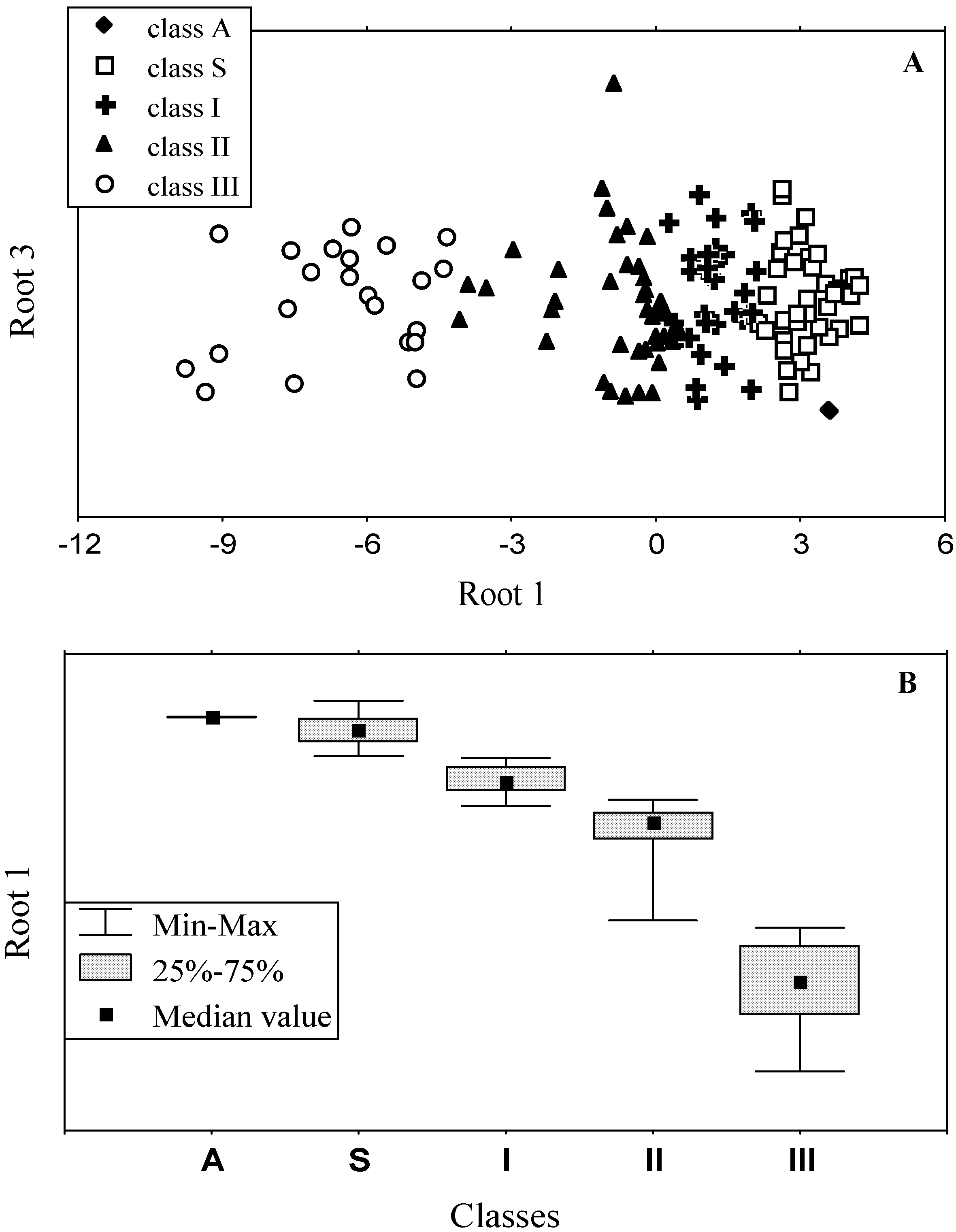
3.3. Accuracy of statistical analysis
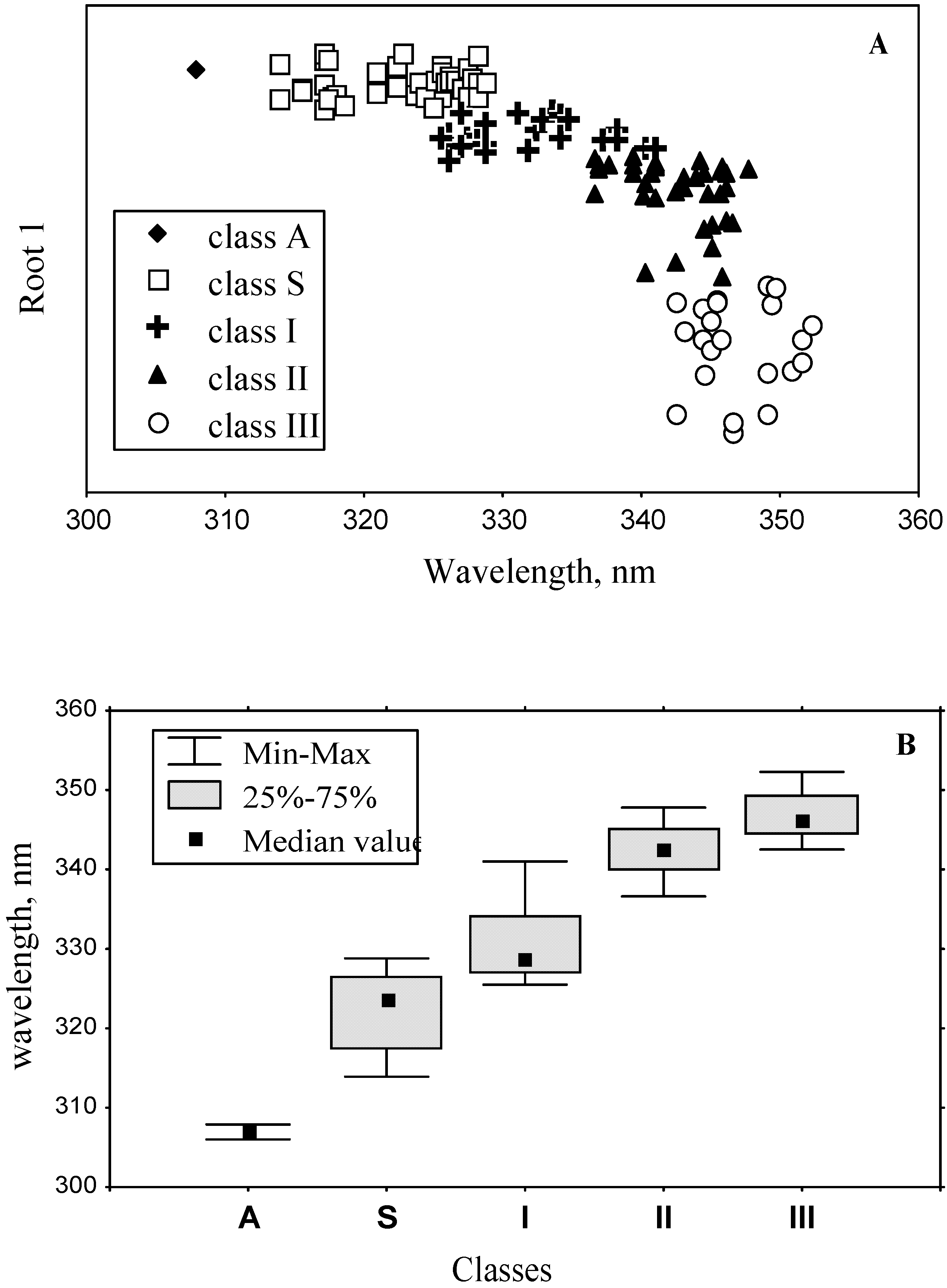
3.4. Model of discrete classes of tryptophan residues in proteins
4. Examples of Application of Spectral and Structural Algorithms for the Study of Protein Structure, Conformation and Dynamics
5. Web-based tool PFAST: Protein Fluorescence And Structural Toolkit
6. Future Direction
References
- Teale, F.W. The ultraviolet fluorescence of proteins in neutral solution. Biochem. J. 1960, 76, 381–388. [Google Scholar] [PubMed]
- Weber, G. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J. 1960, 75, 335–345. [Google Scholar] [PubMed]
- Konev, S.V. Fluorescence and Phosphorescence of Proteins and Nucleic Acids; Plenum Press: New York, NY, USA, 1967. [Google Scholar]
- Longworth, J.W. Excited States of Proteins and Nucleic Acids; Pergamon Press: New York, NY, USA, 1971. [Google Scholar]
- Burstein, E.A. Luminescence of protein chromophores (model studies). In Advances in Science and Technology, Ser. Biophysics; VINITI: Moscow, Russia, 1976; Vol. 6. (in Russian) [Google Scholar]
- Burstein, E.A. Intrinsic protein luminescence. the nature and application. In Advances in Science and Technology, Ser. Biophysics; VINITI: Moscow, Russia, 1977; Vol. 7. (in Russian) [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd Ed. ed; Kluwer Academic/Plenum: New York, NY, USA, 1999. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd Ed. ed; Springer: New York, NY, USA, 2006. [Google Scholar]
- Demchenko, A.P. Ultraviolet Spectroscopy of Proteins.; Springer: Berlin, Germany, 1986. [Google Scholar]
- Callis, P.R. 1La and 1Lb transitions of tryptophan: applications of theory and experimental observations to fluorescence of proteins. Methods Enzymol. 1997, 278, 113–150. [Google Scholar] [PubMed]
- Vivian, J.T.; Callis, P.R. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef]
- Muiño, P.L.; Callis, P.R. Hybrid simulations of solvation effects on electronic spectra: indoles in water. J. Chem. Phys. 1994, 100, 4093–4109. [Google Scholar] [CrossRef]
- Pierce, D.W.; Boxer, S.G. Stark effect spectroscopy of tryptophan. Biophys. J. 1995, 68, 1583–1591. [Google Scholar] [CrossRef]
- Toptygin, D. Effects of the solvent refractive index and its dispersion on the radiative decay rate and extinction coefficient of a fluorescent solute. J. Fluoresc. 2003, 13, 201–219. [Google Scholar] [CrossRef]
- Tikhonov, A. On the solution of ill-posed problems and the method of regularization. Dokl. Akad. Nauk SSSR (in Russian) 1963, 151, 501–504. [Google Scholar]
- Tikhonov, A. Ill-Posed Problems in the Natural Sciences; GBV: Moscow, Russia, 1987; Vol. 344. [Google Scholar]
- Craig, I.; Brown, J. Inverse Problems in Astronomy; Adam Hilger Ltd.: Bristol, UK, 1986. [Google Scholar]
- Henn, S.; Witsch, K. A multigrid approach for minimizing a nonlinear functional for digital image matching. Computing 2000, 64, 339–348. [Google Scholar] [CrossRef]
- Wentzell, P.D.; Nair, S.S.; Guy, R.D. Three-way analysis of fluorescence spectra of polycyclic aromatic hydrocarbons with quenching by nitromethane. Anal. Chem. 2001, 73, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P. Regularization, GSVD and truncated GSVD. BIT 1989, 29, 491–504. [Google Scholar] [CrossRef]
- Hansen, P. Truncated singular value decomposition solutions to discrete ill-posed problems with ill-determined numerical rank. SIAM J. Sci. Stat. Comput. 1990, 11, 503–518. [Google Scholar] [CrossRef]
- Wahl, P.; Auchet, J.C. Resolutions of fluorescence spectra using the decay measurements. application to the study of human serum albumin. Biochim. Biophys. Acta 1972, 285, 99–117. [Google Scholar] [CrossRef]
- Brochon, J.C.; Wahl, P.; Charlier, M.; Maurizot, J.C.; Helene, C. Time resolved spectroscopy of the tryptophyl fluorescence of the E.coli LAC repressor. Biochem. Biophys. Res. Commun. 1977, 79, 1261–1271. [Google Scholar] [CrossRef]
- Knutson, J.R.; Walbridge, D.G.; Brand, L. Decay-associated fluorescence spectra and the heterogeneous emission of alcohol dehydrogenase. Biochemistry 1982, 21, 4671–4679. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Cherek, H. Phase-sensitive fluorescence spectroscopy: a new method to resolve fluorescence lifetimes or emission spectra of components in a mixture of fluorophores. J. Biochem. Biophys. Methods 1981, 5, 19–35. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Cherek, H. Resolution of heterogeneous fluorescence from proteins and aromatic amino acids by phase-sensitive detection of fluorescence. J. Biol. Chem. 1981, 256, 6348–6353. [Google Scholar] [PubMed]
- Wasylewski, Z.; Eftink, M.R. Frequency-domain fluorescence studies of an extracellular metalloproteinase of staphylococcus aureus. Biochim. Biophys. Acta 1987, 915, 331–341. [Google Scholar] [CrossRef]
- Stryjewski, W.; Wasylewski, Z. The resolution of heterogeneous fluorescence of multitryptophan-containing proteins studied by a fluorescence-quenching method. Eur. J. Biochem. 1986, 158, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Wasylewski, Z.; Kaszycki, P.; Guz, A.; Stryjewski, W. Fluorescence-quenching-resolved spectra of fluorophores in mixtures and micellar solutions. Eur. J. Biochem. 1988, 178, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Koloczek, H.; Wasniowska, A.; Potempa, J.; Wasylewski, Z. The fluorescence quenching resolved spectra and red-edge excitation fluorescence measurements of human alpha1-proteinase inhibitor. Biochim. Biophys. Acta 1991, 1973, 619–625. [Google Scholar] [CrossRef]
- Burstein, E.A. An improved algorithm of resolution of fluorescence spectra into quencher accessibility-associated components. Photochem. Photobiol. 1996, 63, 278–280. [Google Scholar] [CrossRef]
- Lehrer, S.S. The selective quenching of tryptophan fluorescence in proteins by iodide ion: lysozyme in the presence and absence of substrate. Biochem. Biophys. Res. Commun. 1967, 29, 767–772. [Google Scholar] [CrossRef]
- Lehrer, S.S. Solute perturbation of protein fluorescence. the quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 1971, 10, 3254–3263. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Leavis, P.C. Solute quenching of protein fluorescence. Methods Enzymol. 1978, 49, 222–236. [Google Scholar] [PubMed]
- Eftink, M.R.; Ghiron, C.A. Exposure of tryptophanyl residues and protein dynamics. Biochemistry 1977, 16, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Eftink, M.R. Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 1991, 35, 127–205. [Google Scholar] [PubMed]
- Siano, D.B.; Metzler, D.E. Band shapes of the electronic spectra of complex molecules. J. Chem. Phys. 1969, 51, 1856–1961. [Google Scholar] [CrossRef]
- Metzler, C.M.; Viswanath, R.; Metzler, D.E. Equilibria and absorption spectra of tryptophanase. J. Biol. Chem. 1991, 266, 9374–9381. [Google Scholar] [PubMed]
- Djikanovic, D.; Kalauzi, A.; Jeremic, M.; Micic, M.; Radotic, K. Deconvolution of fluorescence spectra: contribution to the structural analysis of complex molecules. Colloids Surf. B Biointerfaces 2007, 54, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Kalauzi, A.; Mutavdzic, D.; Djikanovic, D.; Radotic, K.; Jeremic, M. Application of asymmetric model in analysis of fluorescence spectra of biologically important molecules. J. Fluoresc. 2007, 17, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Metzler, D.E.; Harris, C.; Yang, I.Y.; Siano, D.; Thomson, J.A. Band-shape analysis and display of fine structure in protein spectra: a new approach to perturbation spectroscopy. Biochem. Biophys. Res. Commun. 1972, 46, 1588–1597. [Google Scholar] [CrossRef]
- Metzler, C.M.; Cahill, A.E.; Petty, S.; Metzler, D.E.; Lang, L. The widespread applicability of log-normal curves for the description of absorption spectra. Appl. Spectrosc. 1985, 39, 333–339. [Google Scholar] [CrossRef]
- Burstein, E.A.; Emelyanenko, V.I. Log-normal description of fluorescence spectra of organic fluorophores. Photochem. Photobiol. 1996, 64, 316–320. [Google Scholar] [CrossRef]
- Burstein, E.A.; Abornev, S.M.; Reshetnyak, Y.K. Decomposition of protein tryptophan fluorescence spectra into log-normal components. I. Decomposition algorithms. Biophys. J. 2001, 81, 1699–1709. [Google Scholar] [CrossRef]
- Reshetnyak, Y.K.; Burstein, E.A. Decomposition of protein tryptophan fluorescence spectra into log-normal components. II. The statistical proof of discreteness of tryptophan classes in proteins. Biophys. J. 2001, 81, 1710–1734. [Google Scholar] [CrossRef]
- Emelyanenko, V.I.; Reshetnyak, Y.K.; Andreev, O.A.; Burstein, E.A. Log-normal component analysis of fluorescence spectra of prodan and acrylodan bound to proteins. Biophysics 2000, 45, 207–219. [Google Scholar]
- Antipova-Korotaeva, I.I.; Kazanova, N.N. Mathematical decomposition of composite spectral contours into components with partially known parameters. J. Appl. Spectrosc. 1971, 14, 1093–1096. [Google Scholar]
- Aksenenko, V.M.; Shumskaya, T.N.; Slapochnikova, V.A.; Shein, N.V. Quantitative analysis of multicomponent systems based on the mathematical treatment of vibrational spectra. J. Appl. Spectrosc. 1989, 51, 306–311. [Google Scholar] [CrossRef]
- Tikhonov, A.N.; Arsenin, V.Y. Solution of Ill-posed Problems; Winston: New York, NY, USA, 1977. [Google Scholar]
- Linke, D.; Frank, J.; Pope, M.S.; Soll, J.; Ilkavets, I.; Fromme, P.; Burstein, E.A.; Reshetnyak, Y.K.; Emelyanenko, V.I. Folding kinetics and structure of OEP16. Biophys. J. 2004, 86, 1479–1487. [Google Scholar] [CrossRef]
- Torrent, M.; Cuyás, E.; Carreras, E.; Navarro, S.; López, O.; de la Maza, A.; Nogués, M.V.; Reshetnyak, Y.K.; Boix, E. Topology studies on the membrane interaction mechanism of the eosinopjil cationic protein. Biochemistry 2007, 46, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Koshevnik, Y.; Burstein, E.A. Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 2001, 81, 1735–1758. [Google Scholar] [CrossRef]
- Pelley, R.; Horowitz, P. Fluorimetric studies of tryptophyl exposure in Concanavalin A. Biochim. Biophys. Acta 1976, 427, 359–363. [Google Scholar] [CrossRef]
- Brown, M.F.; Omar, S.; Raubach, R.A.; Schleich, T. Quenching of the tyrosyl and tryptophyl fluorescence of subtilisins carlsberg and novo by iodide. Biochemistry 1977, 16, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Rousslang, K.W.; Thomasson, J.M.; Rose, J.B.; Kwiram, A.L. Triplet state of tryptophan in proteins. 2. Differentiation between tryptophan residues 62 and 108 in lysozyme. Biochemistry 1979, 18, 2296–2300. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M. Polarization of intrinsic fluorescence of proteins. iv. changes in the degree of polarization from the emission spectra. Mol. Biol. (Moscow) 1985, 19, 1321–1331. [Google Scholar]
- Turoverov, K.K.; Kuznetsova, I.M.; Zaitsev, V.N. The environment of the tryptophan residue in pseudomonas aeruginosa azurin and its fluorescence properties. Biophys. Chem. 1985, 23, 79–89. [Google Scholar] [CrossRef]
- Dolashka, P.; Dimov, I.; Genov, N.; Svendsen, I.; Wilson, K.S.; Betzel, C. Fluorescence properties of native and photooxidised proteinase k: the x-ray model in the region of the two tryptophans. Biochim. Biophys. Acta 1992, 1118, 303–312. [Google Scholar] [CrossRef]
- Dolashka, P.; Filippi, B.; Wilson, K.S.; Betzel, C.; Genov, N. Spectroscopic studies on proteinase K and subtilisin DY. relation to X-ray models. Int. J. Pept. Protein Res. 1992, 40, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Yakusheva, T.A.; Turoverov, K.K. Contribution of separate tryptophan residues to intrinsic fluorescence of actin. analysis of 3D structure. FEBS Lett. 1999, 452, 205–210. [Google Scholar] [CrossRef]
- Alston, R.W.; Urbanikova, L.; Sevcik, J.; Lasagna, M.; Reinhart, G.D.; Scholtz, J.M.; Pace, C.N. Contribution of single tryptophan residues to the fluorescence and stability of Ribonuclease A. Biophys. J. 2004, 87, 4036–4047. [Google Scholar] [CrossRef] [PubMed]
- Kosinski-Collins, M.S.; Flaugh, S.L.; King, J. Probing folding and fluorescence quenching in human gamma D crystallin greek key domains using triple tryptophan mutant proteins. Protein Sci. 2004, 13, 2223–2235. [Google Scholar] [CrossRef] [PubMed]
- Bedell, J.L.; Edmondson, S.P.; Shriver, J.W. Role of a surface tryptophan in defining the structure, stability, and DNA binding of the hyperthermophile protein sac7d. Biochemistry 2005, 44, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Duy, C.; Fitter, J. How aggregation and conformational scrambling of unfolded states govern fluorescence emission spectra. Biophys. J. 2006, 90, 3704–3711. [Google Scholar] [CrossRef] [PubMed]
- Legardinier, S.; Raguénès-Nicol, C.; Tascon, C.; Rocher, C.; Hardy, S.; Hubert, J.F.; Le Rumeur, E. Mapping of the lipid-binding and stability properties of the central rod domain of human dystrophin. J. Mol. Biol. 2009, 389, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.M.; Kemp, G.J.; Rawlings, C.J.; Brown, N.P.; Sander, C.; Thornton, J.M.; Orengo, C.M.; Wodak, S.J.; Richelle, J. Macromolecular structure information and databases. The EU BRIDGE database project consortium. Trends Biochem. Sci. 1996, 21, 251–256. [Google Scholar] [PubMed]
- Hogue, C.W.; Ohkawa, H.; Bryant, S.H. A dynamic look at structures: www-entrez and the molecular modeling database. Trends Biochem. Sci. 1996, 21, 226–229. [Google Scholar] [CrossRef]
- Islamov, A.S.; Rumjantsev, A.B.; Skvortsov, V.S.; Archakov, A.I. ONIX: an interactive PC program for the examination of protein 3D structures from PDB. Comput. Appl. Biosci. 1997, 13, 111–113. [Google Scholar]
- Laskowski, R.A.; Hutchinson, E.G.; Michie, A.D.; Wallace, A.C.; Jones, M.L.; Thornton, J.M. PDBsum: a web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci. 1997, 22, 488–490. [Google Scholar] [CrossRef]
- Michalickova, K.; Bader, G.D.; Dumontier, M.; Lieu, H.; Betel, D.; Isserlin, R.; Hogue, C.W. SeqHound: biological sequence and structure database as a platform for bioinformatics research. BMC Bioinformatics 2002, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Hollup, S.M.; Salensminde, G.; Reuter, N. WEBnm@: a wweb application for normal mode analyses of proteins. BMC Bioinformatics 2005, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, E.; Xue, B.; Zhou, Y. Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by guided-learning through a two-layer neural network. Proteins 2009, 74, 847–856. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Rodriguez, C.; Madabhushni, A. Investigating the efficacy of nonlinear dimensionality reduction schemes in classifying gene and protein expression studies. IEEE/ACM Trans. Comput. Biol. Bioinformatics 2008, 5, 368–384. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Menon, R.; Das, D.; Bansal, N.; Nahar, N.; Guduru, N.; Jaegle, S.; Peckham, J.; Reshetnyak, Y.K. The protein fluorescence and structural toolkit (PFAST): database and programs for the analysis of protein fluorescence and structural data. Protein: Struct. Funct. Bioinformatics 2008, 71, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Orlov, N.Y.; Orlova, T.G.; Reshetnyak, Y.K.; Burstein, E.A.; Kimura, N. Comparative study of recombinant rat nucleoside diphosphate kinases alpha and beta by intrinsic protein fluorescence. J. Biomol. Struct. Dyn. 1999, 16, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Hendriks, J.; Hellingwerf, K.J. Tryptophan fluorescence monitors structural changes accompanying signalling state formation in the photocycle of photoactive yellow protein. Photochem. Photobiol. Sci. 2004, 6, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Churbanova, I.Y.; Tronin, A.; Strzalka, J.; Gog, T.; Kuzmenko, I.; Johansson, J.S.; Blasie, J.K. Monolayers of a model anesthetic-binding membrane protein: formation, characterization, and halothane-binding affinity. Biophys. J. 2006, 90, 3255–3266. [Google Scholar] [CrossRef] [PubMed]
- Toh, K.C.; van Stokkum, I.H.; Hendriks, J.; Alexandre, M.T.; Arents, J.C.; Perez, M.A.; van Grondelle, R.; Hellingwerf, K.J.; Kennis, J.T. On the signaling mechanism and the absence of photoreversibility in the AppA BLUF domain. Biophys. J. 2008, 95, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Liu, W.F.; Yan, Y.B. Role of the RRM domain in the activity, structure and stability of poly(A)-specific ribonuclease. Arch Biochem. Biophys. 2007, 461, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Su, J.T.; Kim, S.H.; Yan, Y.B. Dissecting the pretransitional conformational changes in aminoacylase I thermal denaturation. Biophys. J. 2007, 92, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Galka, J.J.; Baturin, S.J.; Manley, D.M.; Kehler, A.J.; O'Neil, J.D. Stability of the glycerol facilitator in detergent solutions. Biochemistry 2008, 47, 3513–3524. [Google Scholar] [CrossRef] [PubMed]
- Verheyden, S.; Sillen, A.; Gils, A.; Declerck, P.J.; Engelborghs, Y. Tryptophan properties in fluorescence and functional stability of plasminogen activator inhibitor 1. Biophys. J. 2003, 85, 501–510. [Google Scholar] [CrossRef]
- Runke, G.; Maier, E.; Summers, W.A.; Bay, D.C.; Benz, R.; Court, D.A. Deletion variants of Neurospora mitochondrial porin: electrophysiological and spectroscopic analysis. Biophys. J. 2006, 90, 3155–3164. [Google Scholar] [CrossRef] [PubMed]
- Maglia, G.; Jonckheer, A.; De Maeyer, M.; Frère, J.M.; Engelborghs, Y. An unusual red-edge excitation and time-dependent Stokes shift in the single tryptophan mutant protein DD-carboxypeptidase from Streptomyces: the role of dynamics and tryptophan rotamers. Protein Sci. 2008, 17, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Kitson, R.P.; Lu, M.; Goldfarb, R.H. Conformational and enzymatic changes of 20S proteasome of rat natural killer cells induced by mono and divalent cations. J. Struct. Biol. 2004, 145, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Orlov, N.Y.; Orlova, T.G.; Reshetnyak, Y.K.; Burstein, E.A.; Kimura, N. Comparative study of recombinant rat nucleoside diphosphate kinases alpha and beta by intrinsic protein fluorescence. J. Biomol. Struct. Dyn. 1999, 16, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Tchedre, K.T.; Nair, M.P.; Pritchard, P.H.; Lacko, A.G. Structural differences between wild-type and fish eye disease mutant of lecithin:cholesterol acyltransferase. J. Biomol. Struct. Dyn. 2006, 24, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Segala, M.; Andreev, O.A.; Engelman, D.M. A monomeric membrane peptide that lives in Three Worlds: in solution, attached to and inserted across lipid bilayers. Biophys. J. 2007, 93, 2363–2672. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hixon, J.; Reshetnyak, Y.K. Algorithm for the Analysis of Tryptophan Fluorescence Spectra and Their Correlation with Protein Structural Parameters. Algorithms 2009, 2, 1155-1176. https://doi.org/10.3390/a2031155
Hixon J, Reshetnyak YK. Algorithm for the Analysis of Tryptophan Fluorescence Spectra and Their Correlation with Protein Structural Parameters. Algorithms. 2009; 2(3):1155-1176. https://doi.org/10.3390/a2031155
Chicago/Turabian StyleHixon, John, and Yana K. Reshetnyak. 2009. "Algorithm for the Analysis of Tryptophan Fluorescence Spectra and Their Correlation with Protein Structural Parameters" Algorithms 2, no. 3: 1155-1176. https://doi.org/10.3390/a2031155
APA StyleHixon, J., & Reshetnyak, Y. K. (2009). Algorithm for the Analysis of Tryptophan Fluorescence Spectra and Their Correlation with Protein Structural Parameters. Algorithms, 2(3), 1155-1176. https://doi.org/10.3390/a2031155