Recent Advances in the Computational Discovery of Transcription Factor Binding Sites

Abstract
:1. Introduction
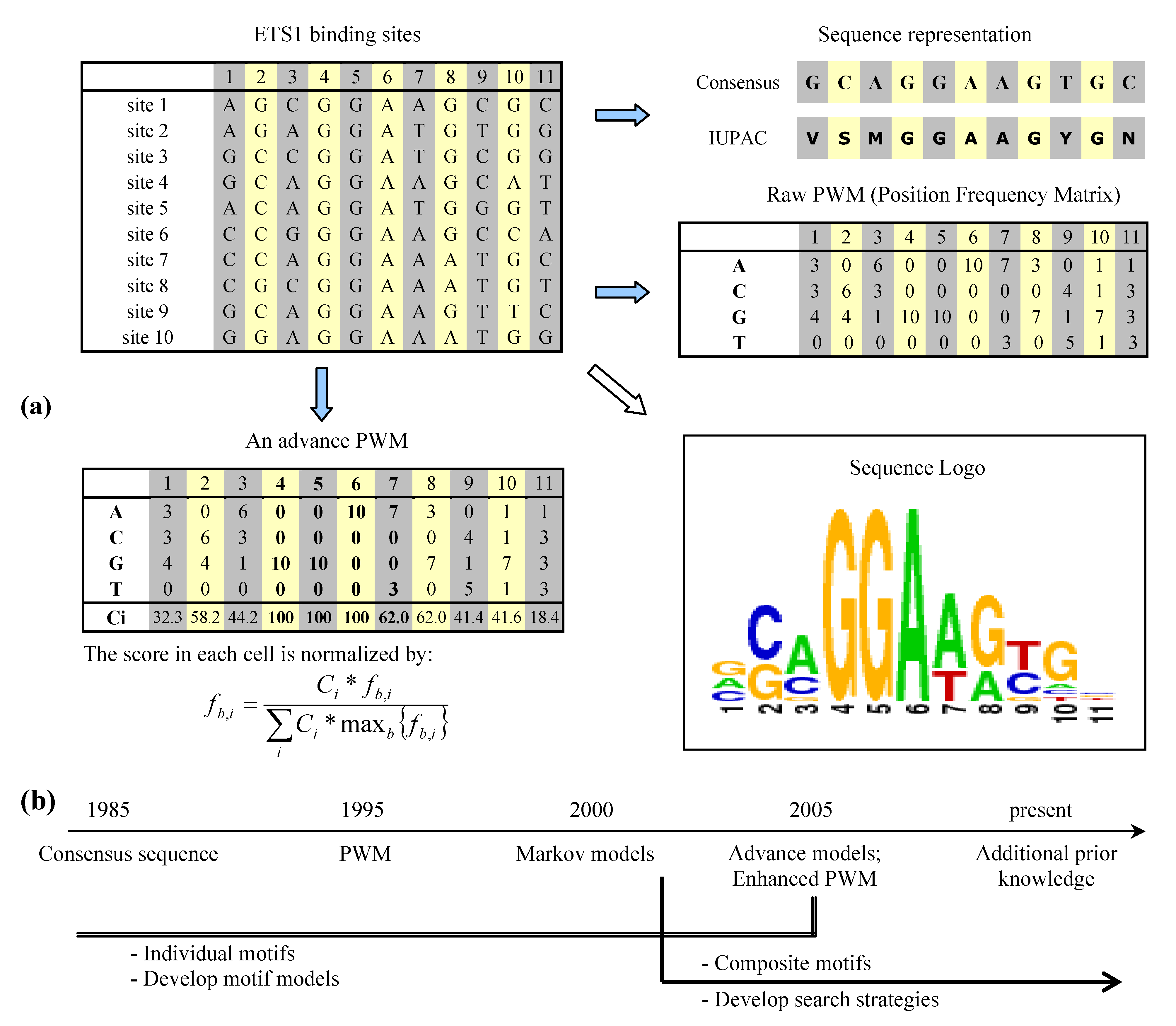
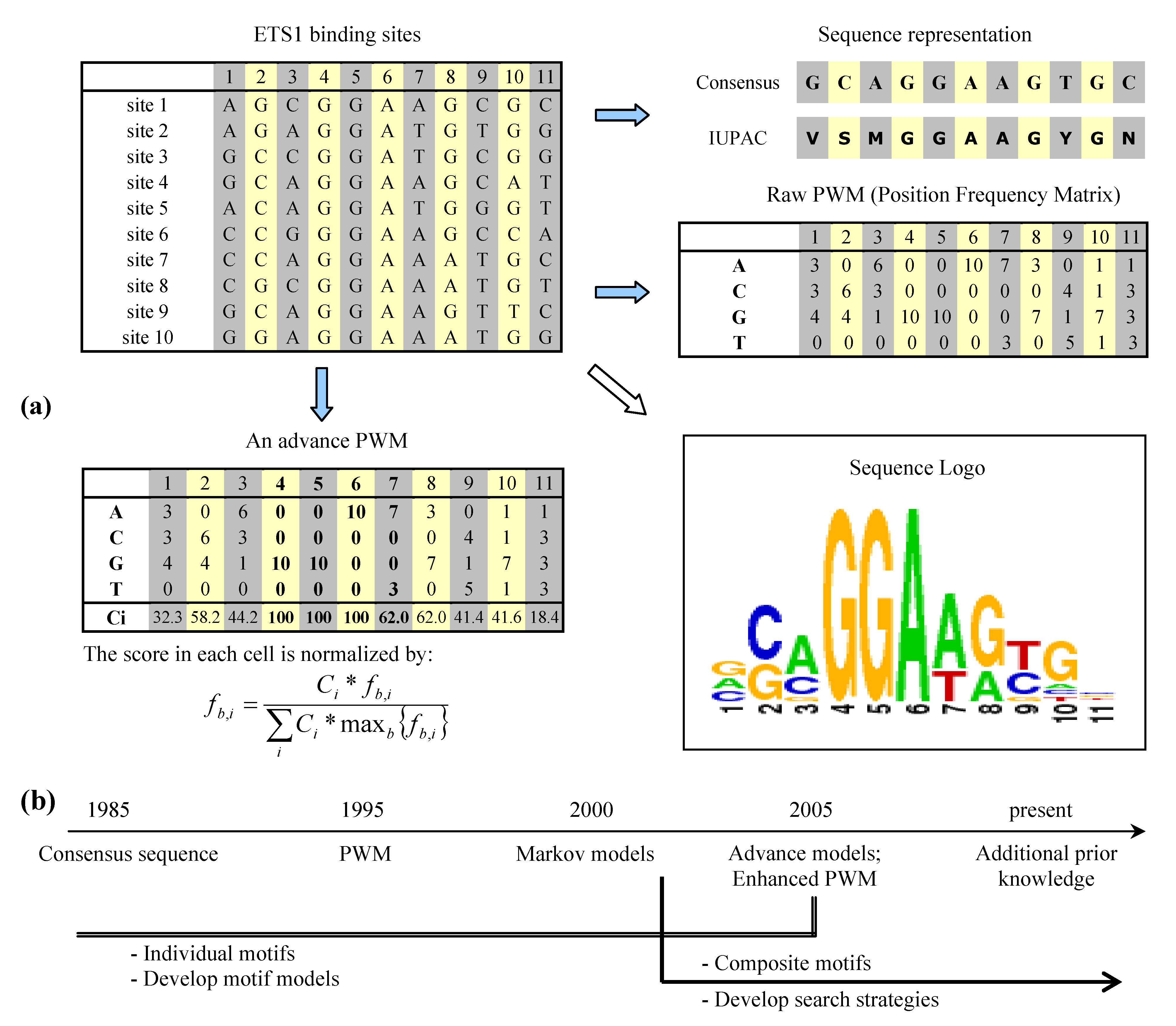
2. Binding site representation
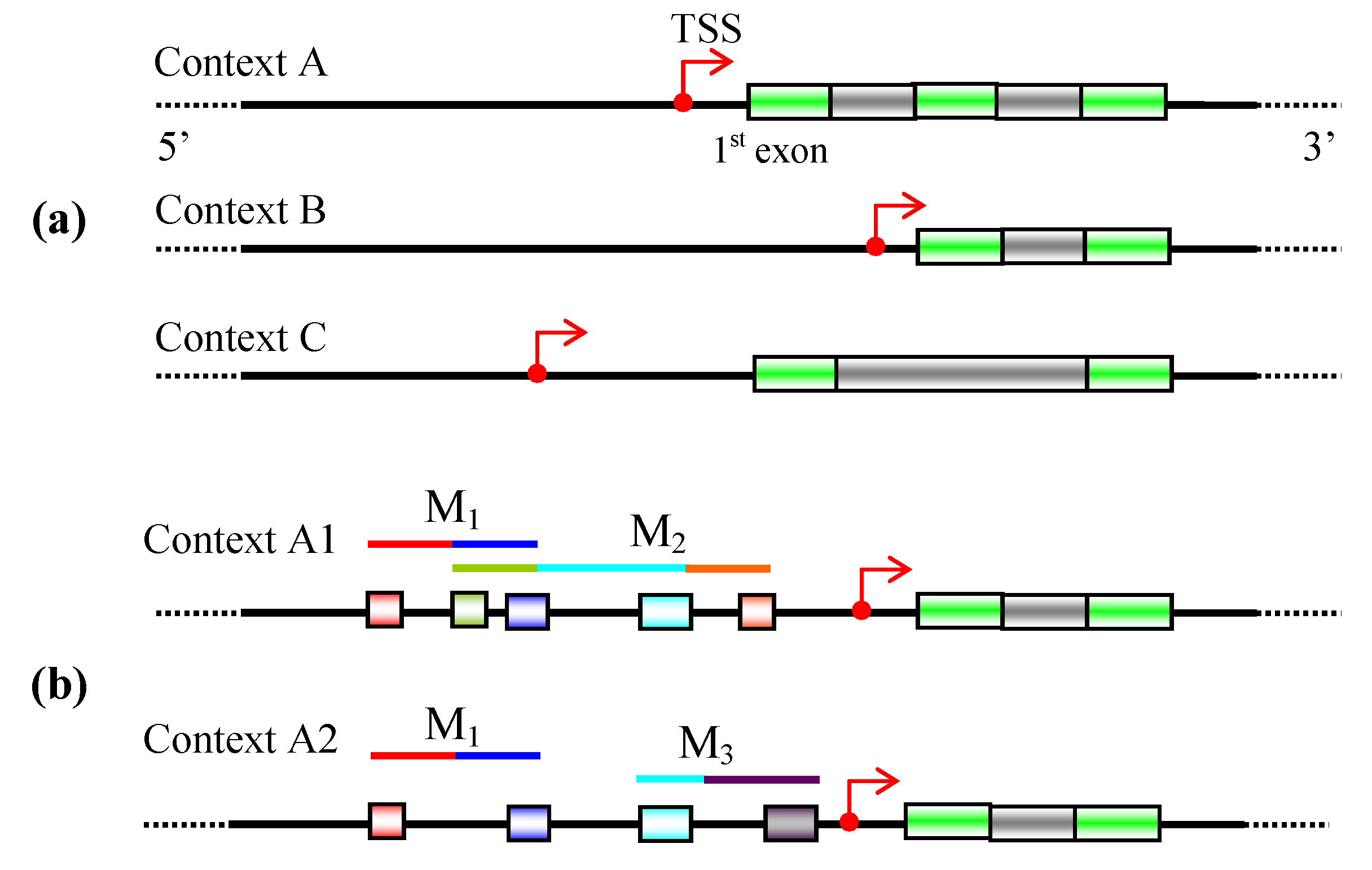
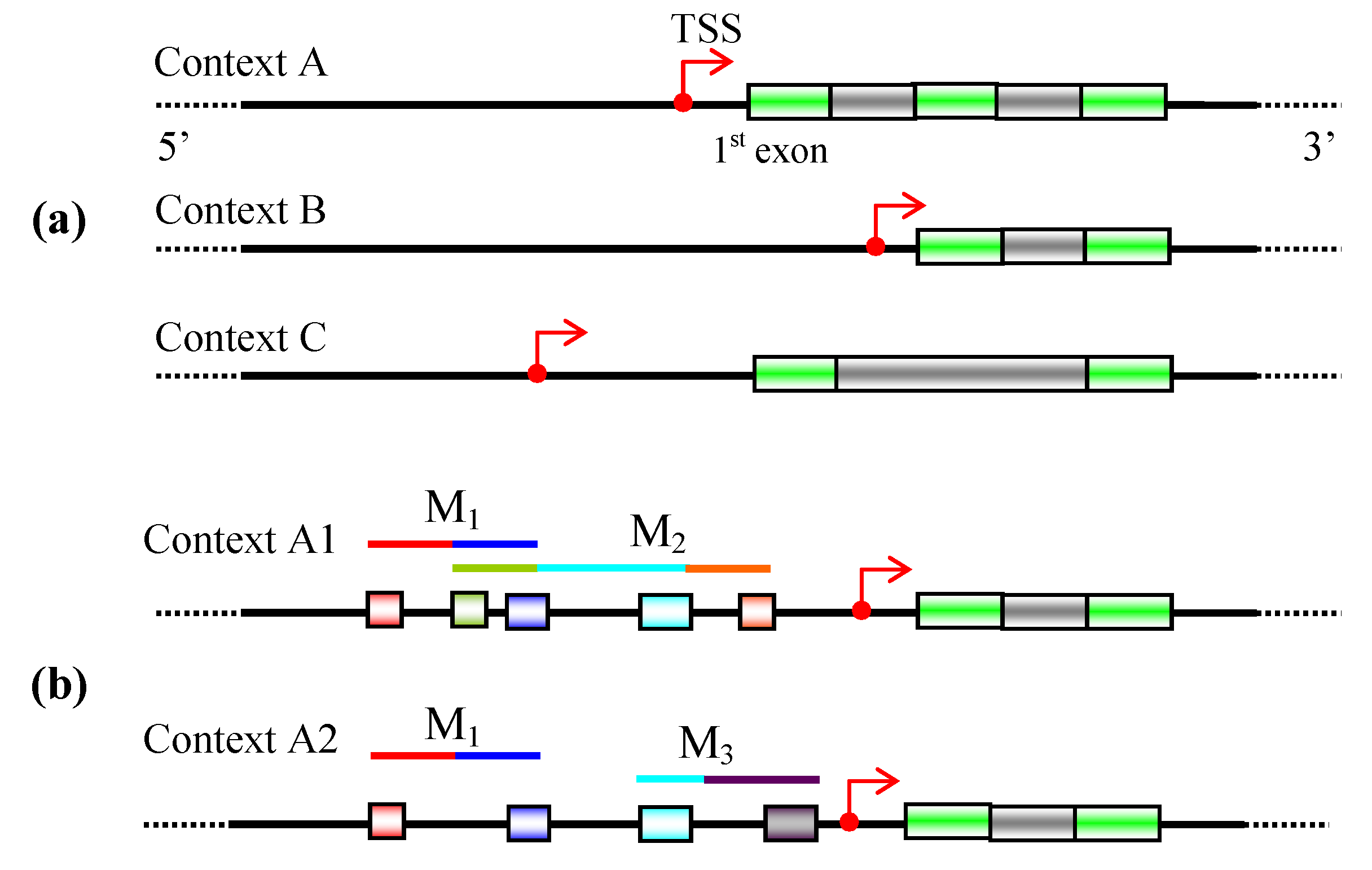
3. Promoter identification
4. Discovery of physical TFBSs

{kind=link}
{kind=link}
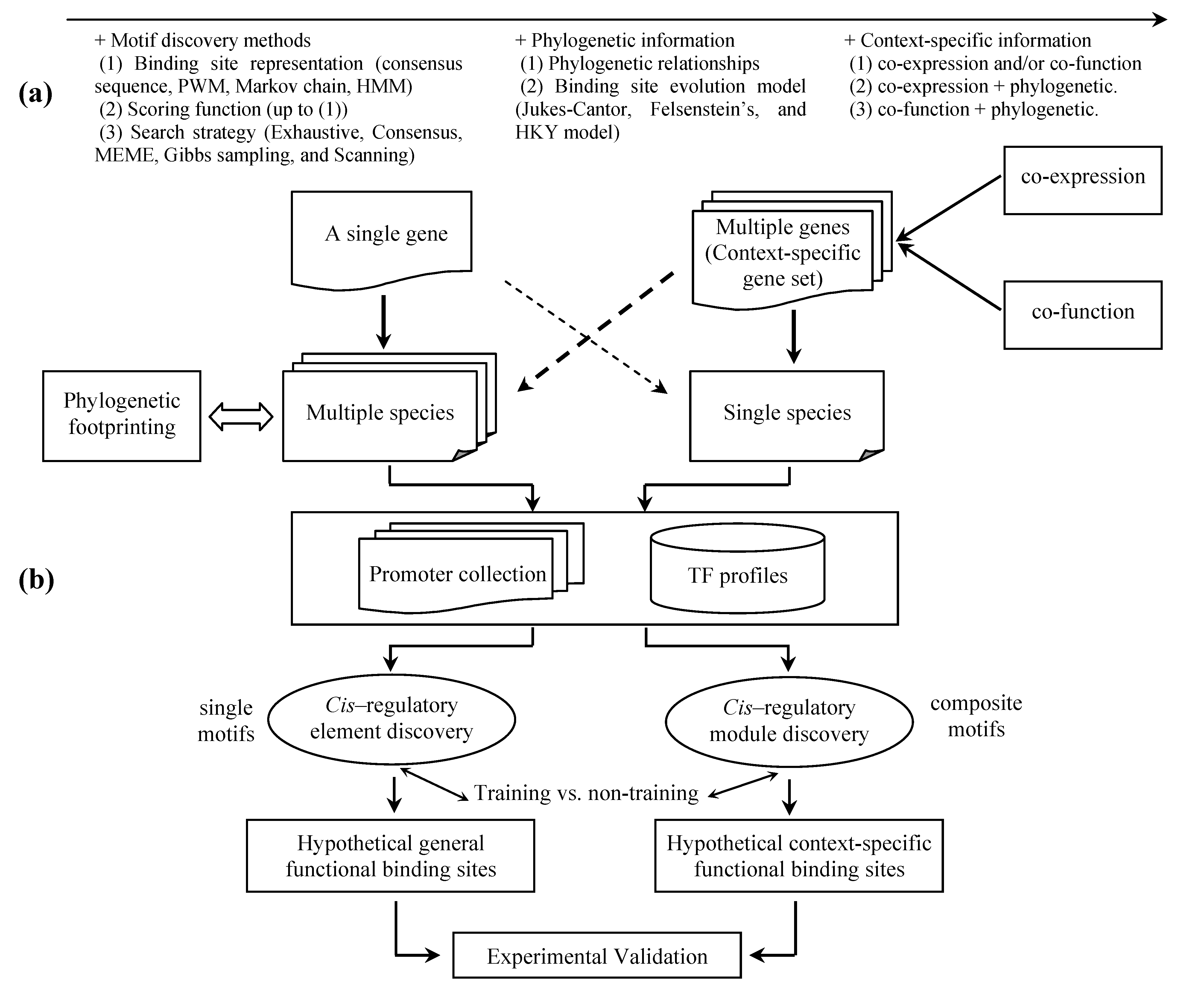{kind=link}
5. Discovery of functional relevant TFBSs
5.1 Phylogenetic footprinting
5.2 Context-specific search
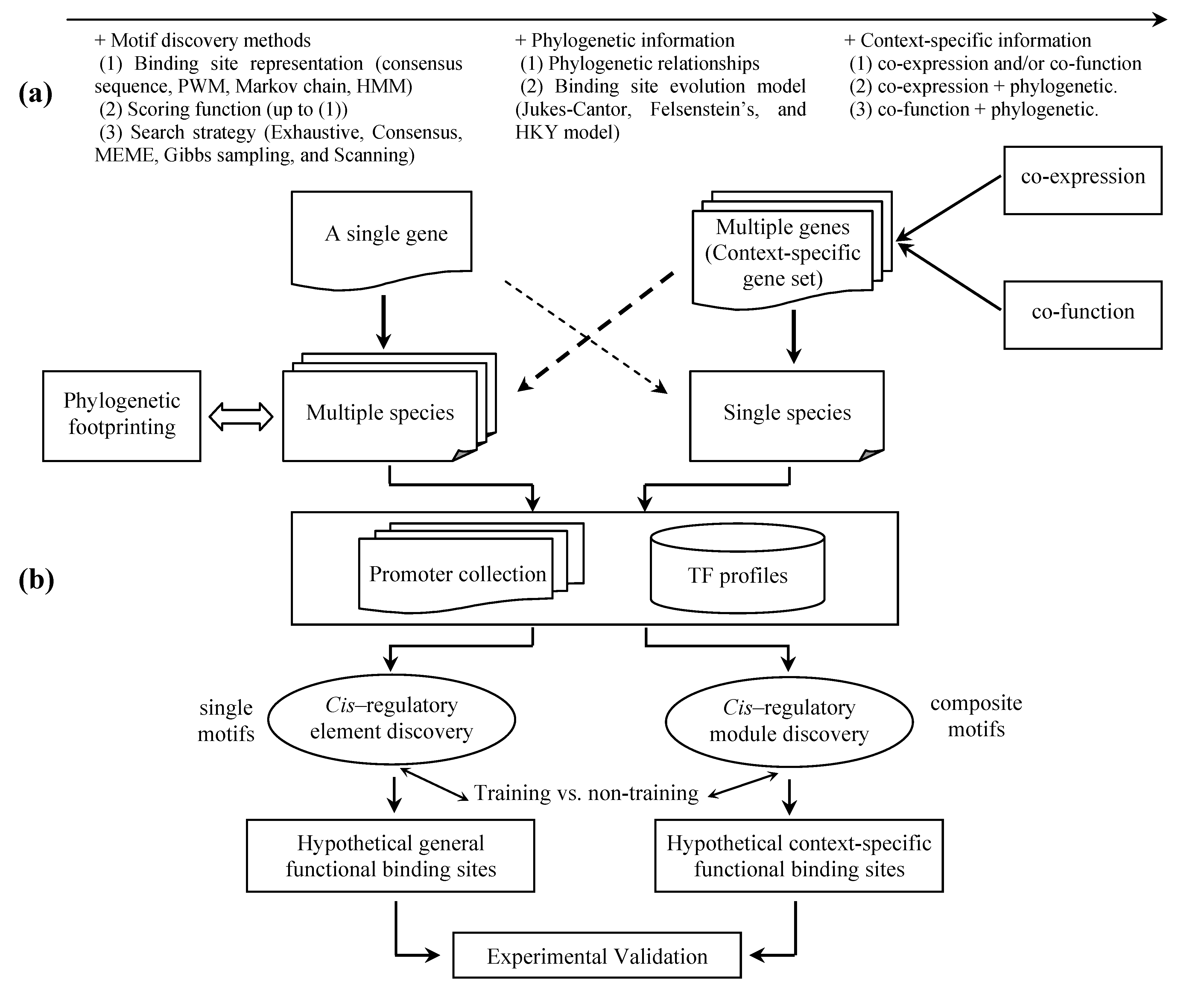
6. Inference of transcriptional regulatory networks
7. Concluding Remarks
Acknowledgements
References and Notes
- Kafatos, F.C. A revolutionary landscape: the restructuring of biology and its convergence with medicine. J Mol Biol 2002, 319(4), 861–867. [Google Scholar] [CrossRef]
- Lemon, B.; Tjian, R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev 2000, 14(20), 2551–2569. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Tjian, R. Transcription regulation and animal diversity. Nature 2003, 424(6945), 147–151. [Google Scholar] [CrossRef] [PubMed]
- van Driel, R.; Fransz, P.F.; Verschure, P.J. The eukaryotic genome: a system regulated at different hierarchical levels. J Cell Sci 2003, 116 Pt 20, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Fessele, S.; Maier, H.; Nelson, P.J. Computer modeling of promoter organization as a tool to study transcriptional coregulation. Faseb J 2003, 17(10), 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.J.; Trinklein, N.D.; Anton, E.D.; Nguyen, L.; Myers, R.M. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res 2006, 16(1), 1–10. [Google Scholar] [CrossRef] [PubMed]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet 2006, 7, 29–59. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B. The gateway to transcription: identifying, characterizing and understanding promoters in the eukaryotic genome. Cell Mol Life Sci 2007, 64(4), 386–400. [Google Scholar] [CrossRef] [PubMed]
- Barrera, L.O.; Ren, B. The transcriptional regulatory code of eukaryotic cells--insights from genome-wide analysis of chromatin organization and transcription factor binding. Curr Opin Cell Biol 2006, 18(3), 291–298. [Google Scholar] [CrossRef] [PubMed]
- Dillon, N. Gene regulation and large-scale chromatin organization in the nucleus. Chromosome Res 2006, 14(1), 117–126. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Langerak, J.; Goetze, S.; Leonhardt, H.; Cremer, T.; van Driel, R.; Lanctot, C. Nuclear architecture: Is it important for genome function and can we prove it? J Cell Biochem 2007, 102(5), 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Grosschedl, R. Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 2007, 21(23), 3027–3043. [Google Scholar] [CrossRef] [PubMed]
- Wray, G.A.; Hahn, M.W.; Abouheif, E.; Balhoff, J.P.; Pizer, M.; Rockman, M.V.; Romano, L.A. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 2003, 20(9), 1377–1419. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.R.; Mager, D.L.; Wilhelm, B.T. Complex controls: the role of alternative promoters in mammalian genomes. Trends Genet 2003, 19(11), 640–648. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.A.; Wu, J.; Yan, P.; Plass, C.; Huang, T.H.; Davuluri, R.V. Genome-wide analysis of alternative promoters of human genes using a custom promoter tiling array. BMC Genomics 2008, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- Sandve, G.K.; Drablos, F. A survey of motif discovery methods in an integrated framework. Biol Direct 2006, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Bulyk, M.L. Computational prediction of transcription-factor binding site locations. Genome Biol 2003, 5(1), 201. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Rolfe, A.; MacIsaac, K.D.; Gerber, G.K.; Pokholok, D.; Zeitlinger, J.; Danford, T.; Dowell, R.D.; Fraenkel, E.; Jaakkola, T.S.; Young, R.A.; Gifford, D.K. High-resolution computational models of genome binding events. Nat Biotechnol 2006, 24(8), 963–970. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Robert, F.; Wyrick, J.J.; Aparicio, O.; Jennings, E.G.; Simon, I.; Zeitlinger, J.; Schreiber, J.; Hannett, N.; Kanin, E.; Volkert, T.L.; Wilson, C.J.; Bell, S.P.; Young, R.A. Genome-wide location and function of DNA binding proteins. Science 2000, 290(5500), 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Roulet, E.; Busso, S.; Camargo, A.A.; Simpson, A.J.; Mermod, N.; Bucher, P. High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat Biotechnol 2002, 20(8), 831–835. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX--a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol Eng 2007, 24(4), 381–403. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, B.; Kihara, D. Limitations and potentials of current motif discovery algorithms. Nucleic Acids Res 2005, 33(15), 4899–4913. [Google Scholar] [CrossRef] [PubMed]
- Sandve, G.K.; Abul, O.; Walseng, V.; Drablos, F. Improved benchmarks for computational motif discovery. BMC Bioinformatics 2007, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M.; Li, N.; Bailey, T.L.; Church, G.M.; De Moor, B.; Eskin, E.; Favorov, A.V.; Frith, M.C.; Fu, Y.; Kent, W.J.; Makeev, V.J.; Mironov, A.A.; Noble, W.S.; Pavesi, G.; Pesole, G.; Regnier, M.; Simonis, N.; Sinha, S.; Thijs, G.; van Helden, J.; Vandenbogaert, M.; Weng, Z.; Workman, C.; Ye, C.; Zhu, Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 2005, 23(1), 137–144. [Google Scholar] [CrossRef] [PubMed]
- Klepper, K.; Sandve, G.K.; Abul, O.; Johansen, J.; Drablos, F. Assessment of composite motif discovery methods. BMC Bioinformatics 2008, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Das, M.K.; Dai, H.K. A survey of DNA motif finding algorithms. BMC Bioinformatics 2007, 8 Suppl 7, S21. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Hata, N.; Banerjee, N.; Futcher, B.; Zhang, M.Q. Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biol 2004, 5(8), R56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. A new framework for identifying combinatorial regulation of transcription factors: a case study of the yeast cell cycle. J Biomed Inform 2007, 40(6), 707–725. [Google Scholar] [CrossRef] [PubMed]
- Brazma, A.; Jonassen, I.; Eidhammer, I.; Gilbert, D. Approaches to the automatic discovery of patterns in biosequences. J Comput Biol 1998, 5(2), 279–305. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, G.; Mauri, G.; Pesole, G. In silico representation and discovery of transcription factor binding sites. Brief Bioinform 2004, 5(3), 217–236. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet 2004, 5(4), 276–287. [Google Scholar] [CrossRef] [PubMed]
- Elnitski, L.; Jin, V.X.; Farnham, P.J.; Jones, S.J. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res 2006, 16(12), 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res 1985, 13(9), 3021–3030. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D. Consensus patterns in DNA. Methods Enzymol 1990, 183, 211–221. [Google Scholar] [PubMed]
- Quandt, K.; Frech, K.; Karas, H.; Wingender, E.; Werner, T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res 1995, 23(23), 4878–4884. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.; Stanway, C.; Tsang, J.S.; Henry, Y.; Kingsman, A.J.; Kingsman, S.M. ARS binding factor 1 binds adjacent to RAP1 at the UASs of the yeast glycolytic genes PGK and PYK1. Nucleic Acids Res 1990, 18(18), 5393–5399. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D. DNA binding sites: representation and discovery. Bioinformatics 2000, 16(1), 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gossling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 2003, 31(13), 3576–3579. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L. A method for identifying splice sites and translational start sites in eukaryotic mRNA. Comput Appl Biosci 1997, 13(4), 365–376. [Google Scholar] [CrossRef] [PubMed]
- Bulyk, M.L.; Johnson, P.L.; Church, G.M. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Res 2002, 30(5), 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Man, T.K.; Stormo, G.D. Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res 2001, 29(12), 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Ellrott, K.; Yang, C.; Sladek, F.M.; Jiang, T. Identifying transcription factor binding sites through Markov chain optimization. Bioinformatics 2002, 18 Suppl 2, S100–109. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997, 268(1), 78–94. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Acids. Cambridge University Press, 1998. [Google Scholar]
- Thijs, G.; Lescot, M.; Marchal, K.; Rombauts, S.; De Moor, B.; Rouze, P.; Moreau, Y. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics 2001, 17(12), 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Ben-Gal, I.; Shani, A.; Gohr, A.; Grau, J.; Arviv, S.; Shmilovici, A.; Posch, S.; Grosse, I. Identification of transcription factor binding sites with variable-order Bayesian networks. Bioinformatics 2005, 21(11), 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21(13), 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Chekmenev, D.S.; Haid, C.; Kel, A.E. P-Match: transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res 2005, 33(Web Server issue), W432–437. [Google Scholar] [CrossRef] [PubMed]
- Gershenzon, N.I.; Stormo, G.D.; Ioshikhes, I.P. Computational technique for improvement of the position-weight matrices for the DNA/protein binding sites. Nucleic Acids Res 2005, 33(7), 2290–2301. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Wasserman, W.W. Constrained binding site diversity within families of transcription factors enhances pattern discovery bioinformatics. J Mol Biol 2004, 338(2), 207–215. [Google Scholar] [CrossRef] [PubMed]
- Hannenhalli, S.; Wang, L.S. Enhanced position weight matrices using mixture models. Bioinformatics 2005, 21 Suppl 1, i204–212. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132(2), 311–322. [Google Scholar] [CrossRef] [PubMed]
- Genomatix. http://www.genomatix.de/.
- Scherf, M.; Klingenhoff, A.; Werner, T. Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: a novel context analysis approach. J Mol Biol 2000, 297(3), 599–606. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.B.; Seah, S.H. Dragon gene start finder: an advanced system for finding approximate locations of the start of gene transcriptional units. Genome Res 2003, 13(8), 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Won, H.H.; Kim, M.J.; Kim, S.; Kim, J.W. EnsemPro: an ensemble approach to predicting transcription start sites in human genomic DNA sequences. Genomics 2008, 91(3), 259–266. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.B.; Brent, M.R.; Brown, R.H.; Frankish, A.; Harrow, J.; Ohler, U.; Solovyev, V.V.; Tan, S.L. Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment. Genome Biol 2006, 7 Suppl 1, S3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A.G.; Baldi, P.; Chauvin, Y.; Brunak, S. The biology of eukaryotic promoter prediction--a review. Comput Chem 1999, 23(3-4), 191–207. [Google Scholar] [CrossRef]
- Qiu, P. Recent advances in computational promoter analysis in understanding the transcriptional regulatory network. Biochem Biophys Res Commun 2003, 309(3), 495–501. [Google Scholar] [CrossRef] [PubMed]
- Werner, T. The state of the art of mammalian promoter recognition. Brief Bioinform 2003, 4(1), 22–30. [Google Scholar] [CrossRef] [PubMed]
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Plass, C.; Huang, T.H. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet 2008, 24(4), 167–177. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet 2007, 8(6), 413–423. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Carninci, P.; Lenhard, B.; Ponjavic, J.; Hayashizaki, Y.; Hume, D.A. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet 2007, 8(6), 424–436. [Google Scholar] [CrossRef] [PubMed]
- Hertz, G.Z.; Hartzell, G.W., 3rd; Stormo, G.D. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput Appl Biosci 1990, 6(2), 81–92. [Google Scholar] [PubMed]
- Lawrence, C.E.; Altschul, S.F.; Boguski, M.S.; Liu, J.S.; Neuwald, A.F.; Wootton, J.C. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science 1993, 262(5131), 208–214. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 1994, 2, 28–36. [Google Scholar] [PubMed]
- Tung, N.T.; Yang, E.; Androulakis, I.P. Machine learning approaches in promoter sequence analysis. In Machine Learning Research Progress; Peters, H., Vogel, Mia, Eds.; Nova Science Publishers, Inc, 2008. [Google Scholar]
- Marsan, L.; Sagot, M.F. Algorithms for extracting structured motifs using a suffix tree with an application to promoter and regulatory site consensus identification. J Comput Biol 2000, 7(3-4), 345–362. [Google Scholar] [CrossRef] [PubMed]
- Hertz, G.Z.; Stormo, G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 1999, 15(7-8), 563–577. [Google Scholar] [CrossRef]
- Vlieghe, D.; Sandelin, A.; De Bleser, P.J.; Vleminckx, K.; Wasserman, W.W.; van Roy, F.; Lenhard, B. A new generation of JASPAR, the open-access repository for transcription factor binding site profiles. Nucleic Acids Res 2006, 34(Database issue), D95–97. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Dietze, P.; Karas, H.; Knuppel, R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 1996, 24(1), 238–241. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; Gocayne, J.D.; Amanatides, P.; Ballew, R.M.; Huson, D.H.; Wortman, J.R.; Zhang, Q.; Kodira, C.D.; Zheng, X.H.; Chen, L.; Skupski, M.; Subramanian, G.; Thomas, P.D.; Zhang, J.; Gabor Miklos, G.L.; Nelson, C.; Broder, S.; Clark, A.G.; Nadeau, J.; McKusick, V.A.; Zinder, N.; Levine, A.J.; Roberts, R.J.; Simon, M.; Slayman, C.; Hunkapiller, M.; Bolanos, R.; Delcher, A.; Dew, I.; Fasulo, D.; Flanigan, M.; Florea, L.; Halpern, A.; Hannenhalli, S.; Kravitz, S.; Levy, S.; Mobarry, C.; Reinert, K.; Remington, K.; Abu-Threideh, J.; Beasley, E.; Biddick, K.; Bonazzi, V.; Brandon, R.; Cargill, M.; Chandramouliswaran, I.; Charlab, R.; Chaturvedi, K.; Deng, Z.; Di Francesco, V.; Dunn, P.; Eilbeck, K.; Evangelista, C.; Gabrielian, A.E.; Gan, W.; Ge, W.; Gong, F.; Gu, Z.; Guan, P.; Heiman, T.J.; Higgins, M.E.; Ji, R.R.; Ke, Z.; Ketchum, K.A.; Lai, Z.; Lei, Y.; Li, Z.; Li, J.; Liang, Y.; Lin, X.; Lu, F.; Merkulov, G.V.; Milshina, N.; Moore, H.M.; Naik, A.K.; Narayan, V.A.; Neelam, B.; Nusskern, D.; Rusch, D.B.; Salzberg, S.; Shao, W.; Shue, B.; Sun, J.; Wang, Z.; Wang, A.; Wang, X.; Wang, J.; Wei, M.; Wides, R.; Xiao, C.; Yan, C.; Yao, A.; Ye, J.; Zhan, M.; Zhang, W.; Zhang, H.; Zhao, Q.; Zheng, L.; Zhong, F.; Zhong, W.; Zhu, S.; Zhao, S.; Gilbert, D.; Baumhueter, S.; Spier, G.; Carter, C.; Cravchik, A.; Woodage, T.; Ali, F.; An, H.; Awe, A.; Baldwin, D.; Baden, H.; Barnstead, M.; Barrow, I.; Beeson, K.; Busam, D.; Carver, A.; Center, A.; Cheng, M.L.; Curry, L.; Danaher, S.; Davenport, L.; Desilets, R.; Dietz, S.; Dodson, K.; Doup, L.; Ferriera, S.; Garg, N.; Gluecksmann, A.; Hart, B.; Haynes, J.; Haynes, C.; Heiner, C.; Hladun, S.; Hostin, D.; Houck, J.; Howland, T.; Ibegwam, C.; Johnson, J.; Kalush, F.; Kline, L.; Koduru, S.; Love, A.; Mann, F.; May, D.; McCawley, S.; McIntosh, T.; McMullen, I.; Moy, M.; Moy, L.; Murphy, B.; Nelson, K.; Pfannkoch, C.; Pratts, E.; Puri, V.; Qureshi, H.; Reardon, M.; Rodriguez, R.; Rogers, Y.H.; Romblad, D.; Ruhfel, B.; Scott, R.; Sitter, C.; Smallwood, M.; Stewart, E.; Strong, R.; Suh, E.; Thomas, R.; Tint, N.N.; Tse, S.; Vech, C.; Wang, G.; Wetter, J.; Williams, S.; Williams, M.; Windsor, S.; Winn-Deen, E.; Wolfe, K.; Zaveri, J.; Zaveri, K.; Abril, J.F.; Guigo, R.; Campbell, M.J.; Sjolander, KV.; Karlak, B.; Kejariwal, A.; Mi, H.; Lazareva, B.; Hatton, T.; Narechania, A.; Diemer, K.; Muruganujan, A.; Guo, N.; Sato, S.; Bafna, V.; Istrail, S.; Lippert, R.; Schwartz, R.; Walenz, B.; Yooseph, S.; Allen, D.; Basu, A.; Baxendale, J.; Blick, L.; Caminha, M.; Carnes-Stine, J.; Caulk, P.; Chiang, Y.H.; Coyne, M.; Dahlke, C.; Mays, A.; Dombroski, M.; Donnelly, M.; Ely, D.; Esparham, S.; Fosler, C.; Gire, H.; Glanowski, S.; Glasser, K.; Glodek, A.; Gorokhov, M.; Graham, K.; Gropman, B.; Harris, M.; Heil, J.; Henderson, S.; Hoover, J.; Jennings, D.; Jordan, C.; Jordan, J.; Kasha, J.; Kagan, L.; Kraft, C.; Levitsky, A.; Lewis, M.; Liu, X.; Lopez, J.; Ma, D.; Majoros, W.; McDaniel, J.; Murphy, S.; Newman, M.; Nguyen, T.; Nguyen, N.; Nodell, M.; Pan, S.; Peck, J.; Peterson, M.; Rowe, W.; Sanders, R.; Scott, J.; Simpson, M.; Smith, T.; Sprague, A.; Stockwell, T.; Turner, R.; Venter, E.; Wang, M.; Wen, M.; Wu, D.; Wu, M.; Xia, A.; Zandieh, A.; Zhu, X. The sequence of the human genome. Science 2001, 291(5507), 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Friberg, M.; von Rohr, P.; Gonnet, G. Scoring functions for transcription factor binding site prediction. BMC Bioinformatics 2005, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Tompa, M. Analysis of computational approaches for motif discovery. Algorithms Mol Biol 2006, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Doniger, S.W.; Huh, J.; Fay, J.C. Identification of functional transcription factor binding sites using closely related Saccharomyces species. Genome Res 2005, 15(5), 701–709. [Google Scholar] [CrossRef] [PubMed]
- Cliften, P.; Sudarsanam, P.; Desikan, A.; Fulton, L.; Fulton, B.; Majors, J.; Waterston, R.; Cohen, B.A.; Johnston, M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 2003, 301(5629), 71–76. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Weinstock, G.M.; Metzker, M.L.; Muzny, D.M.; Sodergren, E.J.; Scherer, S.; Scott, G.; Steffen, D.; Worley, K.C.; Burch, P.E.; Okwuonu, G.; Hines, S.; Lewis, L.; DeRamo, C.; Delgado, O.; Dugan-Rocha, S.; Miner, G.; Morgan, M.; Hawes, A.; Gill, R.; Celera; Holt, R.A.; Adams, M.D.; Amanatides, P.G.; Baden-Tillson, H.; Barnstead, M.; Chin, S.; Evans, C.A.; Ferriera, S.; Fosler, C.; Glodek, A.; Gu, Z.; Jennings, D.; Kraft, C.L.; Nguyen, T.; Pfannkoch, C.M.; Sitter, C.; Sutton, G.G.; Venter, J.C.; Woodage, T.; Smith, D.; Lee, H.M.; Gustafson, E.; Cahill, P.; Kana, A.; Doucette-Stamm, L.; Weinstock, K.; Fechtel, K.; Weiss, R.B.; Dunn, D.M.; Green, E.D.; Blakesley, R.W.; Bouffard, G.G.; De Jong, P.J.; Osoegawa, K.; Zhu, B.; Marra, M.; Schein, J.; Bosdet, I.; Fjell, C.; Jones, S.; Krzywinski, M.; Mathewson, C.; Siddiqui, A.; Wye, N.; McPherson, J.; Zhao, S.; Fraser, C.M.; Shetty, J.; Shatsman, S.; Geer, K.; Chen, Y.; Abramzon, S.; Nierman, W.C.; Havlak, P.H.; Chen, R.; Durbin, K.J.; Egan, A.; Ren, Y.; Song, X.Z.; Li, B.; Liu, Y.; Qin, X.; Cawley, S.; Worley, K.C.; Cooney, A.J.; D'Souza, L.M.; Martin, K.; Wu, J.Q.; Gonzalez-Garay, M.L.; Jackson, A.R.; Kalafus, K.J.; McLeod, M.P.; Milosavljevic, A.; Virk, D.; Volkov, A.; Wheeler, D.A.; Zhang, Z.; Bailey, J.A.; Eichler, E.E.; Tuzun, E.; Birney, E.; Mongin, E.; Ureta-Vidal, A.; Woodwark, C.; Zdobnov, E.; Bork, P.; Suyama, M.; Torrents, D.; Alexandersson, M.; Trask, B.J.; Young, J.M.; Huang, H.; Wang, H.; Xing, H.; Daniels, S.; Gietzen, D.; Schmidt, J.; Stevens, K.; Vitt, U.; Wingrove, J.; Camara, F.; Mar Alba, M.; Abril, J.F.; Guigo, R.; Smit, A.; Dubchak, I.; Rubin, E.M.; Couronne, O.; Poliakov, A.; Hubner, N.; Ganten, D.; Goesele, C.; Hummel, O.; Kreitler, T.; Lee, Y.A.; Monti, J.; Schulz, H.; Zimdahl, H.; Himmelbauer, H.; Lehrach, H.; Jacob, H.J.; Bromberg, S.; Gullings-Handley, J.; Jensen-Seaman, M.I.; Kwitek, AE.; Lazar, J.; Pasko, D.; Tonellato, P.J.; Twigger, S.; Ponting, C.P.; Duarte, J.M.; Rice, S.; Goodstadt, L.; Beatson, S.A.; Emes, R.D.; Winter, E.E.; Webber, C.; Brandt, P.; Nyakatura, G.; Adetobi, M.; Chiaromonte, F.; Elnitski, L.; Eswara, P.; Hardison, R.C.; Hou, M.; Kolbe, D.; Makova, K.; Miller, W.; Nekrutenko, A.; Riemer, C.; Schwartz, S.; Taylor, J.; Yang, S.; Zhang, Y.; Lindpaintner, K.; Andrews, T.D.; Caccamo, M.; Clamp, M.; Clarke, L.; Curwen, V.; Durbin, R.; Eyras, E.; Searle, S.M.; Cooper, G.M.; Batzoglou, S.; Brudno, M.; Sidow, A.; Stone, E.A.; Venter, J.C.; Payseur, B.A.; Bourque, G.; Lopez-Otin, C.; Puente, X.S.; Chakrabarti, K.; Chatterji, S.; Dewey, C.; Pachter, L.; Bray, N.; Yap, V.B.; Caspi, A.; Tesler, G.; Pevzner, P.A.; Haussler, D.; Roskin, K.M.; Baertsch, R.; Clawson, H.; Furey, T.S.; Hinrichs, A.S.; Karolchik, D.; Kent, W.J.; Rosenbloom, K.R.; Trumbower, H.; Weirauch, M.; Cooper, D.N.; Stenson, P.D.; Ma, B.; Brent, M.; Arumugam, M.; Shteynberg, D.; Copley, R.R.; Taylor, M.S.; Riethman, H.; Mudunuri, U.; Peterson, J.; Guyer, M.; Felsenfeld, A.; Old, S.; Mockrin, S.; Collins, F. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 2004, 428(6982), 493–521. [Google Scholar] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res 2003, 13(4), 721–731. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22(22), 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, B. DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 1999, 15(3), 211–218. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000, 302(1), 205–217. [Google Scholar] [CrossRef] [PubMed]
- Siddharthan, R. Sigma: multiple alignment of weakly-conserved non-coding DNA sequence. BMC Bioinformatics 2006, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Cliften, P.F.; Hillier, L.W.; Fulton, L.; Graves, T.; Miner, T.; Gish, W.R.; Waterston, R.H.; Johnston, M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res 2001, 11(7), 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M. Identifying functional elements by comparative DNA sequence analysis. Genome Res 2001, 11(7), 1143–1144. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Tompa, M. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res 2002, 12(5), 739–748. [Google Scholar] [CrossRef] [PubMed]
- McCue, L.; Thompson, W.; Carmack, C.; Ryan, M.P.; Liu, J.S.; Derbyshire, V.; Lawrence, C.E. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res 2001, 29(3), 774–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Stormo, G.D. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 2003, 19(18), 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Guryev, V.; Plasterk, R.H.; Cuppen, E. CONREAL: conserved regulatory elements anchored alignment algorithm for identification of transcription factor binding sites by phylogenetic footprinting. Genome Res 2004, 14(1), 170–178. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Tompa, M. FootPrinter: A program designed for phylogenetic footprinting. Nucleic Acids Res 2003, 31(13), 3840–3842. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.M.; Chiang, D.Y.; Eisen, M.B. Phylogenetic motif detection by expectation-maximization on evolutionary mixtures. Pac Symp Biocomput 2004, 324–335. [Google Scholar]
- Jukes, T.H.C.R.C. Evolution of protein molecules. In Mammalian protein metabolism; Munro, H.N., Ed.; Academic Press: New York, 1969; pp. 21–123. [Google Scholar]
- Sinha, S. PhyME: a software tool for finding motifs in sets of orthologous sequences. Methods Mol Biol 2007, 395, 309–318. [Google Scholar] [PubMed]
- Sinha, S.; Blanchette, M.; Tompa, M. PhyME: a probabilistic algorithm for finding motifs in sets of orthologous sequences. BMC Bioinformatics 2004, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Siddharthan, R. PhyloGibbs-MP: module prediction and discriminative motif-finding by Gibbs sampling. PLoS Comput Biol 2008, 4(8), e1000156. [Google Scholar] [CrossRef] [PubMed]
- Siddharthan, R.; Siggia, E.D.; van Nimwegen, E. PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny. PLoS Comput Biol 2005, 1(7), e67. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; van Nimwegen, E.; Siggia, E.D. A probabilistic method to detect regulatory modules. Bioinformatics 2003, 19 Suppl 1, i292–301. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 1981, 17(6), 368–376. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.M.; Chiang, D.Y.; Pollard, D.A.; Iyer, V.N.; Eisen, M.B. MONKEY: identifying conserved transcription-factor binding sites in multiple alignments using a binding site-specific evolutionary model. Genome Biol 2004, 5(12), R98. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 1985, 22(2), 160–174. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Fay, J.C.; Cohen, B.A. Phylogeny based discovery of regulatory elements. BMC Bioinformatics 2006, 7, 266. [Google Scholar] [CrossRef] [PubMed]
- Carmack, C.S.; McCue, L.A.; Newberg, L.A.; Lawrence, C.E. PhyloScan: identification of transcription factor binding sites using cross-species evidence. Algorithms Mol Biol 2007, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Harbison, C.T.; Gordon, D.B.; Lee, T.I.; Rinaldi, N.J.; Macisaac, K.D.; Danford, T.W.; Hannett, N.M.; Tagne, J.B.; Reynolds, D.B.; Yoo, J.; Jennings, E.G.; Zeitlinger, J.; Pokholok, D.K.; Kellis, M.; Rolfe, P.A.; Takusagawa, K.T.; Lander, E.S.; Gifford, D.K.; Fraenkel, E.; Young, R.A. Transcriptional regulatory code of a eukaryotic genome. Nature 2004, 431(7004), 99–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Lee, H.S.; Jeon, S.H.; Chung, T.H.; Lim, Y.S.; Huh, W.K. High-resolution analysis of condition-specific regulatory modules in Saccharomyces cerevisiae. Genome Biol 2008, 9, R2. [Google Scholar] [CrossRef] [PubMed]
- McCord, R.P.; Berger, M.F.; Philippakis, A.A.; Bulyk, M.L. Inferring condition-specific transcription factor function from DNA binding and gene expression data. Mol Syst Biol 2007, 3, 100. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Sumazin, P.; Zhang, M.Q. Tissue-specific regulatory elements in mammalian promoters. Mol Syst Biol 2007, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lin, J.; Zack, D.J.; Qian, J. Identification of tissue-specific cis-regulatory modules based on interactions between transcription factors. BMC Bioinformatics 2007, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Fessele, S.; Maier, H.; Zischek, C.; Nelson, P.J.; Werner, T. Regulatory context is a crucial part of gene function. Trends Genet 2002, 18(2), 60–63. [Google Scholar] [CrossRef]
- Allocco, D.J.; Kohane, I.S.; Butte, A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics 2004, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Liu, H.; Hahn, C.; Sumazin, P.; Zhang, M.Q.; Zilberstein, A. Genome-wide prediction and analysis of function-specific transcription factor binding sites. In Silico Biol 2004, 4(4), 395–410. [Google Scholar] [PubMed]
- Frech, K.; Danescu-Mayer, J.; Werner, T. A novel method to develop highly specific models for regulatory units detects a new LTR in GenBank which contains a functional promoter. J Mol Biol 1997, 270(5), 674–687. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C.; Li, M.C.; Weng, Z. Cluster-Buster: Finding dense clusters of motifs in DNA sequences. Nucleic Acids Res 2003, 31(13), 3666–3668. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wong, W.H. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc Natl Acad Sci U S A 2004, 101(33), 12114–12119. [Google Scholar] [CrossRef] [PubMed]
- Alkema, W.B.; Johansson, O.; Lagergren, J.; Wasserman, W.W. MSCAN: identification of functional clusters of transcription factor binding sites. Nucleic Acids Res 2004, 32(Web Server issue), W195–198. [Google Scholar] [CrossRef] [PubMed]
- Pierstorff, N.; Bergman, C.M.; Wiehe, T. Identifying cis-regulatory modules by combining comparative and compositional analysis of DNA. Bioinformatics 2006, 22(23), 2858–2864. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, P.; Aerts, S.; Thienpont, B.; De Moor, B.; Moreau, Y.; Marynen, P. ModuleMiner - improved computational detection of cis-regulatory modules: are there different modes of gene regulation in embryonic development and adult tissues? Genome Biol 2008, 9(4), R66. [Google Scholar] [CrossRef] [PubMed]
- Gotea, V.; Ovcharenko, I. DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res 2008, 36(Web Server issue), W133–139. [Google Scholar] [CrossRef] [PubMed]
- Waleev, T.; Shtokalo, D.; Konovalova, T.; Voss, N.; Cheremushkin, E.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E.; Kel, A. Composite Module Analyst: identification of transcription factor binding site combinations using genetic algorithm. Nucleic Acids Res 2006, 34(Web Server issue), W541–545. [Google Scholar] [CrossRef] [PubMed]
- Roth, F.P.; Hughes, J.D.; Estep, P.W.; Church, G.M. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol 1998, 16(10), 939–945. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.; Hughes, J.D.; Campbell, M.J.; Cho, R.J.; Church, G.M. Systematic determination of genetic network architecture. Nat Genet 1999, 22(3), 281–285. [Google Scholar] [PubMed]
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405(6788), 827–836. [Google Scholar] [CrossRef] [PubMed]
- Flintoft, L. Gene regulation: The many paths to coexpression. Nature Reviews Genetics 2007, 8, 827. [Google Scholar] [CrossRef]
- Choi, D.; Fang, Y.; Mathers, W.D. Condition-specific coregulation with cis-regulatory motifs and modules in the mouse genome. Genomics 2006, 87(4), 500–508. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wallqvist, A.; Covell, D.G. Comprehensive analysis of pathway or functionally related gene expression in the National Cancer Institute's anticancer screen. Genomics 2006, 87(3), 315–328. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.; Shapira, M.; Regev, A.; Pe'er, D.; Botstein, D.; Koller, D.; Friedman, N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 2003, 34(2), 166–176. [Google Scholar] [CrossRef] [PubMed]
- Elkon, R.; Linhart, C.; Sharan, R.; Shamir, R.; Shiloh, Y. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res 2003, 13(5), 773–780. [Google Scholar] [CrossRef] [PubMed]
- Cora, D.; Herrmann, C.; Dieterich, C.; Di Cunto, F.; Provero, P.; Caselle, M. Ab initio identification of putative human transcription factor binding sites by comparative genomics. BMC Bioinformatics 2005, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Defrance, M.; Touzet, H. Predicting transcription factor binding sites using local over-representation and comparative genomics. BMC Bioinformatics 2006, 7, 396. [Google Scholar] [CrossRef] [PubMed]
- Monsieurs, P.; Thijs, G.; Fadda, A.A.; De Keersmaecker, S.C.; Vanderleyden, J.; De Moor, B.; Marchal, K. More robust detection of motifs in coexpressed genes by using phylogenetic information. BMC Bioinformatics 2006, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Vandepoele, K.; Casneuf, T.; Van de Peer, Y. Identification of novel regulatory modules in dicotyledonous plants using expression data and comparative genomics. Genome Biol 2006, 7(11), R103. [Google Scholar] [CrossRef] [PubMed]
- King, D.C.; Taylor, J.; Elnitski, L.; Chiaromonte, F.; Miller, W.; Hardison, R.C. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res 2005, 15(8), 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, D.; Taylor, J.; Elnitski, L.; Eswara, P.; Li, J.; Miller, W.; Hardison, R.; Chiaromonte, F. Regulatory potential scores from genome-wide three-way alignments of human, mouse, and rat. Genome Res 2004, 14(4), 700–707. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Tyekucheva, S.; King, D.C.; Hardison, R.C.; Miller, W.; Chiaromonte, F. ESPERR: learning strong and weak signals in genomic sequence alignments to identify functional elements. Genome Res 2006, 16(12), 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Cheng, Y.; Zhou, Y.; King, D.C.; Taylor, J.; Chiaromonte, F.; Kasturi, J.; Petrykowska, H.; Gibb, B.; Dorman, C.; Miller, W.; Dore, L.C.; Welch, J.; Weiss, M.J.; Hardison, R.C. Experimental validation of predicted mammalian erythroid cis-regulatory modules. Genome Res 2006, 16(12), 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.; Scherf, M.; Epple, A.; Werner, T. Multievidence microarray mining. Trends Genet 2005, 21(10), 553–558. [Google Scholar] [CrossRef] [PubMed]
- Gonye, G.E.; Chakravarthula, P.; Schwaber, J.S.; Vadigepalli, R. From promoter analysis to transcriptional regulatory network prediction using PAINT. Methods Mol Biol 2007, 408, 49–68. [Google Scholar] [PubMed]
- Vadigepalli, R.; Chakravarthula, P.; Zak, D.E.; Schwaber, J.S.; Gonye, G.E. PAINT: a promoter analysis and interaction network generation tool for gene regulatory network identification. Omics 2003, 7(3), 235–252. [Google Scholar] [CrossRef] [PubMed]
- Haverty, P.M.; Frith, M.C.; Weng, Z. CARRIE web service: automated transcriptional regulatory network inference and interactive analysis. Nucleic Acids Res 2004, 32(Web Server issue), W213–216. [Google Scholar] [CrossRef] [PubMed]
- Haverty, P.M.; Hansen, U.; Weng, Z. Computational inference of transcriptional regulatory networks from expression profiling and transcription factor binding site identification. Nucleic Acids Res 2004, 32(1), 179–188. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nguyen, T.T.; Androulakis, I.P. Recent Advances in the Computational Discovery of Transcription Factor Binding Sites. Algorithms 2009, 2, 582-605. https://doi.org/10.3390/a2010582
Nguyen TT, Androulakis IP. Recent Advances in the Computational Discovery of Transcription Factor Binding Sites. Algorithms. 2009; 2(1):582-605. https://doi.org/10.3390/a2010582
Chicago/Turabian StyleNguyen, Tung T., and Ioannis P. Androulakis. 2009. "Recent Advances in the Computational Discovery of Transcription Factor Binding Sites" Algorithms 2, no. 1: 582-605. https://doi.org/10.3390/a2010582
APA StyleNguyen, T. T., & Androulakis, I. P. (2009). Recent Advances in the Computational Discovery of Transcription Factor Binding Sites. Algorithms, 2(1), 582-605. https://doi.org/10.3390/a2010582