Protein-Protein Interaction Analysis by Docking


Abstract
:1. Introduction
1.1. Motivation
2. Results and Discussion
2.1. Quality Check for Docking
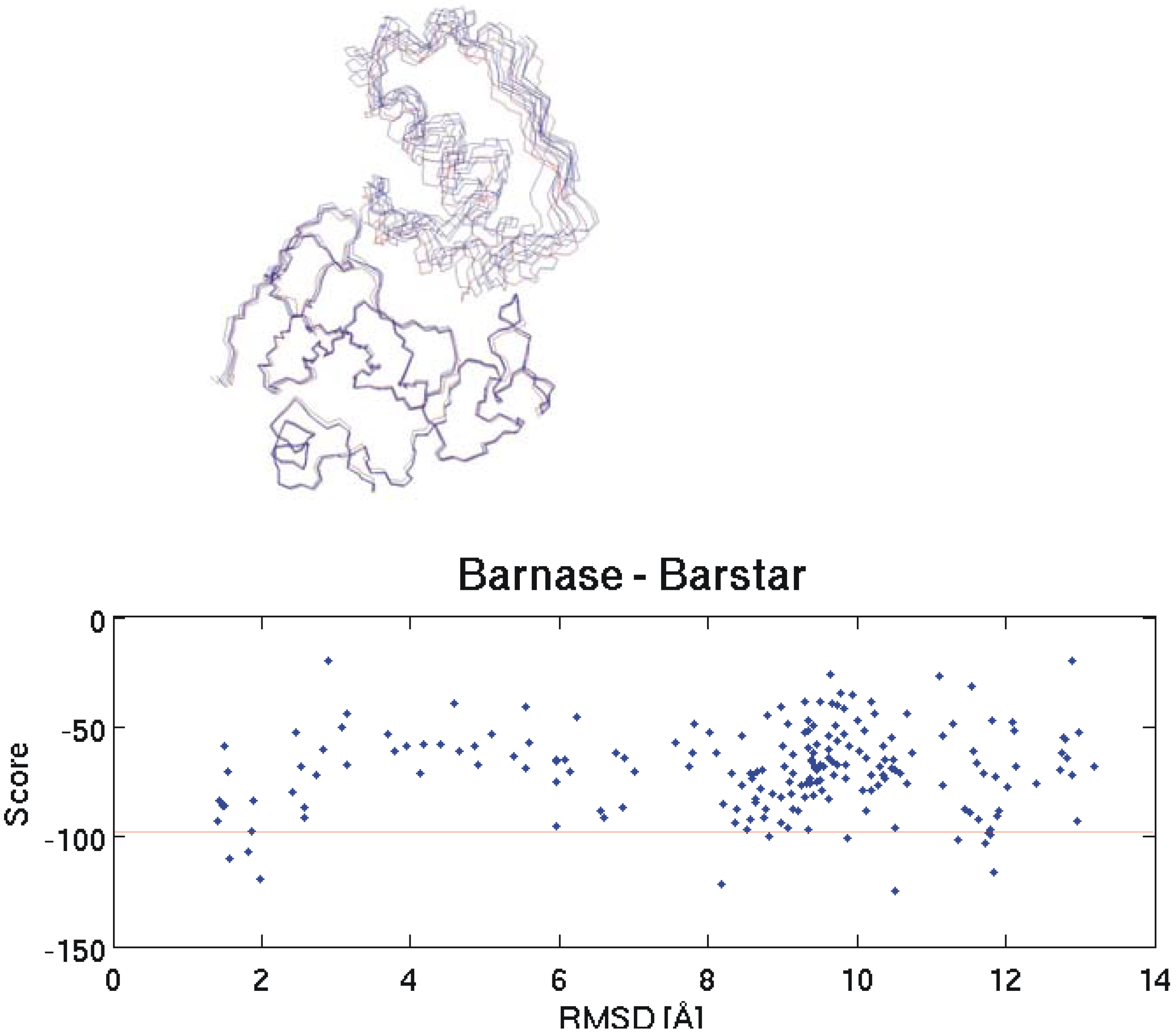
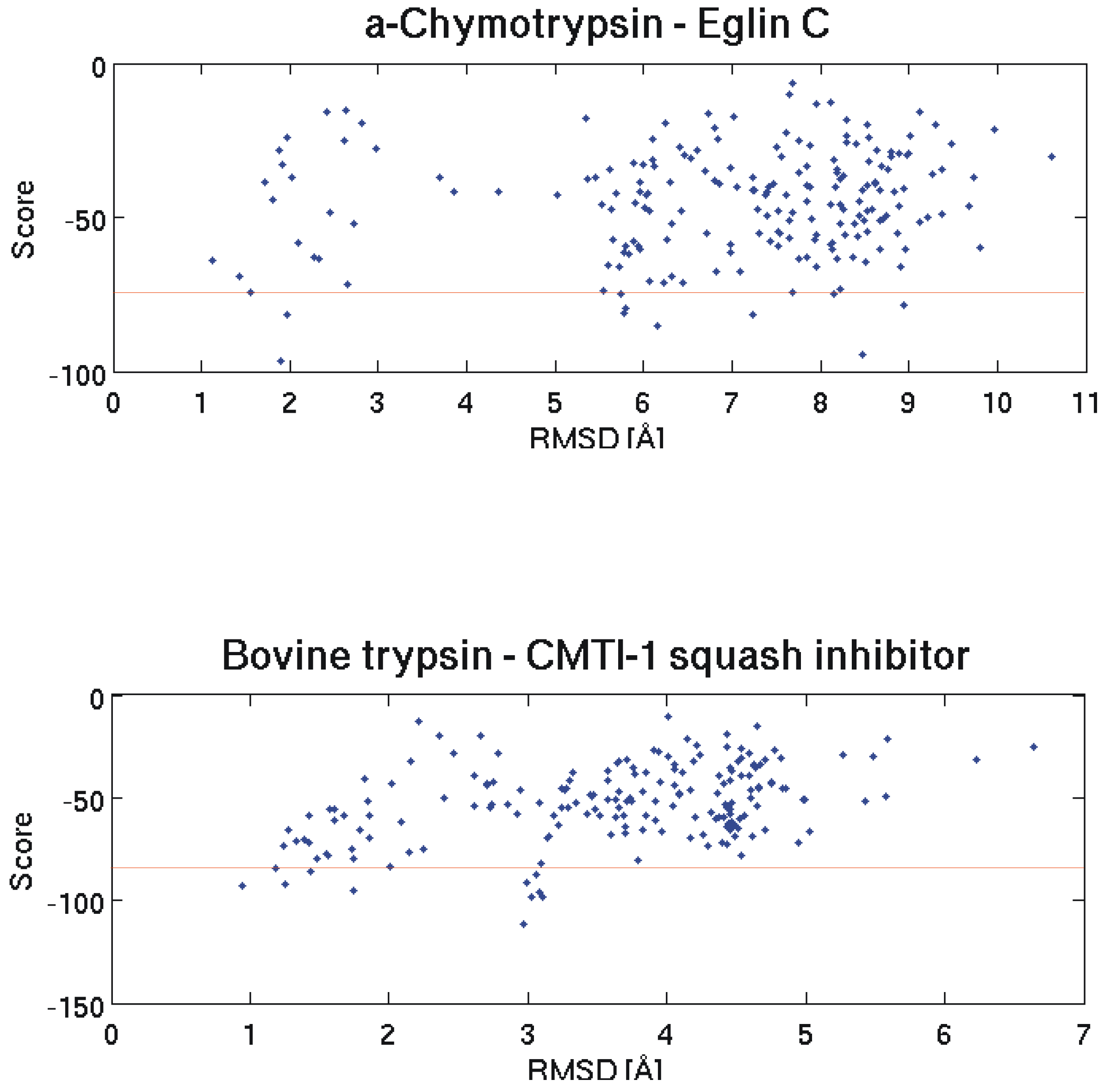
2.2. Discrimination with Interaction Energy

{kind=link}
{kind=link}
| Receptor | Ligand | aEint = EvdW + Eelec [kJ/mol] | bEint = EvdW + Eelec [kJ/mol] | cRank |
|---|---|---|---|---|
| Barnase | Soybean trypsin inhibitor | -670.0 | -1173.9 | 2 |
| Barnase | Ovomucoid 3rd domain | -575.0 | -481.3 | 3 |
| Barnase | Eglin C | -510.6 | -546.6 | 4 |
| Barnase | Pancreatic secretory trypsin inhibitor | -504.7 | -910.7 | 5 |
| Barnase | APPI | -481.3 | -301.3 | 6 |
| α-Chymotrypsin | Barstar | -505.5 | -341.5 | 2 |
| α-Chymotrypsin | APPI | -445.7 | -307.6 | 3 |
| α-Chymotrypsin | Soybean trypsin inhibitor | -364.9 | -469.1 | 4 |
| α-Chymotrypsin | Pancreatic secretory trypsin inhibitor | -306.3 | -397.2 | 5 |
| Bovine trypsin | Glycosylase inhibitor | -761.3 | -876.3 | 1 |
| Bovine trypsin | RAGI inhibitor | -492.2 | -565.0 | 3 |
| Bovine trypsin | Soybean trypsin inhibitor | -436.5 | -513.5 | 4 |
| Bovine trypsin | Streptomyces subtilisin inhibitor | -412.6 | -509.7 | 5 |
| Bovine trypsin | Amicyanin | -323.9 | -352.4 | 6 |
2.3. Conclusions and Outlook
3. Experimental Section
3.1. Docking
Acknowledgements
References
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The Protein Data Bank and structural genomics. Nucl. Acid. Res. 2003, 31, 489–491. [Google Scholar] [CrossRef]
- Aloy, P.; Russell, R.B. Ten thousand interactions for the molecular biologist. Nat. Biotechnol. 2004, 22, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W. Recent progress and future directions in protein-protein docking. Curr. Protein Pept. Sci. 2008, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S. Classification of protein complexes based on docking difficulty. Proteins 2005, 60, 176–180. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Krause, R.; Snel, B.; Cornell, M.; Oliver, S.G.; Fields, S.; Bork, P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 2002, 417, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Aloy, P.; Russell, R.B. Interrogating protein interaction networks through structural biology. Proc. Nat. Acad. Sci. USA. 2002, 99, 5896–5901. [Google Scholar] [CrossRef] [PubMed]
- Cockell, S.J.; Oliva, B.; Jackson, R.M. Structure-based evaluation of in silico predictions of protein-protein interactions using comparative docking. Bioinformatics 2007, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Mintseris, J.; Wiehe, K.; Pierce, B.; Anderson, R.; Chen, R.; Janin, J.; Weng, Z. Protein-protein docking benchmark 2.0: An update. Proteins 2005, 60, 214–216. [Google Scholar]
- Schumann, F.H.; Riepl, H.; Maurer, T.; Gronwald, W.; Neidig, K.P.; Kalbitzer, H.R. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 2007, 39, 275–289. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fink, F.; Ederer, S.; Gronwald, W. Protein-Protein Interaction Analysis by Docking. Algorithms 2009, 2, 429-436. https://doi.org/10.3390/a2010429
Fink F, Ederer S, Gronwald W. Protein-Protein Interaction Analysis by Docking. Algorithms. 2009; 2(1):429-436. https://doi.org/10.3390/a2010429
Chicago/Turabian StyleFink, Florian, Stephan Ederer, and Wolfram Gronwald. 2009. "Protein-Protein Interaction Analysis by Docking" Algorithms 2, no. 1: 429-436. https://doi.org/10.3390/a2010429
APA StyleFink, F., Ederer, S., & Gronwald, W. (2009). Protein-Protein Interaction Analysis by Docking. Algorithms, 2(1), 429-436. https://doi.org/10.3390/a2010429