Pattern Recognition and Pathway Analysis with Genetic Algorithms in Mass Spectrometry Based Metabolomics

Abstract
:1. Introduction
2. Results and Discussion
2.1 General workflow
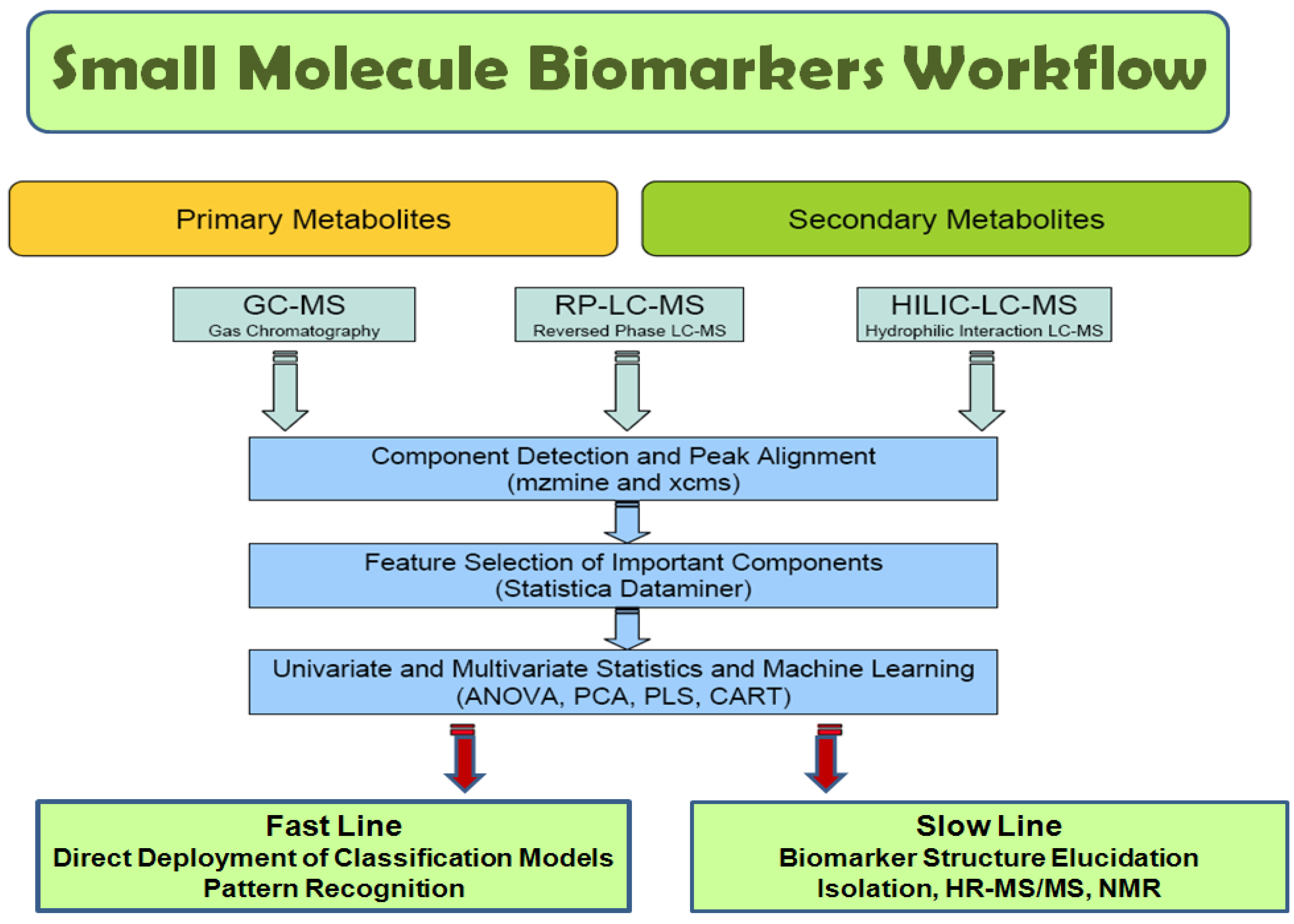
2.1.1 Data acquisition (LC-MS)
2.1.2 Data annotation
2.1.3 Data pre-processing
2.1.4 Unsupervised Analysis without Feature Selection
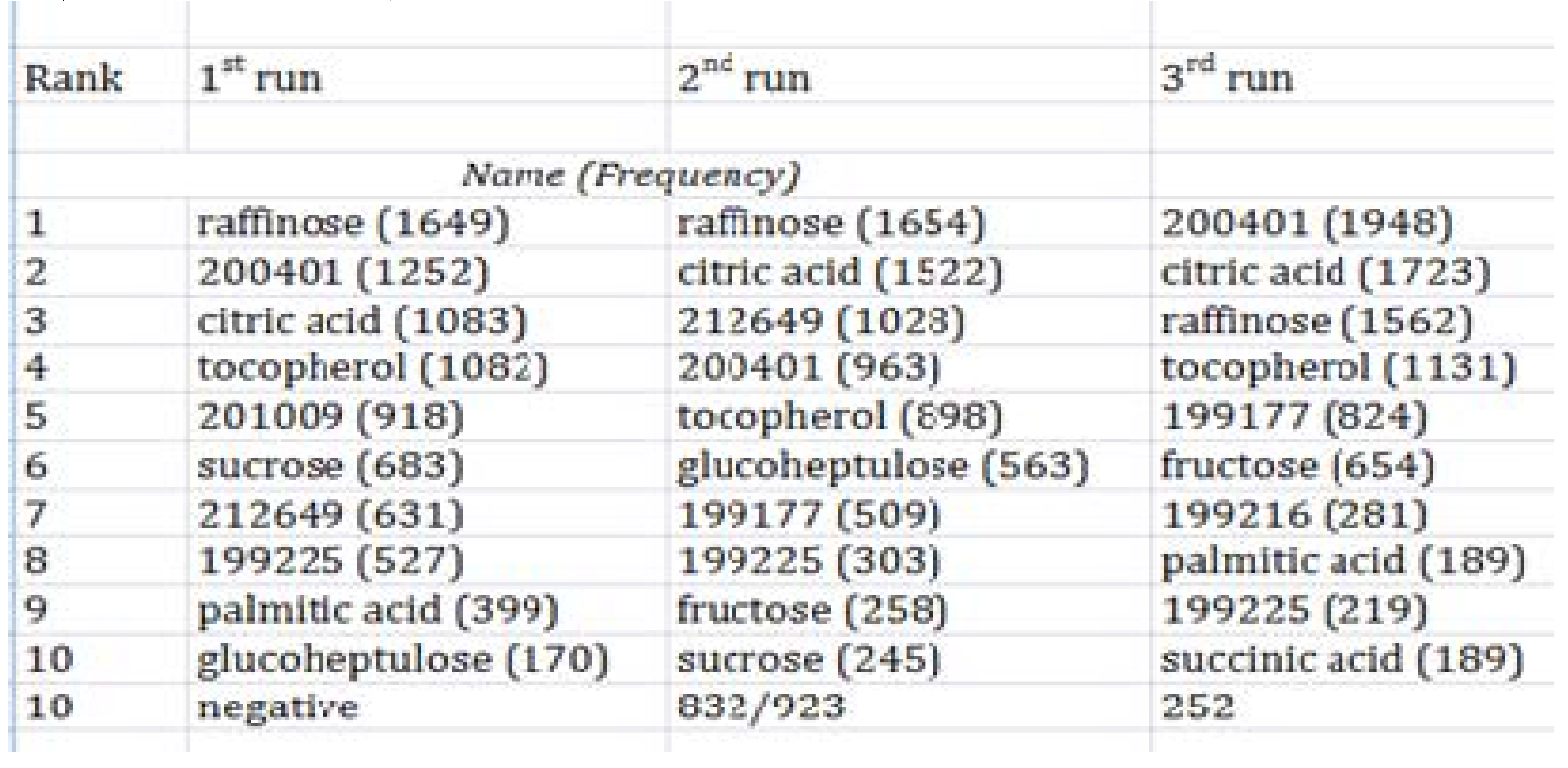
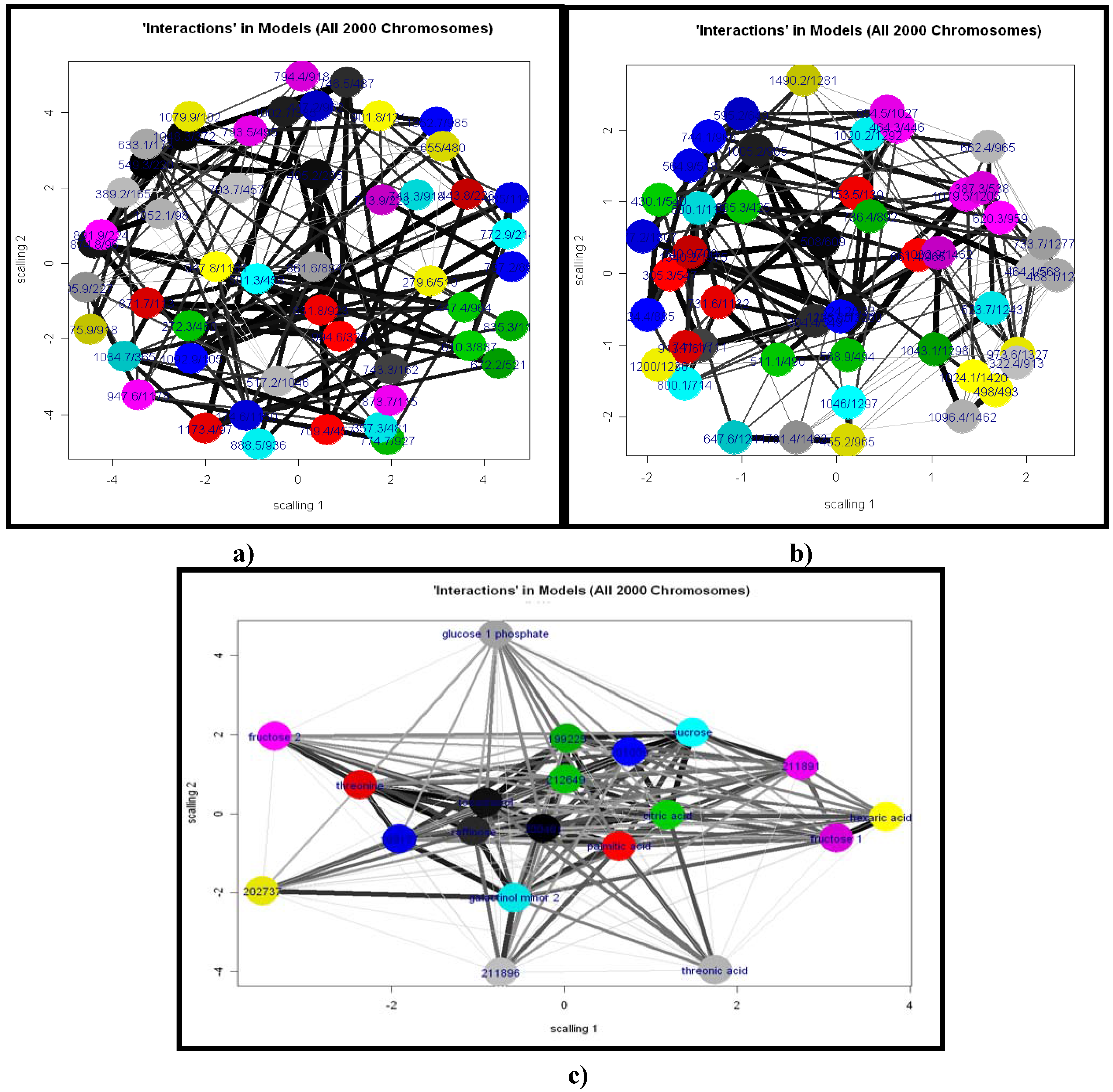
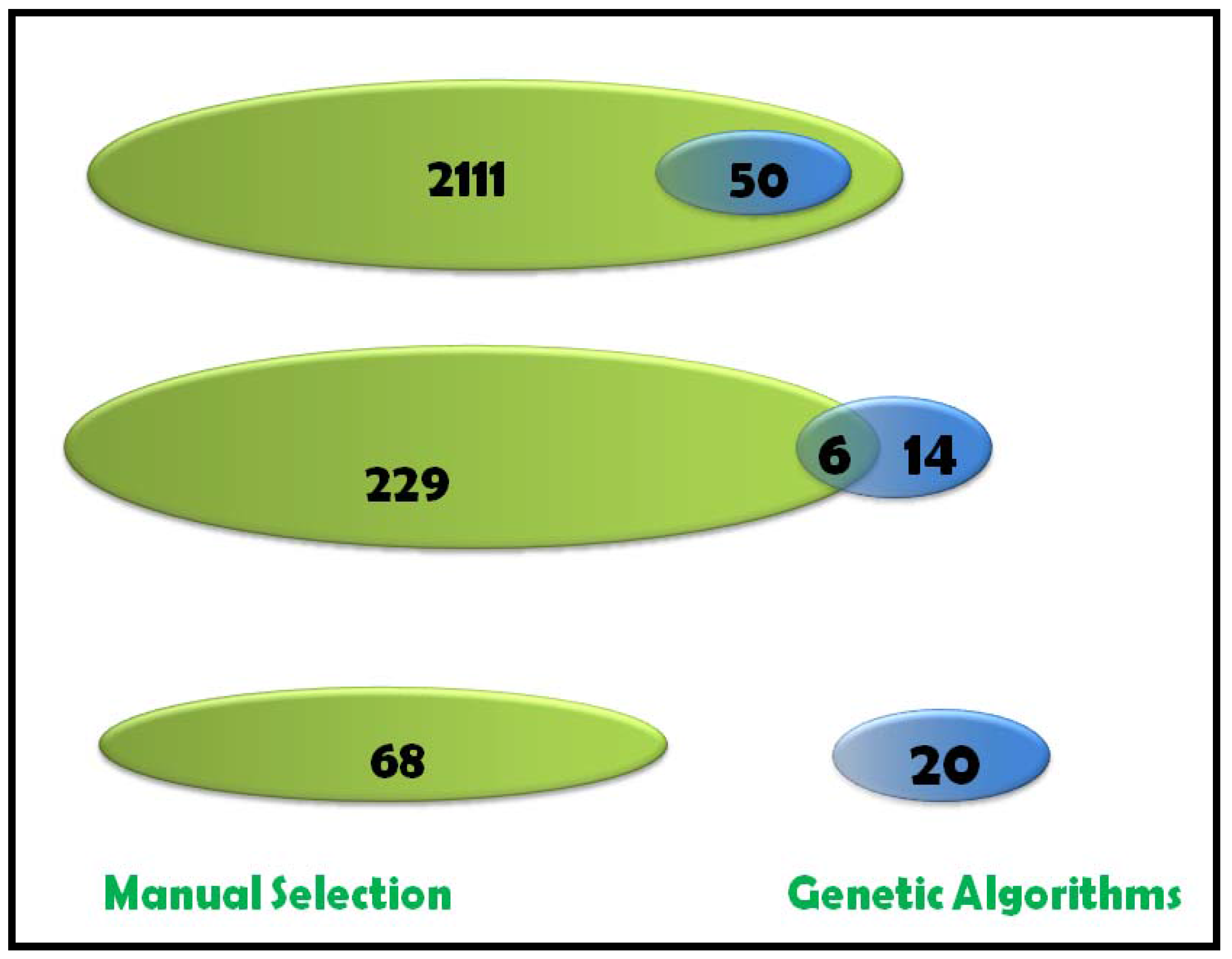
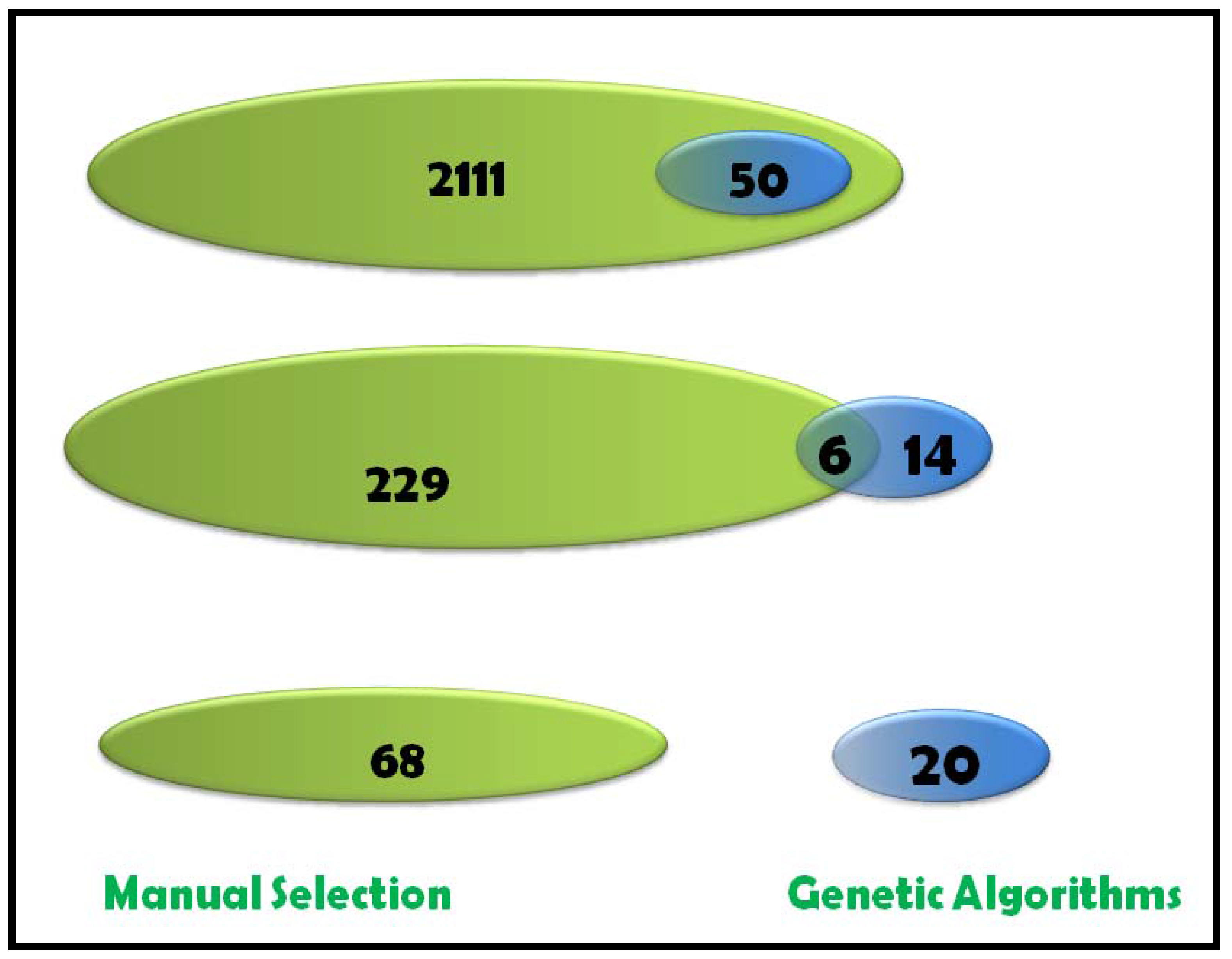
2.1.5 Feature Selection Using GA
- GA procedure initially creates chromosomes that are subsets of variables;
- Fitness of each subset was defined as its ability to predict the group membership of each sample in the dataset, and the GA assigns a score to each subset;
- GA procedure selects the fittest subset; this fittest subset generates more numerous offspring;
- Two randomly selected parent subsets are used to create two new subsets mimicking biological crossover and mutation mechanisms; the process is repeated from stage 2 until an accurate subset is obtained.
2.1.6 Classification and Prediction
2.1.7 Pathway Analysis
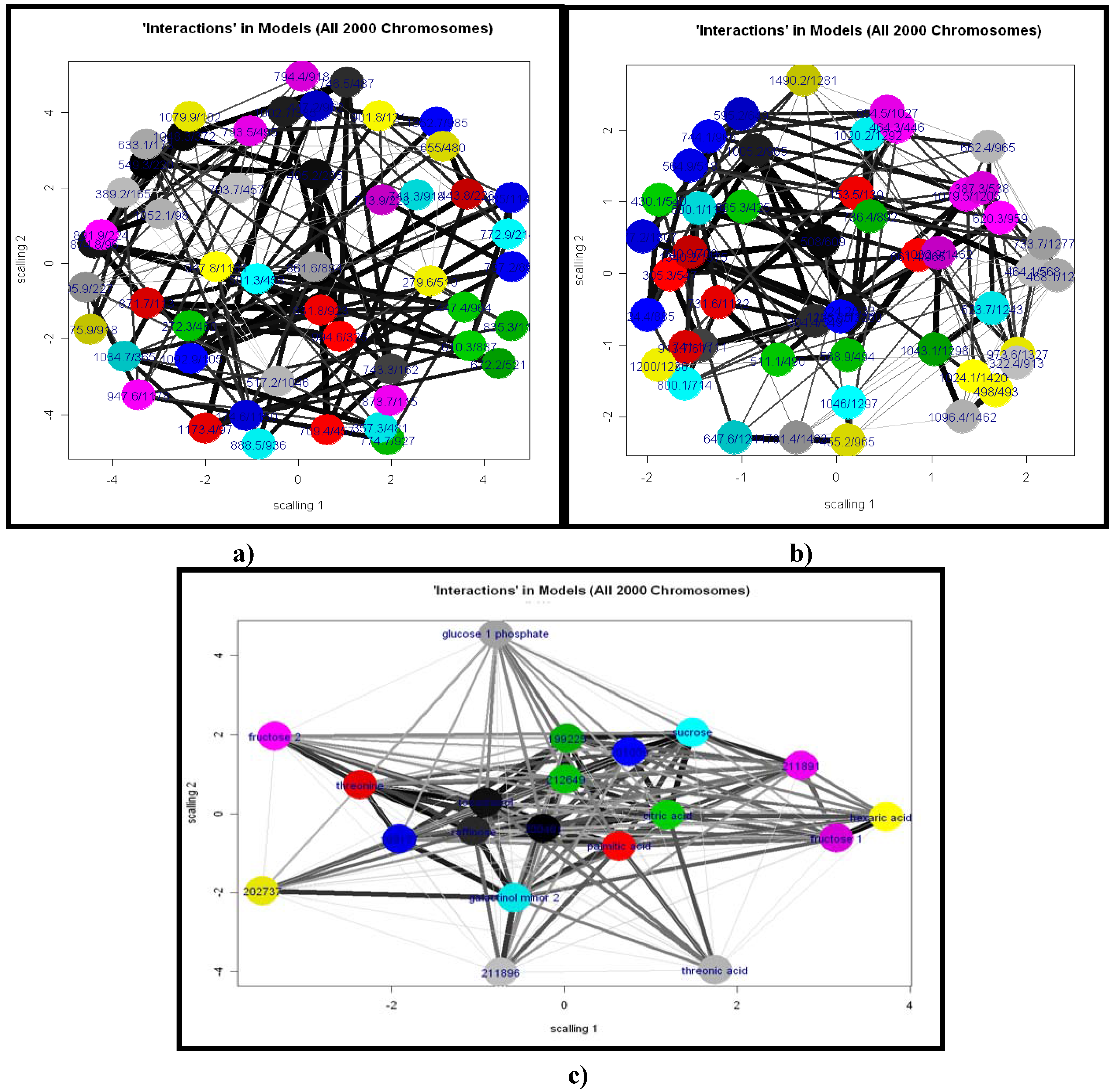
2.1.8 GA Limitations
2.2 Examples
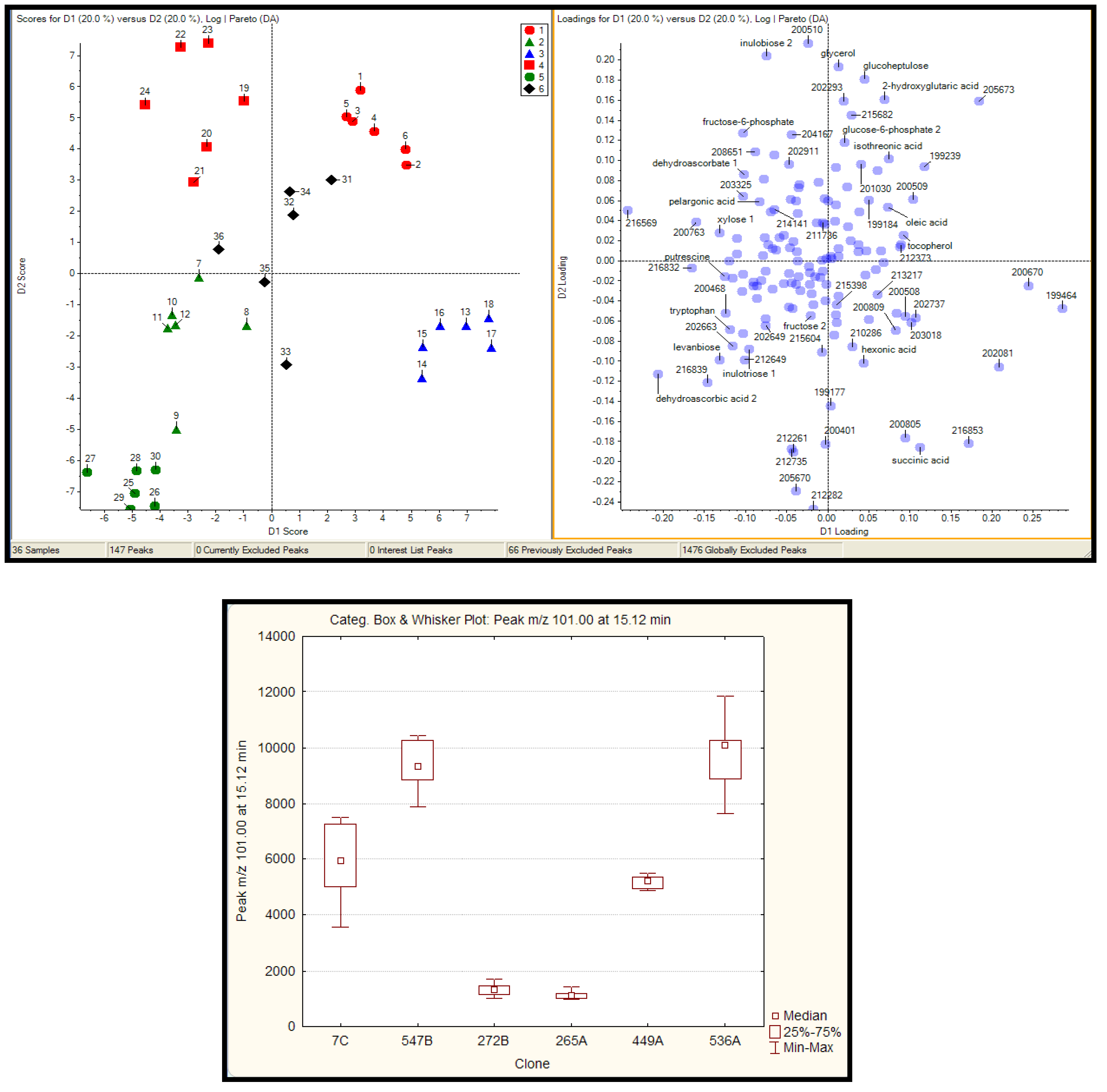
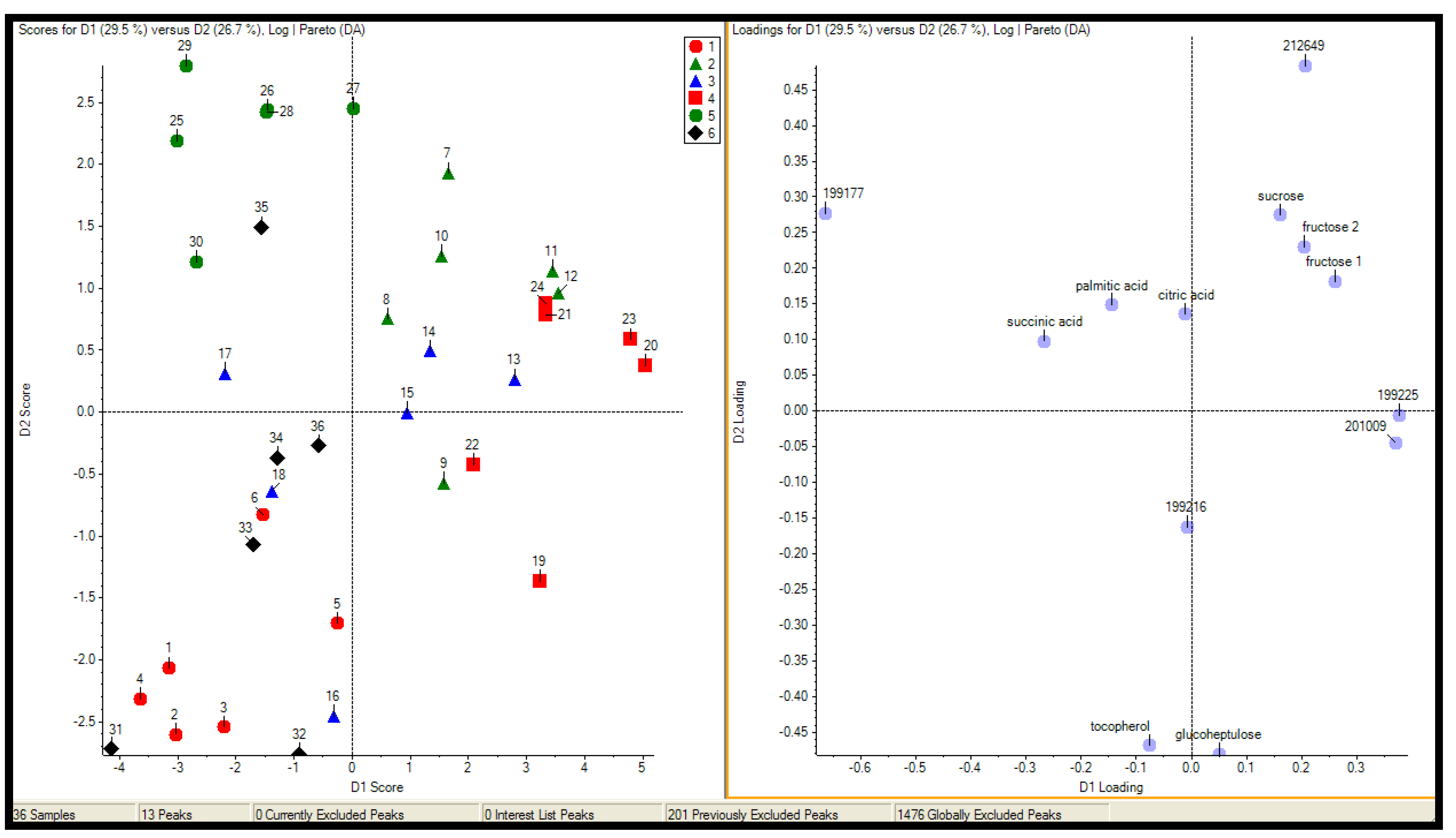
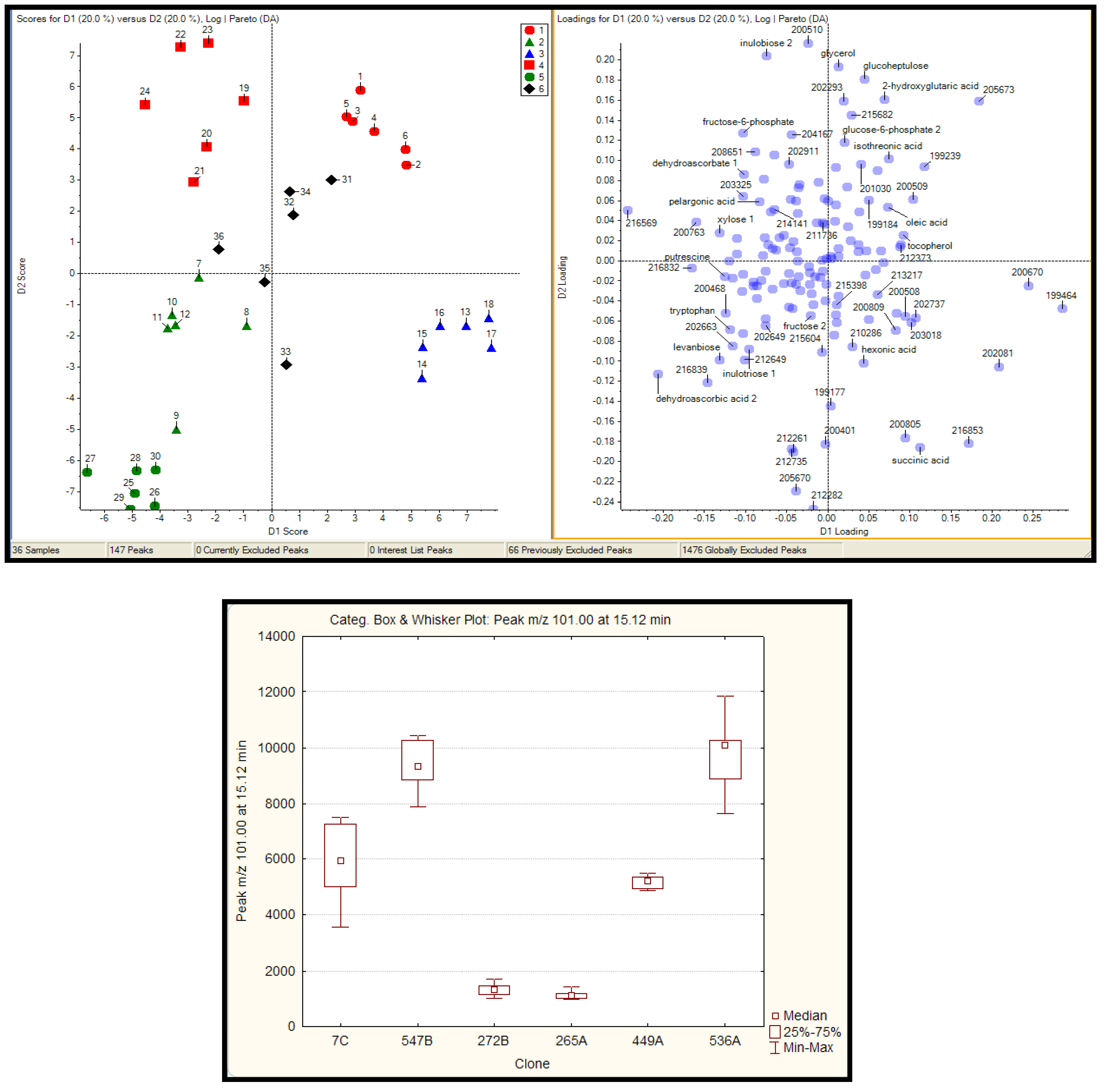
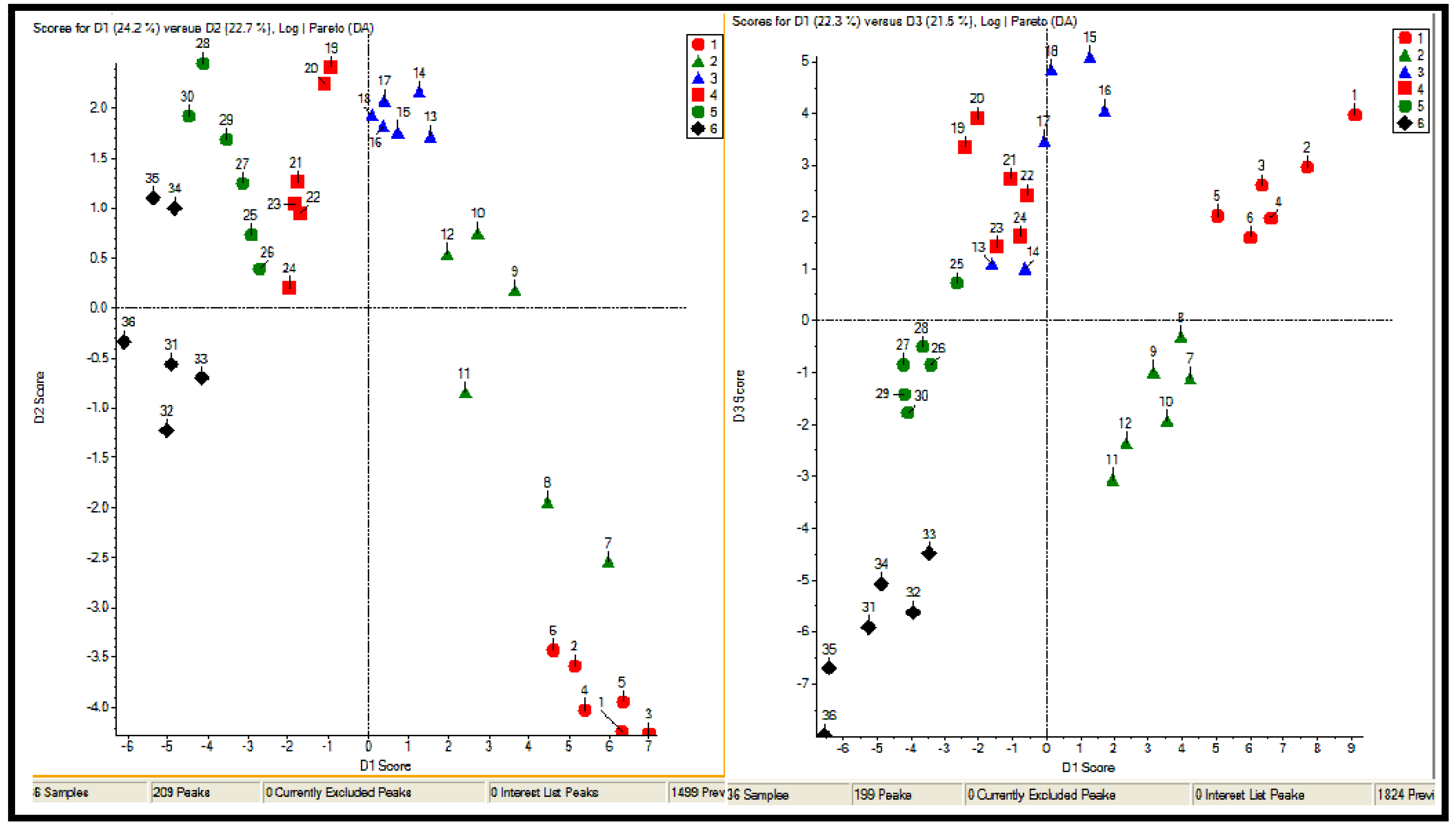
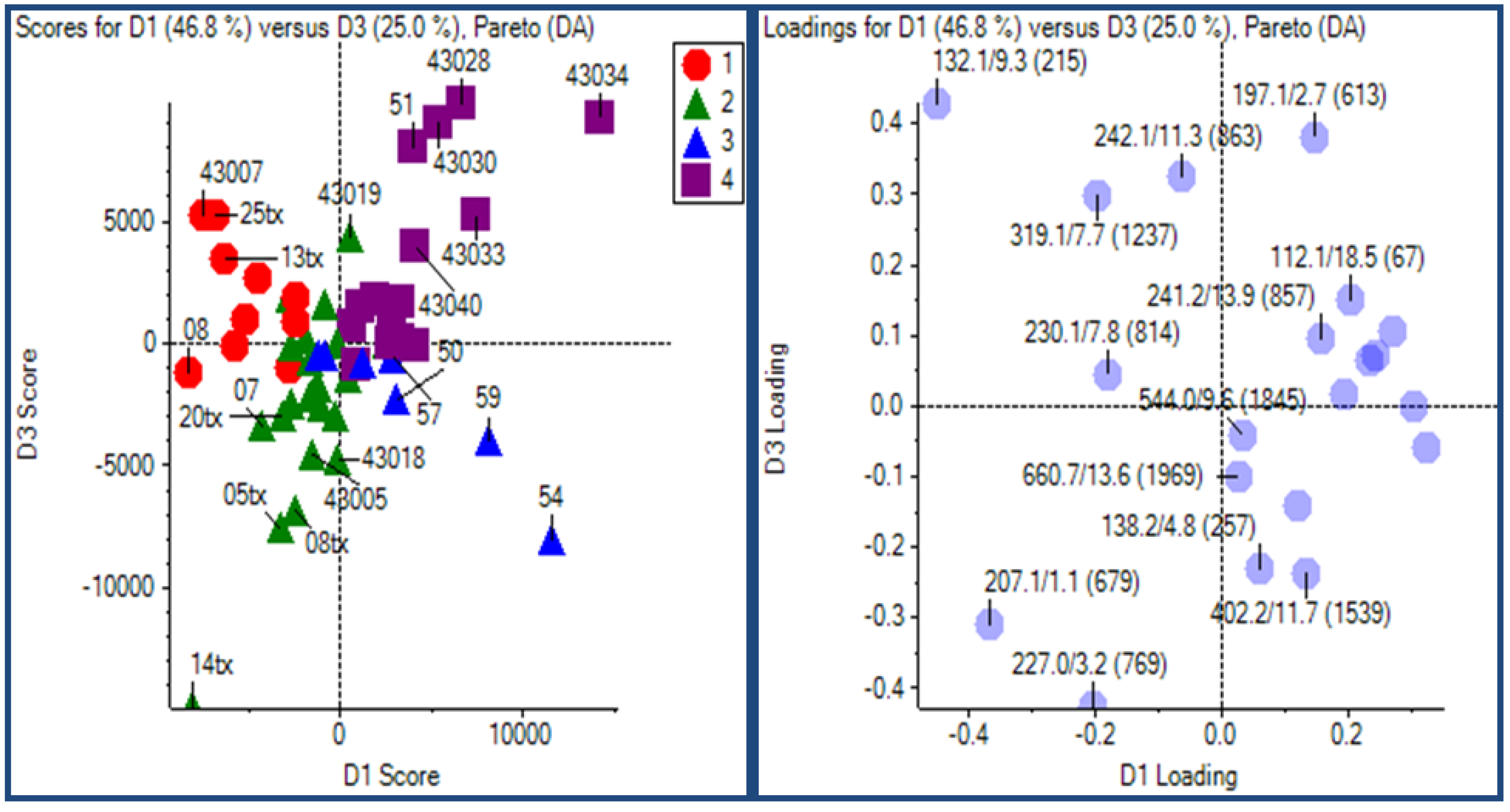
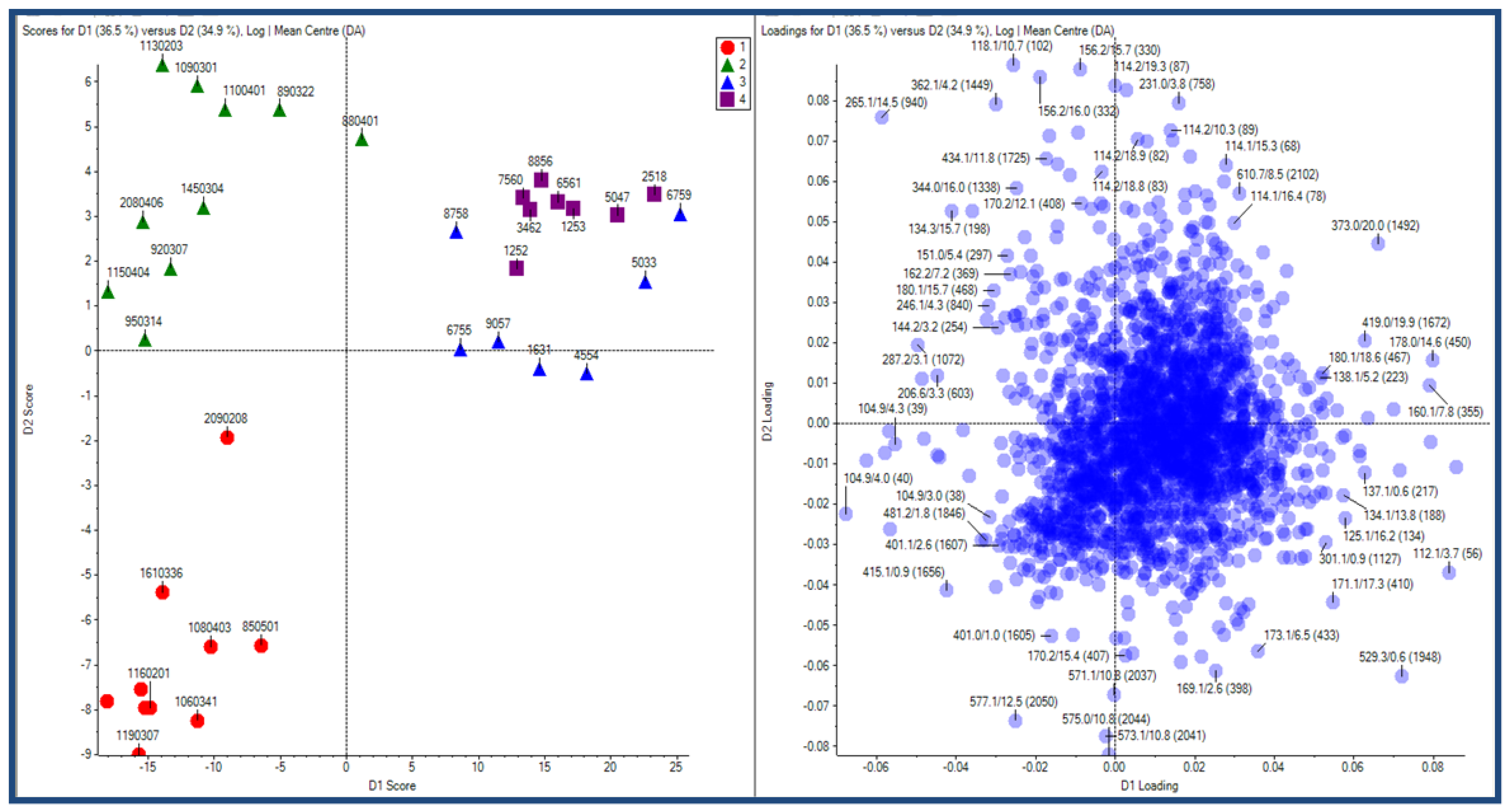
2.2.1 Plant samples




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
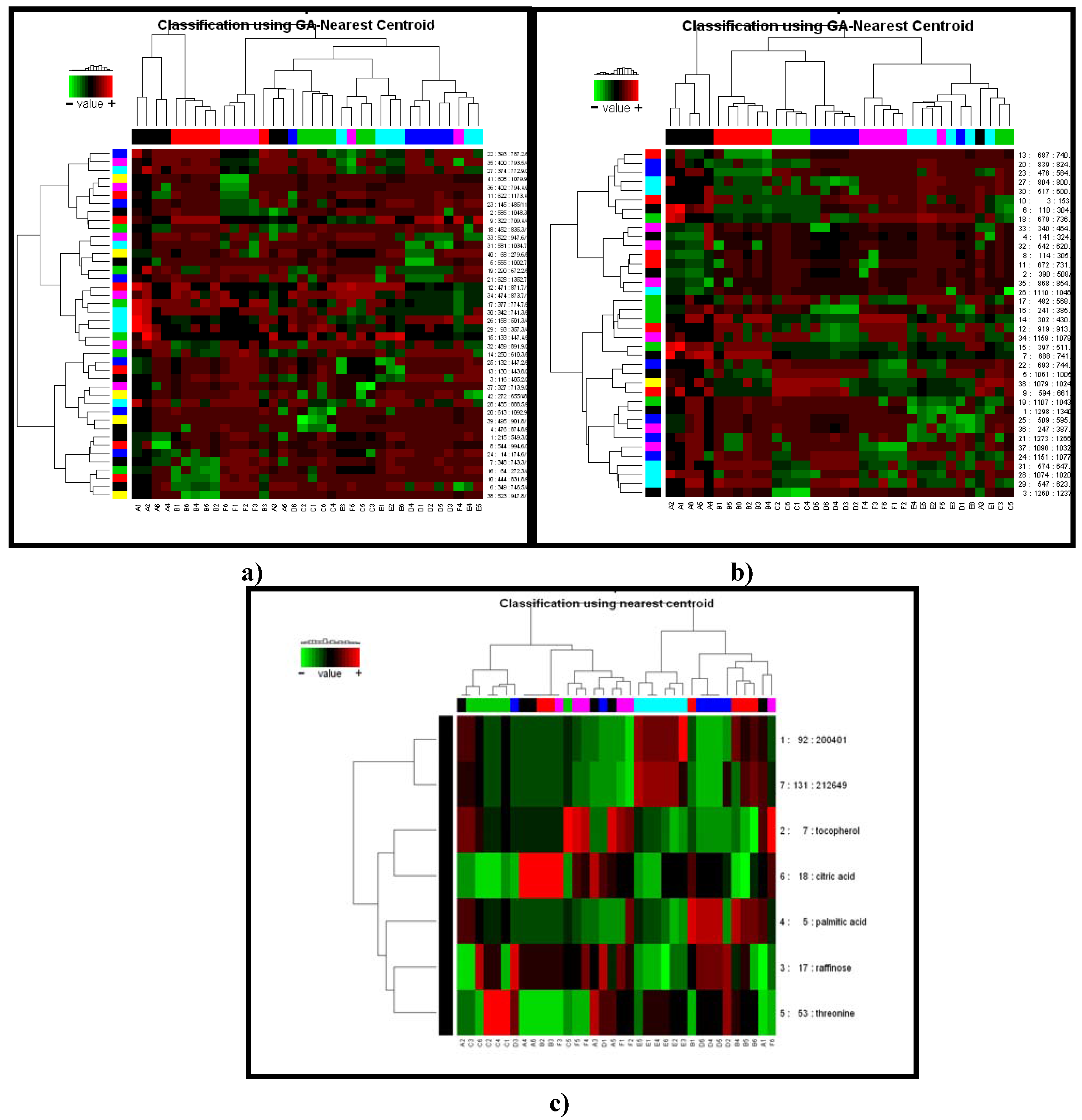{kind=link}
{kind=link}
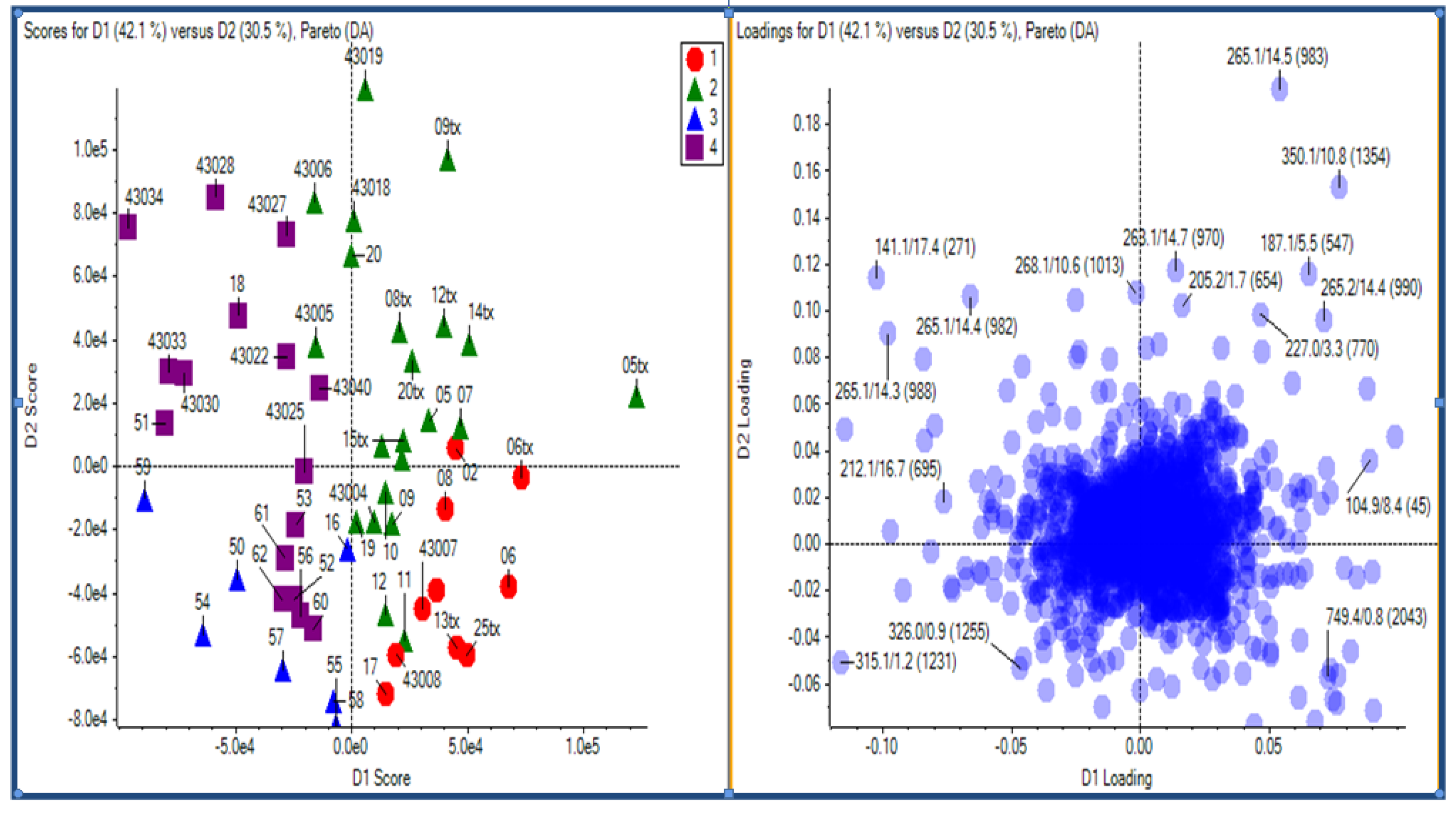{kind=link}
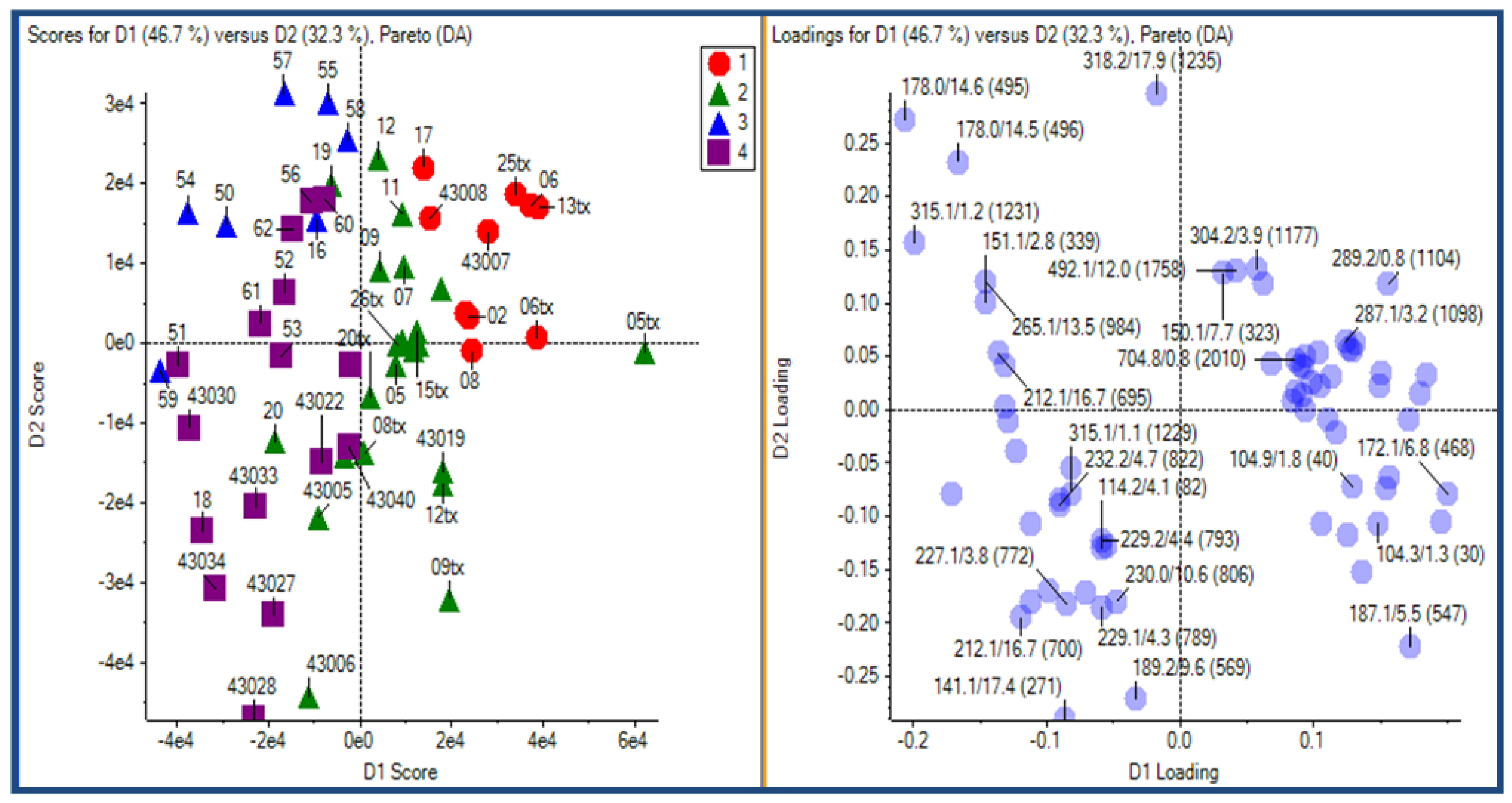{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
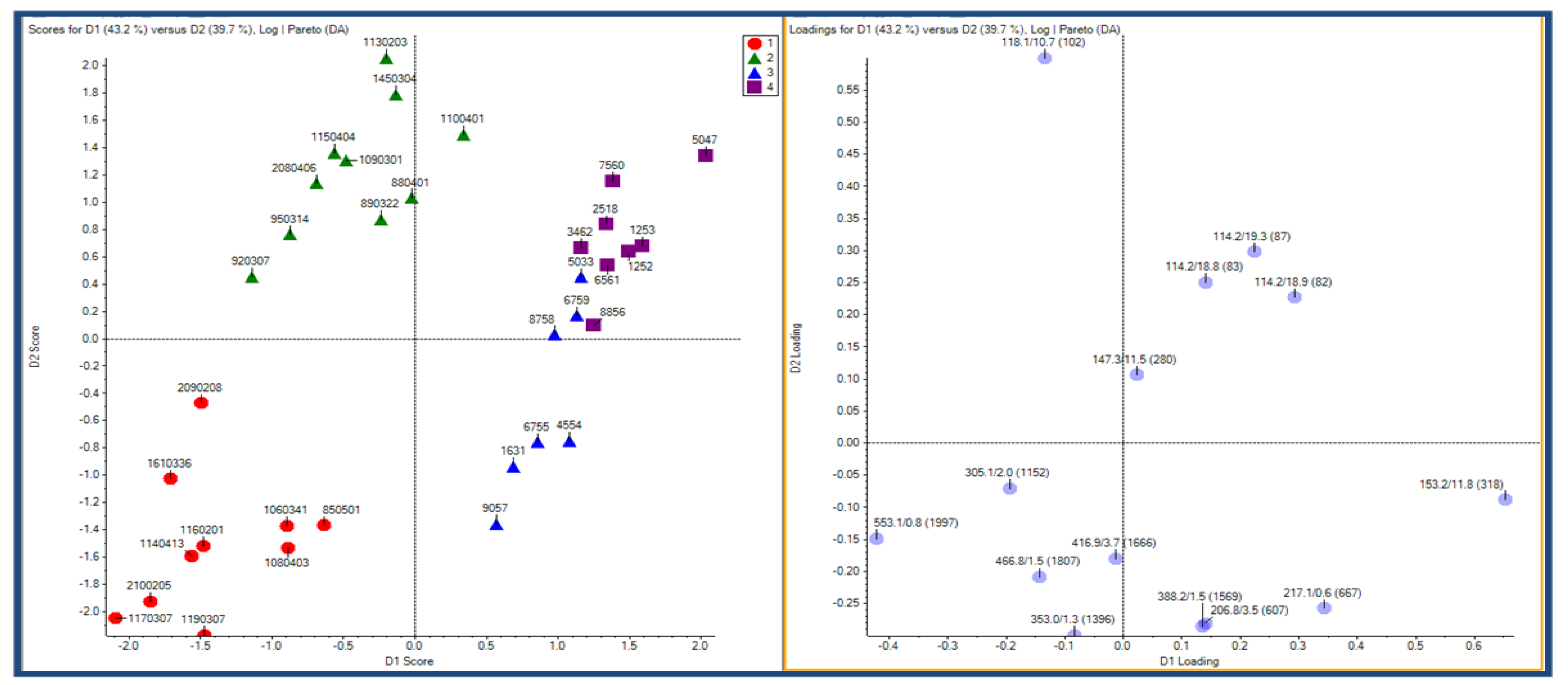{kind=link}
{kind=link}
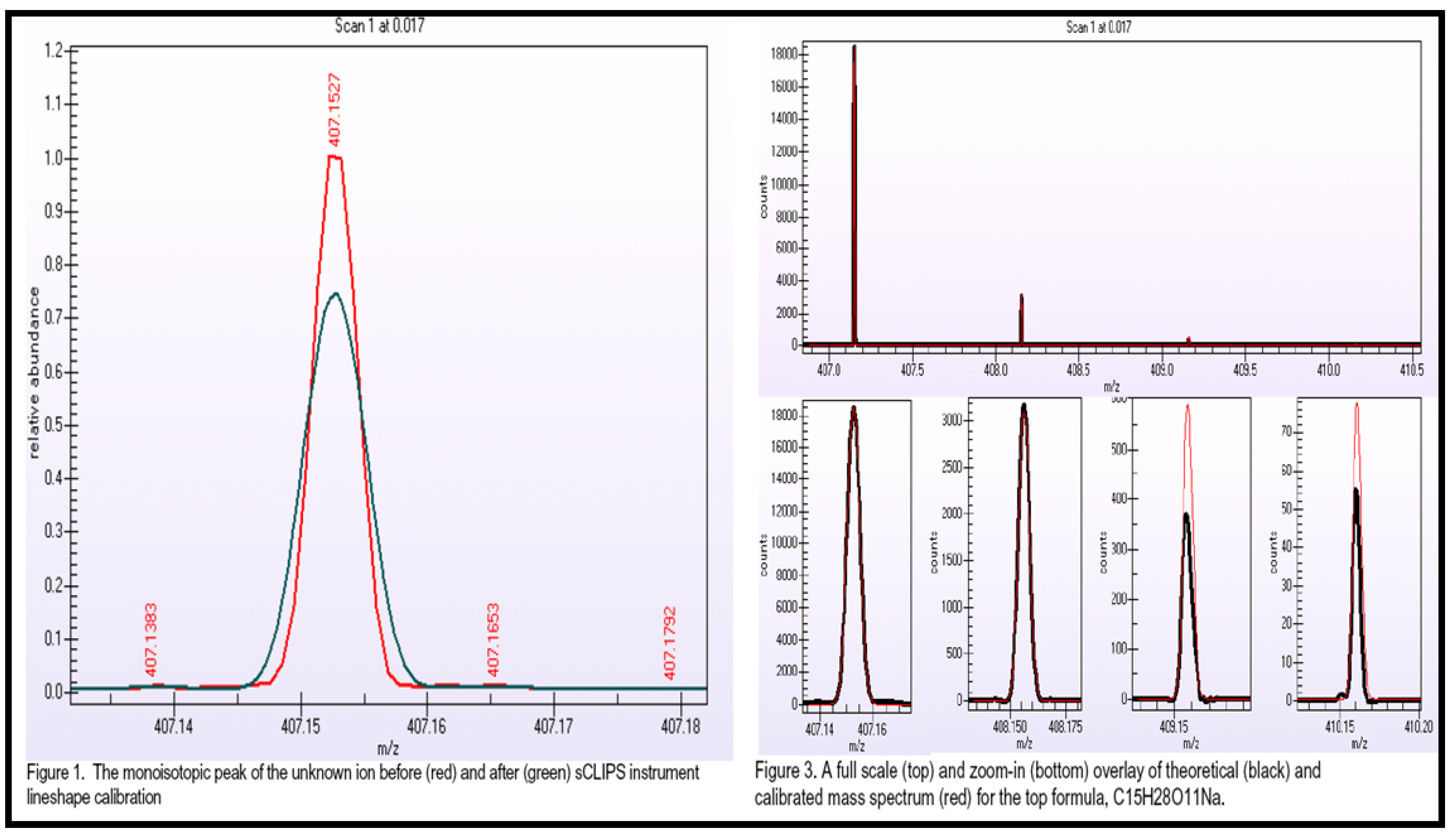{kind=link}
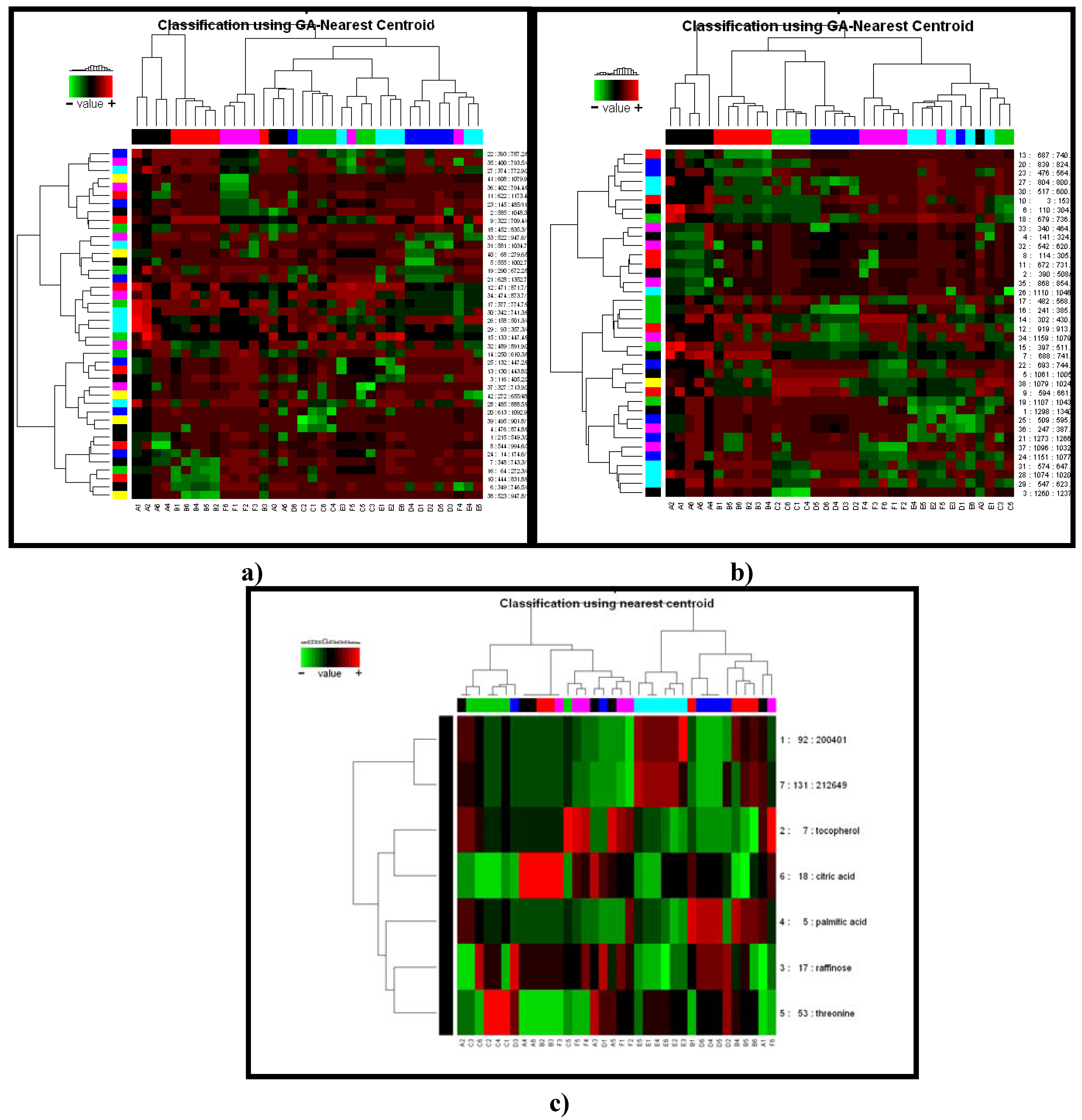
2.2.2 Case Summary
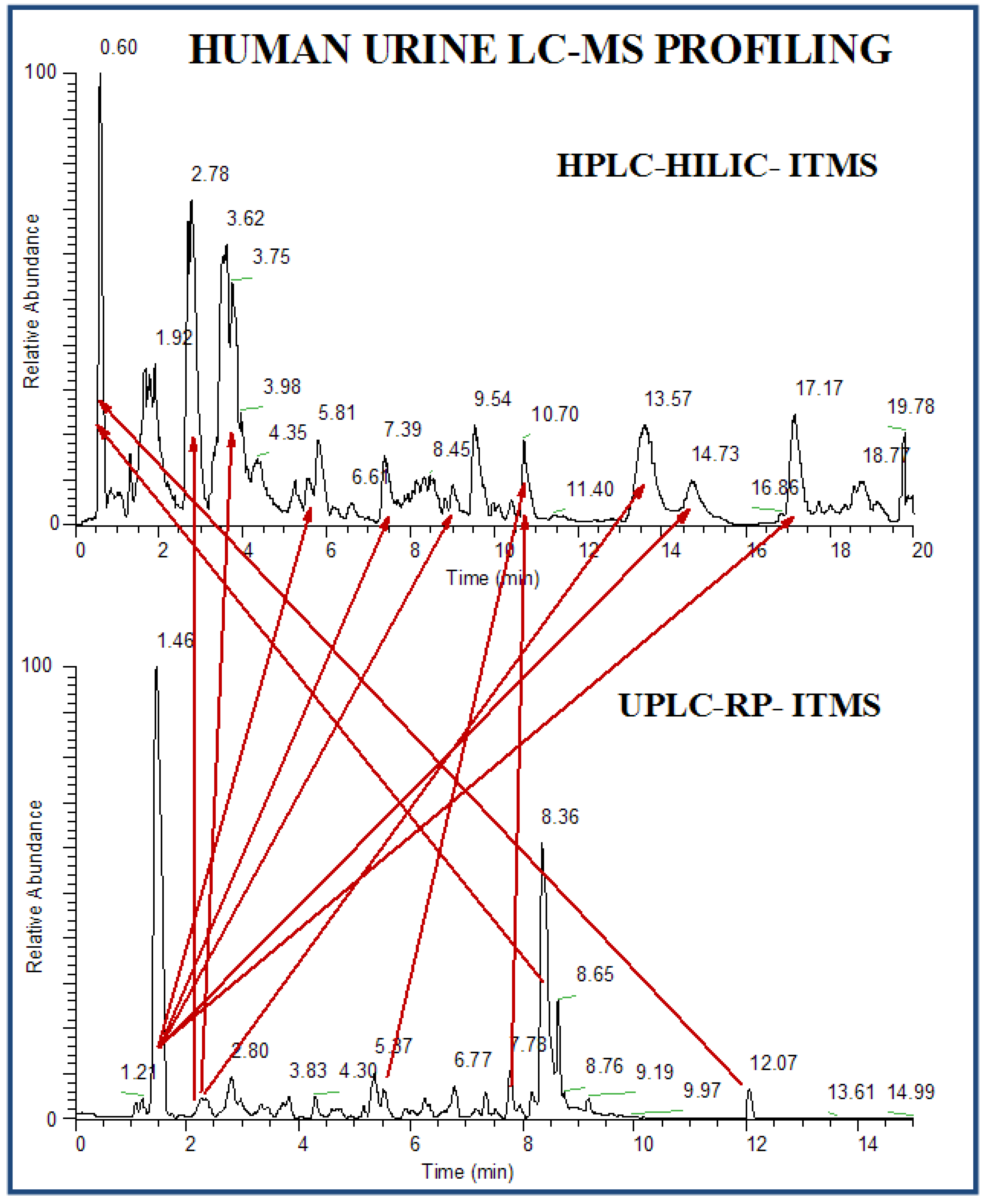
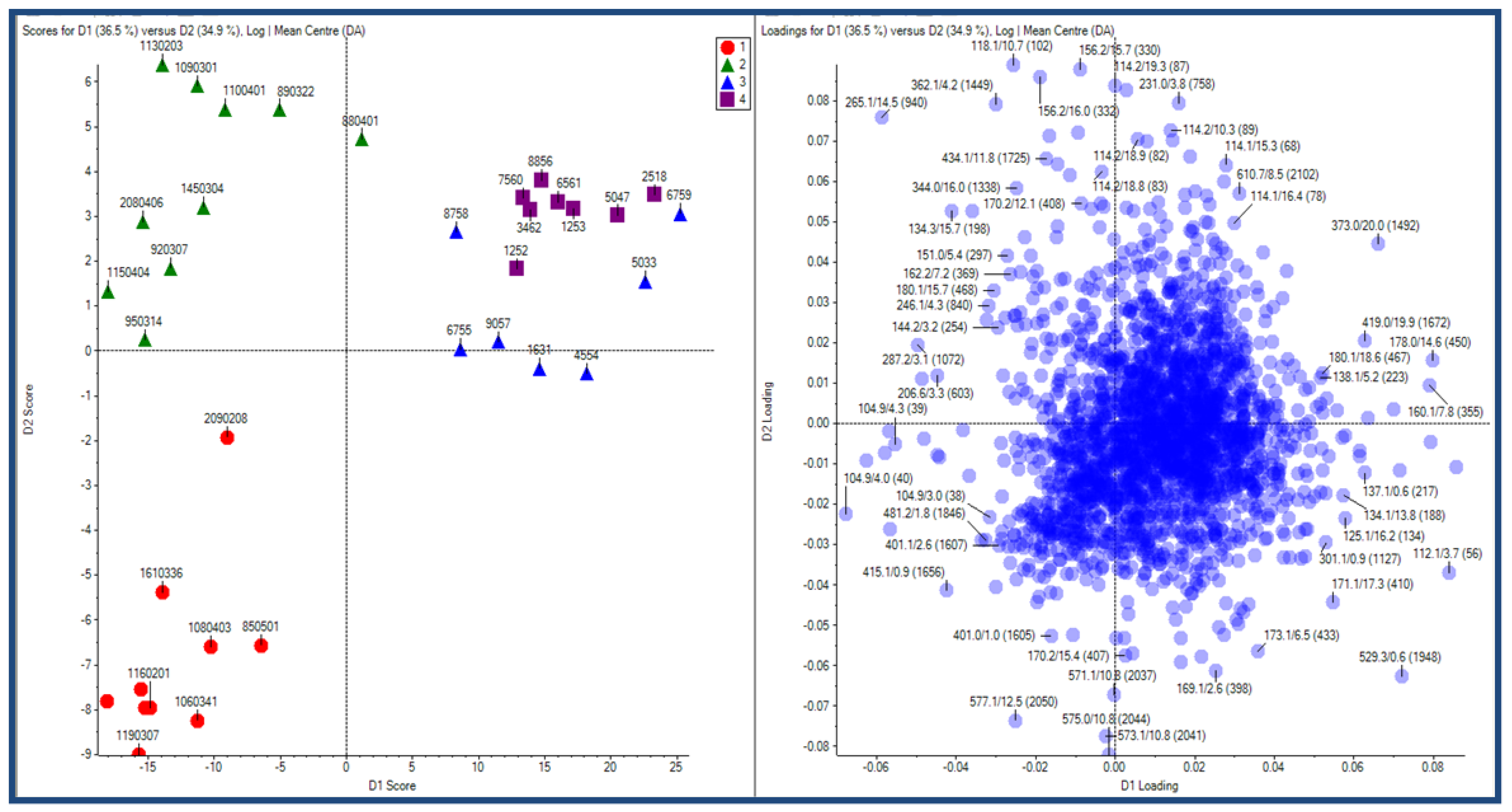
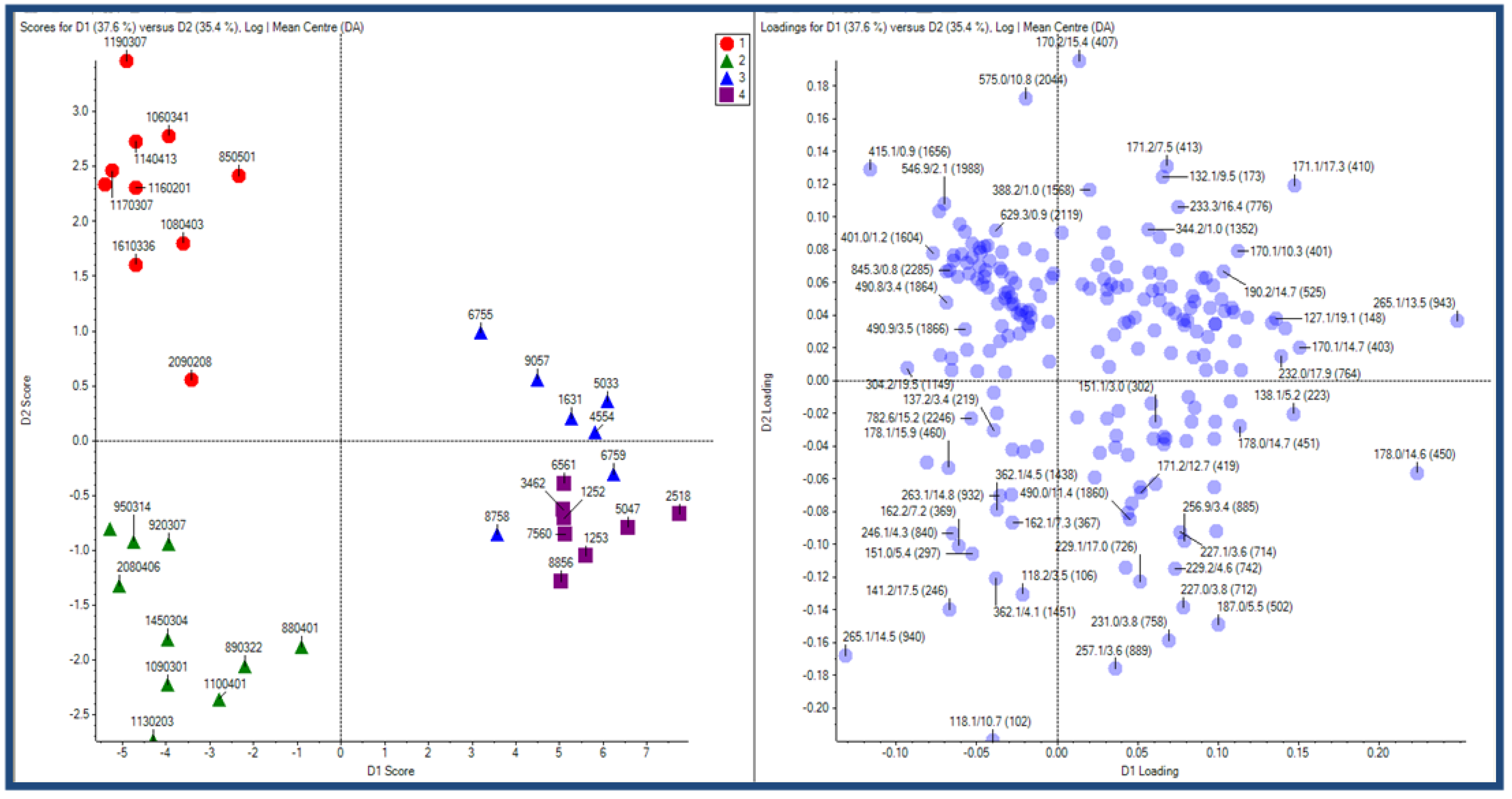
2.2.3 Human urine samples
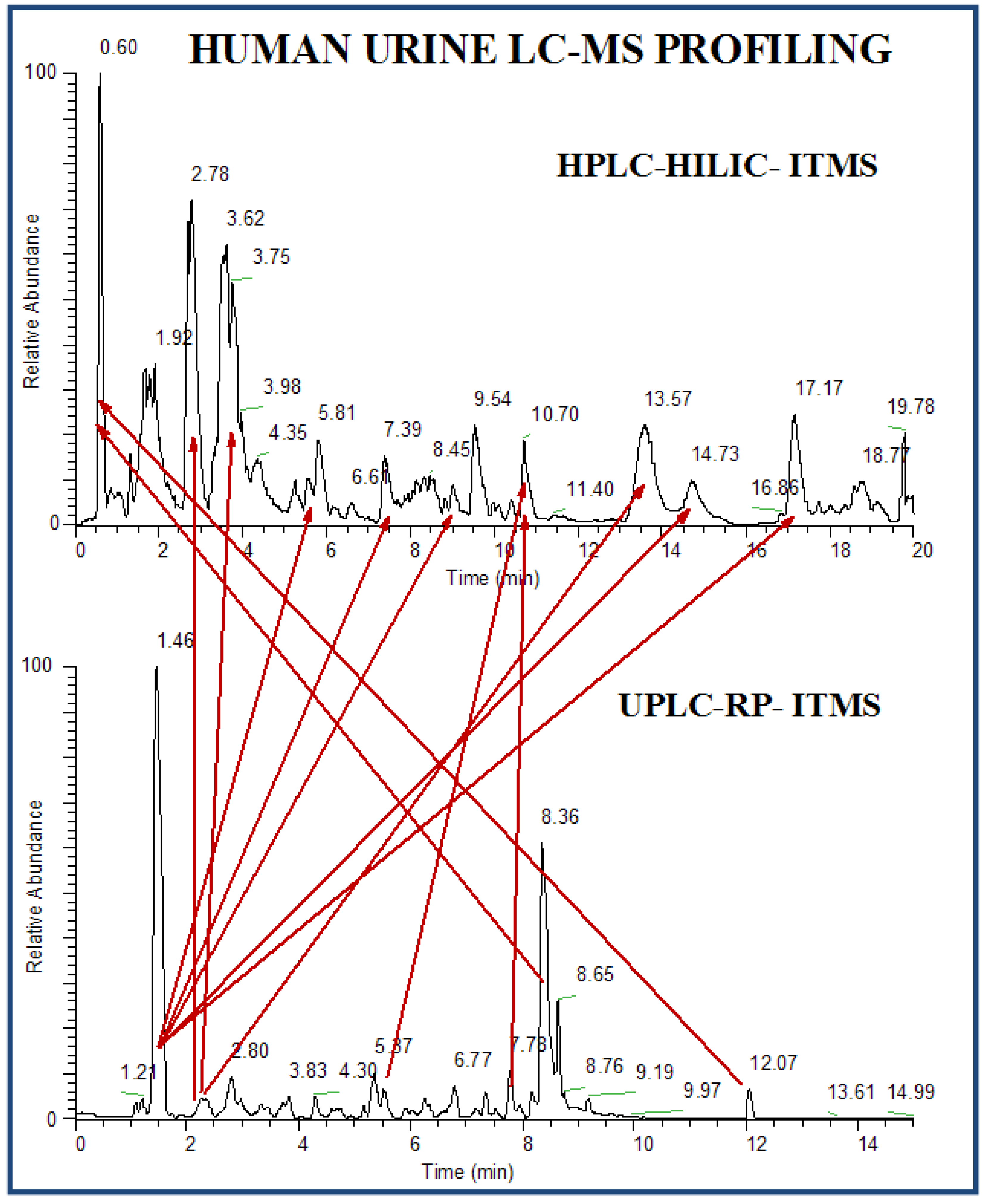
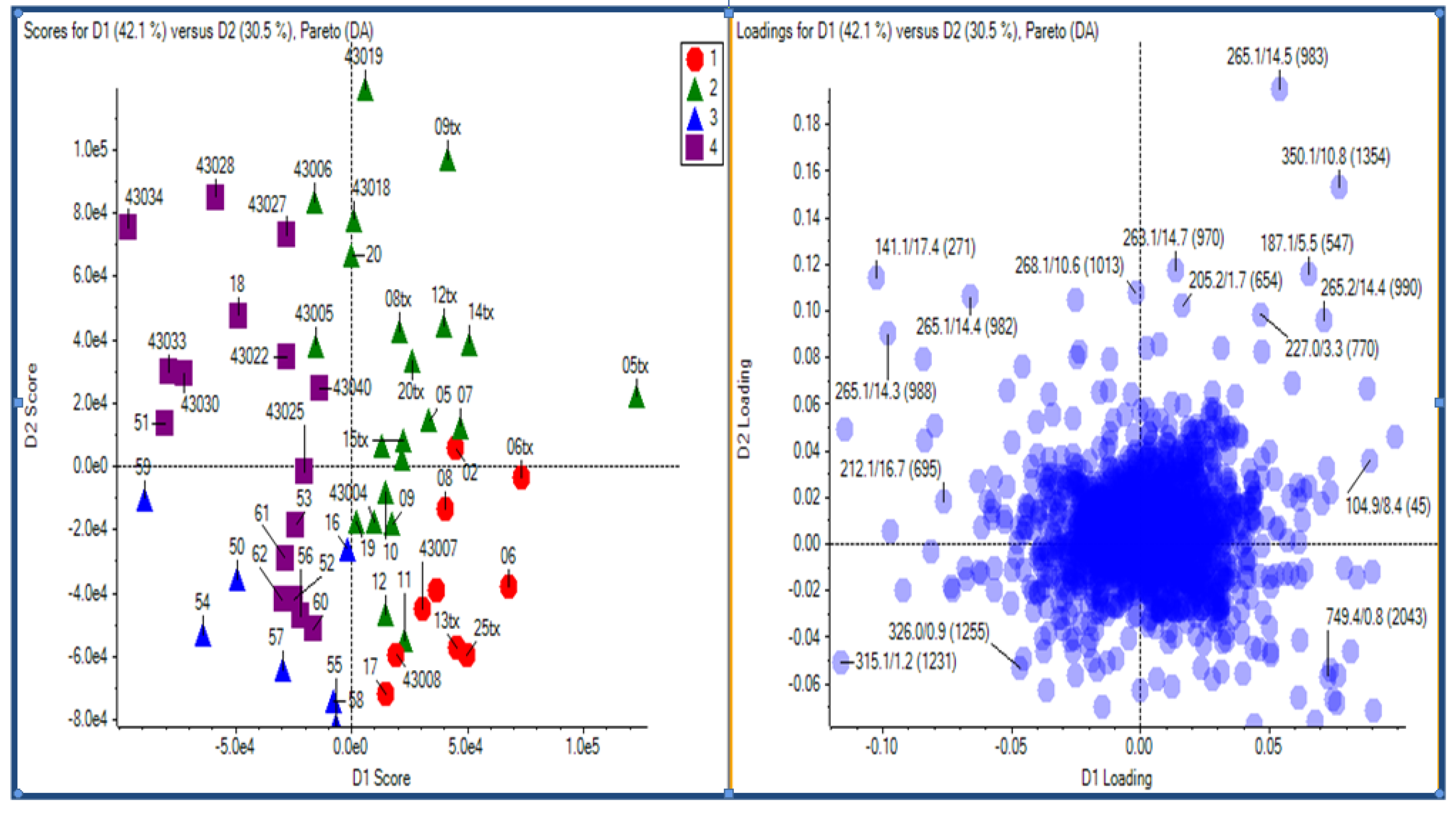
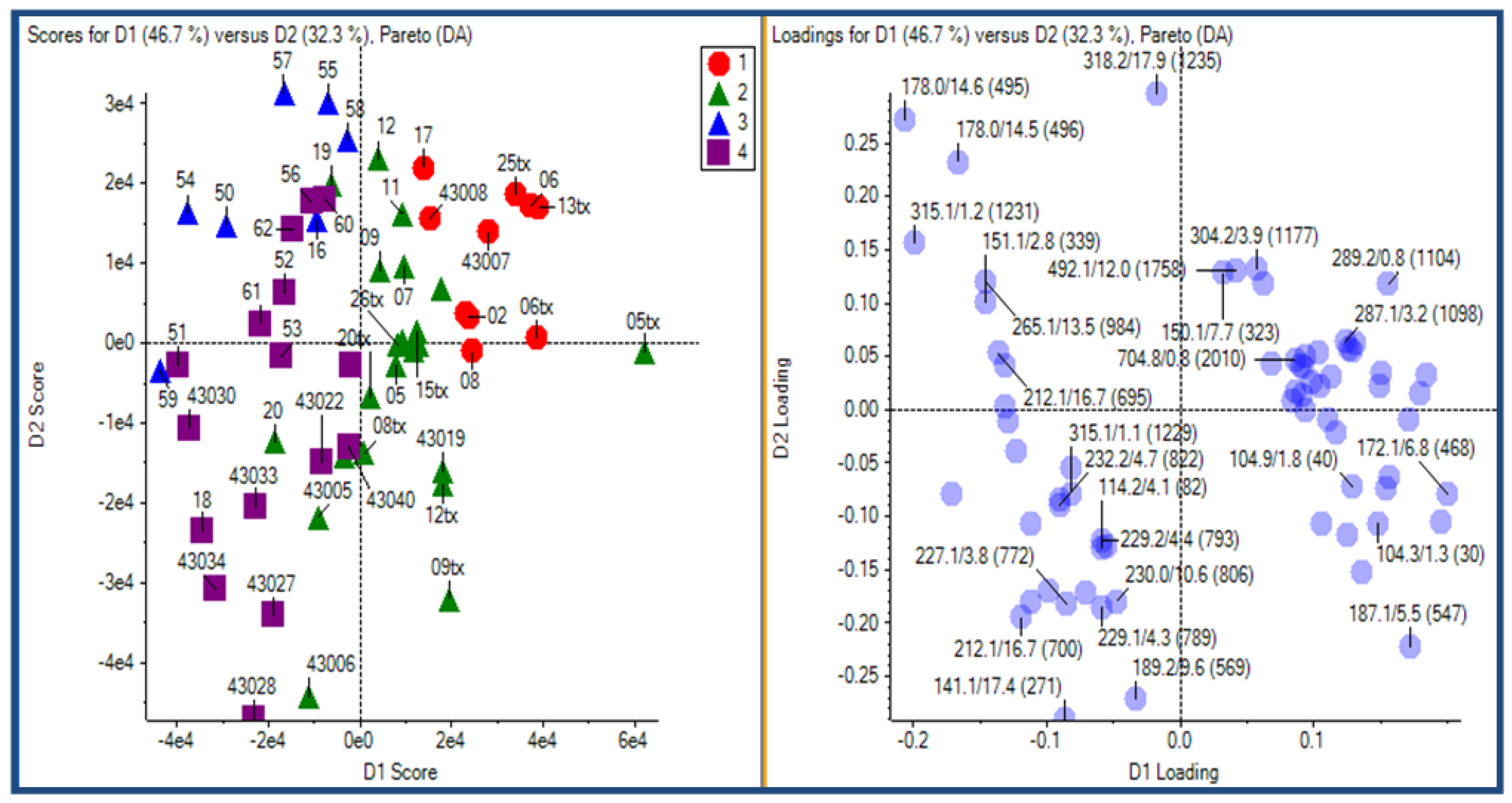

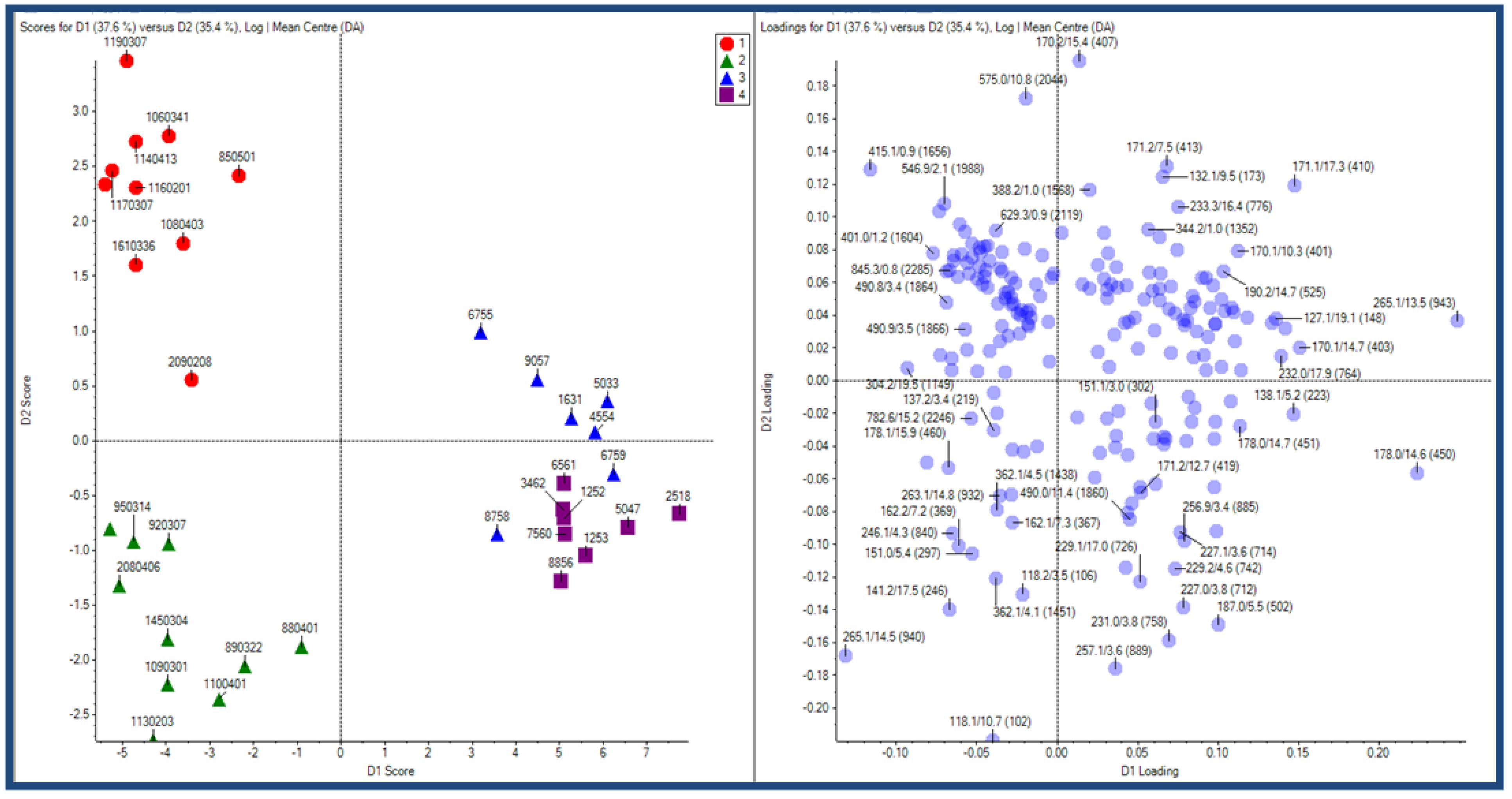


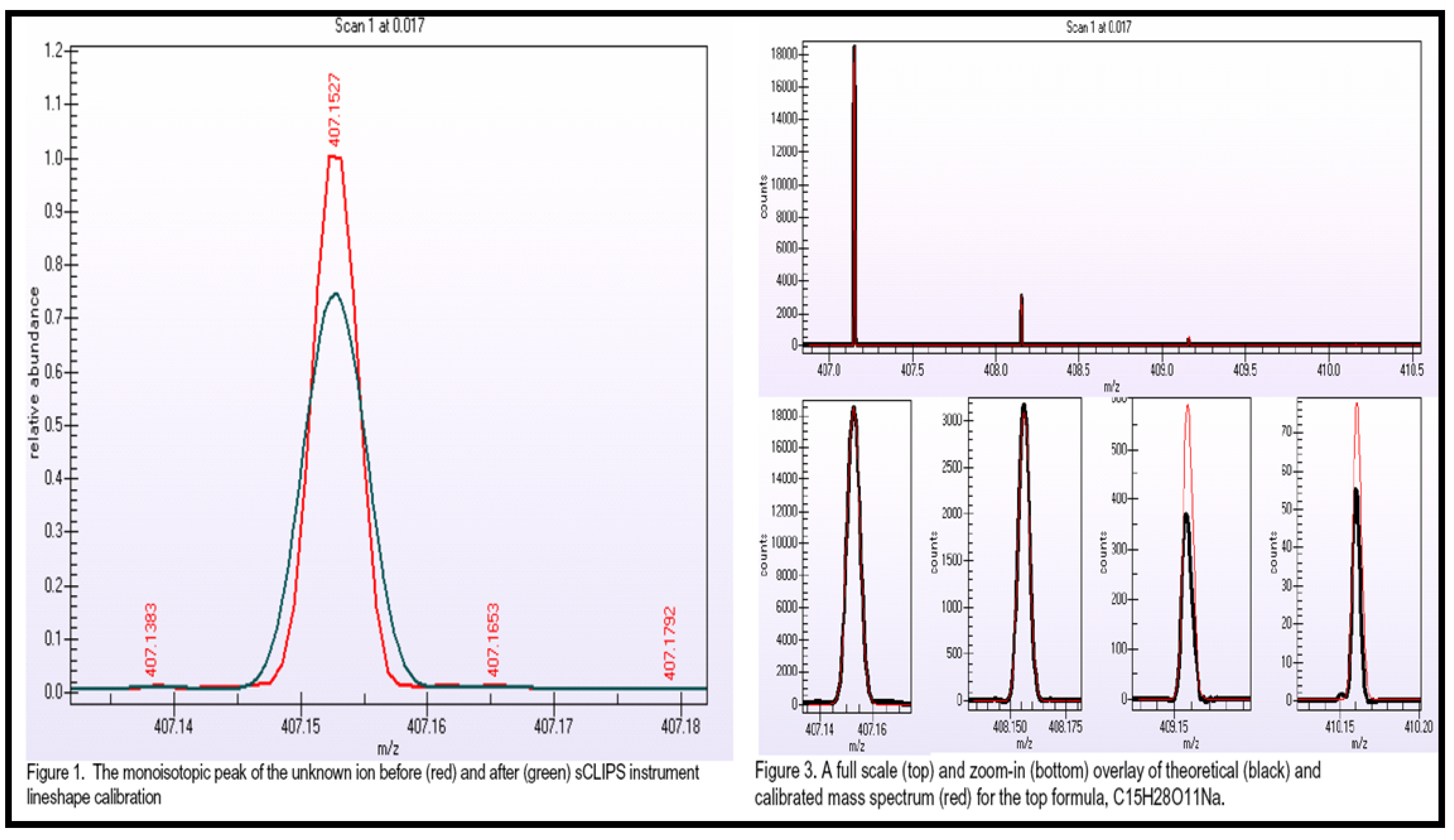
2.2.4 Case Summary
3. Experimental Section
3.1 Reagents and Standards
3.2 Metabolomics Experimental Design and Sample Preparation
3.3 LC–ESI–MS analysis
3.4 GC–TOF–MS analysis
3.5 Raw Data Processing and Statistical Data Mining
3.6 Mass Spectral Structure Elucidation
Conclusions
Acknowledgements
References and Notes
- Bentley, D. R. Genomic sequence information should be released immediately and freely in the public domain. Science 1996, 274, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D. R. Genomes for medicine. Nature 2004, 429, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Kruglyak, L.; Nickerson, D. A. Variation is the spice of life. Nat. Genet. 2001, 27, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Kopka, J.; Trethewey, R. N.; Willmitzer, L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Anal. Chem. 2000, 72, 3573–3580. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Tolstikov, V.; Weckwerth, W.; Fiehn, O.; Fukusaki, H. Micro HPLC for Metabolomics. In Frontier of metabolomic research; Springer-Verlag: Tokyo, 2003; pp. 85–100. [Google Scholar]
- Ikegami, T.; Kobayashi, H.; Kimura, H.; Tolstikov, V.; Fiehn, O.; Tanaka, N. High-Performance Liquid Chromatography for Metabolomics: High-Efficiency Separations Utilizing Monolithic Silica Columns. In Metabolomics. The Frontier of Systems Biology; Springer-Verlag: Tokyo, 2005; pp. 107–126. [Google Scholar]
- Tanaka, N.; Kimura, H.; Tokuda, D.; Hosoya, K.; Ikegami, T.; Ishizuka, N.; Minakuchi, H.; Nakanishi, K.; Shintani, Y.; Furuno, M.; Cabrera, K. Simple and comprehensive two-dimensional reversed-phase HPLC using monolithic silica columns. Anal. Chem. 2004, 76, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kobayashi, H. Monolithic columns for liquid chromatography. Anal. Bioanal. Chem. 2003, 376, 298–301. [Google Scholar] [PubMed]
- Tanaka, N.; Kobayashi, H.; Nakanishi, K.; Minakuchi, H.; Ishizuka, N. Monolithic LC columns. Anal. Chem. 2001, 73, 420A–429A. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V. V.; Fiehn, O.; Tanaka, N. Application of liquid chromatography-mass spectrometry analysis in metabolomics: reversed-phase monolithic capillary chromatography and hydrophilic chromatography coupled to electrospray ionization-mass spectrometry. In Metabolomics, Methods in Molecular Biology; Weckwerth, W., Ed.; Humana Press: Totowa, NJ, 2007; Volume 358, pp. 141–155. [Google Scholar]
- Tolstikov, V. V.; Lommen, A.; Nakanishi, K.; Tanaka, N.; Fiehn, O. Monolithic silica-based capillary reversed-phase liquid chromatography/electrospray mass spectrometry for plant metabolomics. Anal. Chem. 2003, 75, 6737–6740. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R. S.; Granger, J. H.; Stumpf, C. L.; Johnson, K. A.; Smith, B. W.; Gaulitz, S.; Wilson, I. D.; Castro-Perez, J. A rapid screening approach to metabonomics using UPLC and q-TOF mass spectrometry: application to age, gender and diurnal variation in normal/Zucker obese rats and black, white and nude mice. Analyst 2005, 130, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Hemstrom, P.; Irgum, K. Hydrophilic interaction chromatography. J. Sep. Sci. 2006, 29, 1784–1821. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N. Three-dimensional mapping of N-linked oligosaccharides using anion-exchange, hydrophobic and hydrophilic interaction modes of high-performance liquid chromatography. J. Chromatogr. A 1996, 720, 217–225. [Google Scholar] [CrossRef]
- Tolstikov, V. V.; Fiehn, O. Analysis of highly polar compounds of plant origin: combination of hydrophilic interaction chromatography and electrospray ion trap mass spectrometry. Anal. Biochem. 2002, 301, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Alpert, A. J. Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem. 2008, 80, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Mizzen, C. A.; Alpert, A. J.; Levesque, L.; Kruck, T. P.; McLachlan, D. R. Resolution of allelic and non-allelic variants of histone H1 by cation-exchange-hydrophilic-interaction chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2000, 744, 33–46. [Google Scholar] [CrossRef]
- Alpert, A. J.; Shukla, M.; Shukla, A. K.; Zieske, L. R.; Yuen, S. W.; Ferguson, M. A.; Mehlert, A.; Pauly, M.; Orlando, R. Hydrophilic-interaction chromatography of complex carbohydrates. J. Chromatogr. A 1994, 676, 191–122. [Google Scholar] [CrossRef]
- Boutin, J. A.; Ernould, A. P.; Ferry, G.; Genton, A.; Alpert, A. J. Use of hydrophilic interaction chromatography for the study of tyrosine protein kinase specificity. J. Chromatogr. 1992, 583, 137–143. [Google Scholar] [CrossRef]
- Alpert, A. J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolite profiling in Arabidopsis. In Arabidopsis Protocols; Methods in Molecular Biology series; Salinas, J., Sanchez-Serrano, J. J., Eds.; Humana Press: Totowa NJ, 2006; pp. 439–447. [Google Scholar]
- Kind, T.; Tolstikov, V.; Fiehn, O.; Weiss, R. H. A comprehensive urinary metabolomic approach for identifying kidney cancer. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006, 7, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Jain, A. K.; Duin, R. P. W.; Mao, J. Statistical pattern recognition: a review. Trans. Pattern An. Mach. Intell. 2000, 22, 4–37. [Google Scholar] [CrossRef]
- Scholz, M.; Gatzek, S.; Sterling, A.; Fiehn, O.; Selbig, J. Metabolite fingerprinting: detecting biological features by independent component analysis. Bioinformatics 2004, 20, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Sansone, S. A.; Fan, T.; Goodacre, R.; Griffin, J. L.; Hardy, N. W.; Kaddurah-Daouk, R.; Kristal, B. S.; Lindon, J.; Mendes, P.; Morrison, N.; Nikolau, B.; Robertson, D.; Sumner, L. W.; Taylor, C.; van der Werf, M.; van Ommen, B.; Fiehn, O. The metabolomics standards initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H. E.; Broadhurst, D.; Goodacre, R.; Smith, A. R. Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 2003, 62, 919–928. [Google Scholar] [CrossRef]
- Goodacre, R.; York, E. V.; Heald, J. K.; Scott, I. M. Chemometric discrimination of unfractionated plant extracts analyzed by electrospray mass spectrometry. Phytochemistry 2003, 62, 859–863. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Lee, J. W.; Lee, J. B.; Park, M.; Song, S. H. An extensive comparison of recent classification tools applied to microarray data. Comput. Stat. Data An. 2005, 48, 869–885. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.; Shi, Q.; Xu, X.-q.; Leung, H.-c.; Harris, L.; Iglehart, J.; Miron, A.; Liu, J.; Wong, W. Recursive SVM feature selection and sample classification for mass-spectrometry and microarray data. BMC Bioinformatics 2006, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R. Making sense of the metabolome using evolutionary computation: seeing the wood with the trees. J. Exp. Bot. 2005, 56, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Trevino, V.; Falciani, F. GALGO: an R package for multivariate variable selection using genetic algorithms. Bioinformatics 2006, 22, 1154–1156. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Tolstikov, V. V. Probing genetic algorithms for feature selection in comprehensive metabolic profiling approach. Rapid Commun. Mass Spectrom. 2008, 22, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Fiehn, O. SetupX--a public study design database for metabolomic projects. Pac. Symp. Biocomput. 2007, 12, 169–180. [Google Scholar]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M. Setup and Annotation of Metabolomic Experiments by Integrating Biological and Mass Spectrometric Metadata. Data Integration in the Life Sciences: Second International Workshop 2005, DILS. 224–239. [Google Scholar]
- Wagner, C.; Sefkow, M.; Kopka, J. Construction and application of a mass spectral and retention time index database generated from plant GC/EI-TOF-MS metabolite profiles. Phytochemistry Plant Metabolomics 2003, 62, 887–900. [Google Scholar] [CrossRef]
- Smith, C. A.; Want, E. J.; O'Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Ivosev, G.; Burton, L.; Bonner, R. Dimensionality Reduction and Visualization in Principal Component Analysis. J. Anal. Chem. 2008, 80, 4933–4944. [Google Scholar] [CrossRef] [PubMed]
- Burton, L.; Ivosev, G.; Tate, S.; Impey, G.; Wingate, J.; Bonner, R. Instrumental and experimental effects in LC-MS-based metabolomics. J. Chromatogr. B 2008, 871, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, N. O. Performance of a genetic algorithm for mass spectrometry proteomics. BMC Bioinformatics 2004, 5, 180. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V. Metabolic Fingerprinting of Breast Cancer Development. In Biomarker Discovery Summit, Philadelphia, PA, September 29 – October 1; 2008. [Google Scholar]
- Tolstikov, V. Mass Spectrometry-Derived Metabolic Biomarkers and Signatures in Diagnostic Development. In Biomarker Discovery Summit, Philadelphia, PA, September 29 – October 1; 2008. [Google Scholar]
- Kemsley, E. K.; Le Gall, G.; Dainty, J. R.; Watson, A. D.; Harvey, L. J.; Tapp, H. S.; Colquhoun, I. J. Multivariate techniques and their application in nutrition: a metabolomics case study. Br. J. Nutr. 2007, 98, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberge, C.; Wan, Y.; Dao, L. H.; Guidoin, R.; Zhang, Z. A biodegradable electrical bioconductor made of polypyrrole nanoparticle/poly(D,L-lactide) composite: A preliminary in vitro biostability study. J. Biomed. Mater. Res. A 2003, 66, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R. C.; Carey, V. J.; Bates, D. M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; Hornik, K.; Hothorn, T.; Huber, W.; Iacus, S.; Irizarry, R.; Leisch, F.; Li, C.; Maechler, M.; Rossini, A. J.; Sawitzki, G.; Smith, C.; Smyth, G.; Tierney, L.; Yang, J. Y.; Zhang, J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zou, W.; Tolstikov, V.V. Pattern Recognition and Pathway Analysis with Genetic Algorithms in Mass Spectrometry Based Metabolomics. Algorithms 2009, 2, 638-666. https://doi.org/10.3390/a2020638
Zou W, Tolstikov VV. Pattern Recognition and Pathway Analysis with Genetic Algorithms in Mass Spectrometry Based Metabolomics. Algorithms. 2009; 2(2):638-666. https://doi.org/10.3390/a2020638
Chicago/Turabian StyleZou, Wei, and Vladimir V. Tolstikov. 2009. "Pattern Recognition and Pathway Analysis with Genetic Algorithms in Mass Spectrometry Based Metabolomics" Algorithms 2, no. 2: 638-666. https://doi.org/10.3390/a2020638
APA StyleZou, W., & Tolstikov, V. V. (2009). Pattern Recognition and Pathway Analysis with Genetic Algorithms in Mass Spectrometry Based Metabolomics. Algorithms, 2(2), 638-666. https://doi.org/10.3390/a2020638