Abstract
This paper investigates the effectiveness of Word2Vec-based molecular representation learning on SMILES (Simplified Molecular Input Line Entry System) strings for a downstream prediction task related to the market approvability of chemical compounds. Here, market approvability is treated as a proxy classification label derived from approval status, where only the molecular structure is analyzed. We train character-level embeddings using Continuous Bag of Words (CBOW) and Skip-Gram with Negative Sampling architectures and apply the resulting embeddings in a downstream classification task using a multi-layer perceptron (MLP). To evaluate the utility of these lightweight embedding techniques, we conduct experiments on a curated SMILES dataset labeled by approval status under both imbalanced and SMOTE-balanced training conditions. In addition to our Word2Vec-based models, we include a ChemBERTa-based baseline using the pretrained ChemBERTa-77M model. Our findings show that while ChemBERTa achieves a higher performance, the Word2Vec-based models offer a favorable trade-off between accuracy and computational efficiency. This efficiency is especially relevant in large-scale compound screening, where rapid exploration of the chemical space can support early-stage cheminformatics workflows. These results suggest that traditional embedding models can serve as viable alternatives for scalable and interpretable cheminformatics pipelines, particularly in resource-constrained environments.
1. Introduction
Molecular representation learning is a fundamental component of cheminformatics, enabling machine learning models to analyze chemical structures and predict molecular properties. SMILES is a widely used linear notation system that encodes chemical structures as character sequences [1]. Its text-based format allows for the application of NLP techniques to molecular data analysis.
Recent advances in NLP have inspired numerous efforts to learn data-driven vector embeddings for molecules using SMILES strings. Word2Vec, introduced by Mikolov et al. [2], is one such method that learns dense, contextual embeddings from raw text using architectures like CBOW and Skip-Gram with Negative Sampling. In cheminformatics, this approach has been adapted in models such as SMILES2Vec [3] and Mol2Vec [4], which generate embeddings from character sequences and molecular fragments, respectively.
In addition, transformer-based models such as ChemBERTa [5] have emerged as state-of-the-art, utilizing masked language modeling (MLM) to learn contextualized molecular representations. While powerful, these deep models require significant computational resources, long training times, and large-scale datasets.
In this work, we propose a lightweight alternative by repurposing Word2Vec fo prediction in a novel and rarely explored downstream task, market approvability. We use the term market approvability as a proxy task, defined by the historical approval status of compounds. We acknowledge that real-world approval depends on broader clinical, biological, and regulatory considerations beyond the molecular structure, and therefore, our task should be viewed as an early-stage computational screening problem rather than a comprehensive predictor of approval. Framing the problem in this way allows us to benchmark embedding methods based on a meaningful but limited label while avoiding an overstatement of their predictive scope.
The goal of this work is to improve scalability by enabling faster and more efficient exploration of large compound libraries. Such methods can help researchers focus on the most promising candidates for subsequent evaluation, though ultimate confirmation depends on experimental and regulatory studies.
Using a curated SMILES dataset labeled for market approval provided by the miDruglikeness study [6], we train unsupervised character-level Word2Vec embeddings and evaluate them using a downstream MLP classifier. In addition, we introduce a ChemBERTa-based model as a high-capacity baseline to assess the trade-offs in accuracy, training time, and memory usage.
Recent work such as ChemAP [7] has explored approval-oriented predictions using SMILES-based and fragment-level features; however, our contribution lies in directly benchmarking lightweight Word2Vec embeddings against ChemBERTa. Our experiments show that while ChemBERTa achieves the highest predictive performance, the Word2Vec-based models reduce the training time by up to 10% and the embedding memory usage by over 90%, making them more efficient for resource-constrained scenarios. These findings underscore the practical value of efficient embedding models in supporting early cheminformatics pipelines, where scalability and interpretability matter just as much as predictive accuracy.
2. The Background and Related Work
SMILES is a compact, text-based representation of chemical structures introduced by Weininger [1]. It encodes molecules as sequences of characters where atoms, bonds, and structural features are expressed using specific symbols. This allows complex molecular graphs to be represented in a simple, readable string format, making SMILES highly suitable for computational analysis, storage, and machine learning applications, as illustrated in Figure 1.
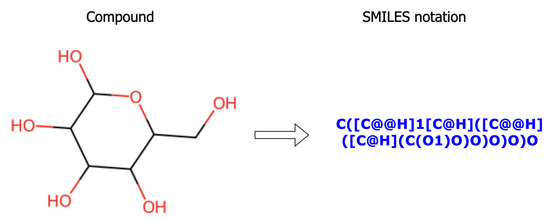
Figure 1.
An illustration of SMILES encoding: a chemical compound (glucose) is converted into a linear text representation for computational use.
Molecular representation learning has been widely explored in recent years through the adaptation of NLP techniques to chemical data. Several works have investigated the use of embedding models to learn meaningful vector representations from SMILES strings or molecular fragments. This section reviews the most relevant works, including Word2Vec [2], SMILES2Vec [3], Mol2Vec [4], and ChemBERTa [5]. Each of these methods has contributed significantly to the field of cheminformatics but differs from this work in terms of the task objectives, embedding strategy, and application scope.
2.1. Word2Vec
Word2Vec, proposed by Mikolov et al. [2], is a seminal model in NLP that learns dense vector representations (embeddings) of words based on their context within a sequence. The model offers two primary architectures: Continuous Bag of Words, which predicts a target word from its surrounding context, and Skip-Gram, which predicts surrounding context words given a target word. Word2Vec was designed for large-scale unsupervised learning of the word embeddings in natural language corpora.
This work draws direct inspiration from Word2Vec [2] by applying its CBOW and Skip-Gram architectures to character-level SMILES strings. However, unlike the original Word2Vec, which operates on natural language, this study applies it to molecular sequences with the novel objective of predicting the market approvability of chemical compounds—a task not explored in the original Word2Vec framework.
2.2. SMILES2Vec
SMILES2Vec, proposed by Goh et al. [3], adapts Word2Vec-style embeddings to chemical property predictions by learning character-level SMILES embeddings in a supervised learning setup. The model directly integrates embedding learning into downstream tasks such as toxicity prediction and solubility estimation. SMILES2Vec demonstrated that SMILES sequences could effectively serve as the input to deep neural networks without requiring handcrafted features.
This work shares a similar character-level SMILES representation approach to that in SMILES2Vec [3]. However, the key difference lies in the learning strategy. While SMILES2Vec learns embeddings jointly with the downstream task, this study trains Word2Vec embeddings in an unsupervised manner and applies them to a separate downstream classification task, specifically market approvability prediction.
2.3. Mol2Vec
Mol2Vec, introduced by Jaeger et al. [4], extends the Word2Vec framework to learning the embeddings of molecular substructures rather than characters. The model uses Morgan fingerprints to extract chemical fragments, treating them as words within a molecule-as-document paradigm. These embeddings have been shown to capture chemical intuition and improve similarity-based tasks.
While Mol2Vec [4] uses fragment-based embeddings, this study focuses on character-level embeddings directly from SMILES strings, eliminating the need for molecular fingerprint generation. Furthermore, Mol2Vec was primarily applied to similarity analysis and clustering, whereas this work targets market approvability prediction as a real-world downstream classification task.
2.4. ChemBERTa
ChemBERTa, proposed by Chithrananda et al. [5], applies transformer-based language models to SMILES strings for molecular representation learning. The model is pretrained on millions of SMILES sequences using masked language modeling, allowing it to capture both local and global structural patterns within molecules. ChemBERTa has shown a strong performance across a range of molecular property prediction tasks, such as solubility estimation, toxicity prediction, and bioactivity classification [5].
In our study, ChemBERTa is employed as a high-capacity baseline model, allowing us to evaluate the relative performance and efficiency of lightweight Word2Vec-based embeddings in comparison to deep-transformer-based representations.
2.5. ChemAP
Recent work such as ChemAP [7] has extended the use of SMILES-based embeddings, including ChemBERTa, to the prediction of drug approval outcomes. ChemAP integrates both transformer-derived representations and fragment- or graph-based structural features, thereby incorporating a richer chemical context than sequence-only approaches. In their study, Cho et al. demonstrated that combining multiple structural views can improve the predictive accuracy for approval-related tasks.
While ChemAP demonstrates the utility of deep and structurally enriched embeddings for approval-oriented prediction, our study differs by directly benchmarking Word2Vec-style shallow embeddings against ChemBERTa. By focusing on these lightweight models, our work provides insight into when computationally efficient embeddings can serve as a viable alternative, particularly for scaling to large datasets in resource-constrained environments.
3. Dataset
This study utilizes the ’market approvability’ dataset, which is the final of three hierarchical classification tasks developed by Cai et al. in the miDruglikeness study [6]. It is important to clarify that ’market approvability’ in this context does not represent a predictive model for the multifaceted regulatory approval process, which is contingent on clinical, economic, and strategic factors beyond molecular structure. Rather, it defines a specific binary classification task designed to distinguish between the molecular structures of compounds based on their historical outcomes in the drug development pipeline. The objective is to assess whether structural information, encoded into SMILES strings, is sufficient for this classification.
3.1. Data Curation and Composition
The dataset was constructed from curated subsets of the ZINC15 database [8], with the classes defined as follows:
- Positive Samples (Class 1) → Compounds sourced from the ZINC15 “world” subset. This set consists of drugs that have received market approval in major jurisdictions, including from the FDA.
- Negative Samples (Class 0) → Compounds sourced from the “investigational-only” subset. This set comprises molecules that have been evaluated in clinical trials but have not been approved as drugs.
The original authors applied a rigorous preprocessing workflow to ensure data quality, which included filtering for organic molecules, standardizing chemical representations, and removing duplicate entries. This curation resulted in a well-defined task of distinguishing between historically successful drugs and late-stage investigational compounds based solely on their molecular features.
3.2. Dataset Statistics
Each molecule is represented as a SMILES string [1], paired with a binary label indicating its class. The complete dataset curated by Cai et al. contains 5706 positive samples and 3680 negative samples, exhibiting a class imbalance ratio of approximately 1.55. For our work, we utilize the specific training and test partitions provided by the original authors. The detailed statistics of these partitions are summarized in Table 1.

Table 1.
The dataset statistics for the market approvability classification task.
4. The Proposed Method
To provide a comprehensive overview of the learning pipeline, two parallel methodologies were developed: one based on Word2Vec-style embedding models and another leveraging the transformer-based architecture. Both approaches start from raw SMILES strings and apply preprocessing to producing chemically meaningful token sequences. In the first pathway, a custom rule-based tokenizer is used to segment the SMILES strings into atomic and structural subunits, which are then embedded using unsupervised learning with context-based models and trained via negative sampling. In the second approach, SMILES strings are processed using ChemBERTa’s pretrained tokenizer and model to obtain contextualized embeddings directly. In both cases, mean pooling is applied to convert variable-length sequences into fixed-size molecular embeddings. These embeddings are then passed to an MLP classifier for binary classification. As illustrated in Figure 2 below, the following architecture outlines the full learning process:
4.1. Preprocessing
All SMILES strings were preprocessed using a custom rule-based tokenizer operating at an atom-level granularity. A regular expression pattern was employed to identify and extract chemically meaningful substructures, including multi-character atoms such as Br and Cl; standard atomic symbols like N; ring closures; bond types; stereochemistry; and charge annotations enclosed in brackets (e.g., [C@@H]). To preserve the stereochemical and ionic information, all bracketed tokens were treated as single entities. An optional filter allowed for the replacement of non-critical bracketed tokens with a generic [UNK] symbol while retaining specific cases such as [C@@H] or [nH]; however, this filter was disabled during our main experiments.
Tokenization was performed independently on the training and test sets to simulate real-world inference scenarios better, where previously unseen molecules may appear. From the training set, a vocabulary was then constructed by computing the token frequencies across all molecules. Each unique token was assigned an integer index according to its frequency rank, ensuring consistent mapping for model training and evaluation.
The resulting vocabulary consisted of 51 unique tokens, which were subsequently used to encode SMILES inputs into integer sequences for the Skip-Gram and CBOW embedding models described in Section 4.2. This preprocessing procedure ensured that tokenized representations captured chemically meaningful substructures while remaining robust to diverse molecular inputs, thereby providing a reliable foundation for embedding training.
4.2. Embedding Learning
In order to transform SMILES strings into fixed-length numerical representations, we trained two unsupervised embedding models: Skip-Gram and CBOW. These models are adapted from natural language processing and learn to map tokens (atoms, bonds, substructures) into a continuous vector space based on their surrounding chemical context.

Figure 2.
An overview of the molecular representation learning process, as illustrated in the two proposed pipelines.
4.2.1. The Skip-Gram Model
To prepare the training data for the Skip-Gram model, we generated context–target pairs in a manner similar to that in standard word embedding tasks. Each SMILES string was tokenized into a sequence of chemically meaningful symbols and treated as a sequence analogous to a sentence in NLP, where each token acts as a subword or chemical subunit. The Skip-Gram approach is designed to predict surrounding context tokens given a central token. For each token in the sequence, a symmetric window of size w is used to define its left and right neighbors. These neighboring tokens serve as positive context examples for the central (target) token. Each token is first mapped to its corresponding vocabulary index. Then, a sliding window of size is applied across the sequence, and for every token position, all valid surrounding tokens within the window are collected to form training pairs. Using a window size of , we generated a total of 1,507,242 pairs. These pairs constitute the core input for training the Skip-Gram model. During training, each pair is used to optimize the embedding vectors such that the target token is similar to its true context tokens and dissimilar to random negatives (see Section 4.3). This method allows the model to learn the local chemical structure and capture co-occurrence statistics between substructures in SMILES, yielding embeddings that are chemically informative and transferable to downstream tasks.
To learn distributed representations of SMILES tokens, we implemented the Skip-Gram model using PyTorch 2.7.1. The model architecture follows the standard two-embedding design, where each token in the vocabulary is assigned two separate embedding vectors: one when the token functions as a target and another when it appears as a context token. The target embedding matrix learns token representations when tokens are presented as central or target words. Conversely, the context embedding matrix is optimized when tokens occur as the surrounding context. This dual-embedding structure is crucial for capturing asymmetric co-occurrence dynamics, which are common in molecular sequences where substructures often appear in specific directional relationships. Both embedding matrices were initialized using Xavier uniform initialization to ensure stable convergence and effective learning of low-dimensional representations.
During training, the model receives three inputs per batch: a list of target token indices, the corresponding context token indices, and a matrix of negative sample indices for each target. In the forward pass, the model computes the dot product between each target and its true context vector, yielding the positive score. It also computes the dot products between each target and a fixed number of randomly sampled unrelated context vectors, resulting in a set of negative scores. These scores are passed to a negative sampling loss function. The loss encourages the model to increase the similarity between valid target–context pairs while decreasing the similarity between targets and randomly sampled negatives. This approach allows for efficient training even with relatively large vocabularies and forms the basis for the learned chemical embeddings used in subsequent classification tasks. The precise loss formulation is presented in Section 4.3.
4.2.2. The CBOW Model
In addition to Skip-Gram, we implemented a CBOW model to learn unsupervised token embeddings from SMILES strings. While Skip-Gram aims to predict surrounding context tokens given a central token, CBOW reverses this task: it predicts the central token based on its surrounding context. Each SMILES sequence was first tokenized and mapped to a list of vocabulary indices. For every token position, a fixed-size window was applied to collecting its neighboring tokens to the left and right. These surrounding tokens formed the context, while the token at the center of the window served as the target. With a window size of 2, each training example included up to four context tokens. This resulted in a total of 389,481 CBOW training samples. To ensure uniform input dimensionality during training, the context lists were shorter than a maximum length of 4. Each training sample was then stored as a fixed-length vector, where the first four positions contained the context tokens and the final position contained the target token. These vectors were passed to the model in batches.
The CBOW model architecture consists of two embedding layers, one for input contexts and one for output targets. The input embedding layer maps each context token to a vector space, and the vectors from all context tokens in the window are summed to form a single representation. The output embedding layer maps the target token to a vector in the same space. Both embedding matrices were initialized using Xavier uniform initialization to encourage stable training. In the forward pass, the dot product between the aggregated context vector and the target token vector is computed to form the positive prediction score. This is then contrasted with scores from negatively sampled targets (see Section 4.3), which are treated as negative examples. The CBOW model, like the Skip-Gram model, is trained using a negative sampling objective. However, unlike Skip-Gram, which considers each context–target pair separately, CBOW aggregates all context tokens into a single vector to predict the center token. The loss formulation and negative sampling procedure used are identical to those described in Section 4.3.
4.3. Negative Sampling and the Loss Function
To efficiently train both the Skip-Gram and CBOW models over a large number of token pairs, we adopted the negative sampling technique. This method provides a scalable alternative to computing a full softmax across the vocabulary at every training step, which would otherwise be computationally prohibitive.
The objective of both embedding models is to assign similar vector representations to tokens that frequently co-occur in SMILES sequences. This is achieved by maximizing the similarity between observed pairs (positive samples) and minimizing the similarity between tokens that do not co-occur (negative samples). Instead of calculating probabilities over the full vocabulary, negative sampling approximates the softmax by drawing a small number of K negative samples per training example.
Loss Formulation:
Let be the input (context) embedding and be the output (target) embedding for a real (context, target) pair. Let denote the output embeddings of K negative samples drawn randomly from the vocabulary. The negative sampling loss for a single training example is defined as
where is the sigmoid function. The first term encourages the model to assign a high dot product (i.e., similarity) to valid token pairs. The second term penalizes the model if randomly sampled (non-contextual) tokens are incorrectly assigned high similarity to the input.
To stabilize numerical computations, a small constant was added during training:
This loss formulation was used identically across both embedding models. For Skip-Gram, the score is computed between a single context and a target. For CBOW, the context vector is formed by summing all embeddings from surrounding tokens and is then used to predict the center token. This shared loss enabled a unified optimization framework across both architectures while accommodating their structural differences.
4.4. The Training Configuration and Hyperparameters
Both the Skip-Gram and CBOW models were trained under a unified training pipeline using consistent hyperparameter settings. The embedding dimensionality was set to 50, providing a compact yet expressive representation of each token. Training was conducted for 5 epochs, with a batch size of 64 and a negative sampling rate of 5 negatives per positive pair. The learning rate was fixed at 0.01, and the Adam optimizer was employed due to its ability to adjust the learning rates and its effectiveness in low-data and noisy gradient scenarios. All training was conducted on a CUDA-enabled NVIDIA GeForce RTX 4090 GPU with 24 GB of GDDR6X VRAM and 128 streaming multiprocessors. The CPU used was an Intel® Core™ Ultra 9 285K CPU (24 cores). The loss values were averaged across batches to monitor the convergence behavior over time.
The training loop followed a consistent pattern for both models. In each iteration, a minibatch of data was sampled—either Skip-Gram pairs or CBOW context–target tuples. For every training instance, five negative samples were drawn uniformly from the vocabulary. A forward pass then computed the dot products between the embeddings of true and negative token pairs. The negative sampling loss, described in Section 4.3, was used to calculate the error. A backward pass followed by a parameter update completed the iteration.
Each batch included one target token and one context token per sample, along with five negative context tokens. The model architecture employed two separate embedding matrices: one for tokens appearing as targets and another for tokens in context positions. Each example used up to four context tokens surrounding a central target token, with the context tokens summed to form a single context representation. Context lists shorter than four tokens were right-padded with zeros. During training, each batch included these summed context vectors, one target token per sample, and five negative targets for contrastive learning. As with Skip-Gram, the CBOW model maintained separate embeddings for the input and output tokens.
After training, each SMILES string was embedded by averaging the learned token embeddings. In the case of CBOW, these embeddings were extracted from the input embedding matrix, while for Skip-Gram, the target embedding matrix was used.
4.5. ChemBERTa Molecular Representation
To investigate the potential of transformer-based architectures for SMILES representation, the ChemBERTa model was used. Specifically, the DeepChem/ChemBERTa-77M-MLM checkpoint from Hugging Face was employed. SMILES strings from the training and test datasets were tokenized using ChemBERTa’s tokenizer with a maximum sequence length of 512 tokens. The tokenization outputs were subsequently fed into the pretrained model in evaluation mode.
4.6. Embedding Generation
For each input sequence, contextualized token embeddings were obtained from the final hidden layer. To convert variable-length token sequences into fixed-length molecular representations, the mean pooling strategy was applied. Specifically, the embeddings were averaged across the sequence dimension, yielding a single feature vector per molecule.
5. The Classification Model
To evaluate the power of the learned SMILES embeddings, we trained downstream classifiers. The classifier was trained twice—once using the original imbalanced training data and once using a SMOTE-balanced variant.
We implemented a multi-layer perceptron using PyTorch to perform binary classification on fixed-length SMILES embeddings derived from either the Skip-Gram or CBOW model. The network accepts embedding vectors of various dimensions (50, 100, 300, and 512) as the input. The 512-dimensional embedding was included to allow for a direct comparison with the fixed-length embeddings generated by ChemBERTa, ensuring consistent input sizes for the comparative analysis. The MLP consists of two hidden layers. The first layer contains 128 units with ReLU activation, followed by dropout with a rate of 0.1. The second hidden layer includes 64 units, again followed by ReLU and dropout. The final output layer is a single neuron with sigmoid activation, producing a probability score for binary classification.
Training was carried out using the Binary Cross-Entropy Loss (BCE) function, optimized via the Adam algorithm with a learning rate of . A batch size of 64 was used.
6. Results
Model training was conducted using stratified K-fold cross-validation (k = 5) on the designated training dataset. Within each fold, the model was trained on a portion of the training data, while a dedicated validation subset was used solely to monitor the validation loss during training. Both original and SMOTE-resampled data were employed, with SMOTE applied specifically to the training subset of each fold. For the performance evaluation, an independent test set, unseen throughout the cross-validation process, was utilized to assess the model trained in each fold. The mean performance metrics and their corresponding 95% confidence intervals were then calculated by averaging the results from each of the K model evaluations on this external test set. Training and validation loss curves were plotted for each run to monitor training progression.
We evaluated the learned representations using an MLP classifier, both with and without SMOTE. The analysis was performed in all four embedding dimensions to understand how the representational dimension and data balancing affected the classification performance.
6.1. The CBOW Model
The training duration for generating the CBOW embeddings varied across different embedding dimensions, as summarized in Table 2. An initial increase in the training time was observed, with the 50-dimension model requiring 120.2 ± 7.0 s, which increased to 130.3 ± 9.0 s for the 100-dimension embeddings. With further increases in the dimension, the training times then slightly decreased, reducing to 128.8 ± 9.5 s for 300 dimensions and 127.2 ± 9.5 s for 512 dimensions. Across all tested configurations, the embedding generation training times had standard deviations ranging from 7.0 to 9.5 s. The classification performance metrics for the MLP models utilizing the generated CBOW embeddings are summarized in Table 3.
Table 2.
Computational times for CBOW embedding training.
Table 3.
Condensed performance metrics and training times (F1-score, accuracy, ROC-AUC) for MLP models–CBOW.
Across all of the CBOW-based experiments, the baseline MLP models without SMOTE achieved a solid overall accuracy (approximately 69.9% to 71.5%) yet struggled with the minority class, whose F1-score remained below 61.2% (compared to 75.2–77.6% for the majority class), resulting in modest macro F1-scores between 67.9% and 69.6%. Introducing SMOTE markedly improved the class balance: the F1-score for Class 0 increased to 63.5–65.5%, the macro F1-score rose to 68.6–70.9%, and the ROC-AUC values reached 75.6–79.0%. These gains came at the cost of slightly longer training times, ranging from approximately 60.2 to 63.7 s. The optimal configuration was the MLP model trained on 50-dimensional CBOW embeddings with SMOTE, which achieved the highest accuracy of 71.9 ± 1.1%, the highest macro F1-score of 70.9 ± 1.3%, and the highest ROC-AUC of 79.0%. The relatively narrow confidence intervals for these metrics further underscore the stability and effectiveness of this configuration. Overall, the results demonstrate that SMOTE, when combined with appropriately dimensioned embeddings, substantially mitigates class imbalance and enhances the classification performance. Figure 3 displays the ROC-AUC curve for the CBOW MLP model.
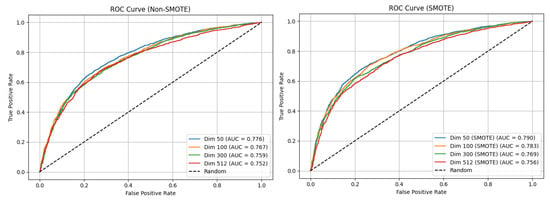
Figure 3.
The ROC-AUC curves for the original CBOW MLP model. The left plot shows the performance without resampling, while the right plot shows the performance with SMOTE applied to the training data.
6.2. The Skip-Gram Model
An analysis of the Skip-Gram embedding generation training time, as summarized in Table 4, revealed a general increase with the embedding dimension, particularly in the lower to mid-range, before leveling off. The 50-dimension model required 531.7 ± 20.1 s for training, which increased to 606.2 ± 23.1 s for the 100-dimension model and peaked at 703.1 ± 24.4 s for the 300-dimension model. For the 512-dimension model, the training duration slightly decreased to 700.6 ± 23.0 s. Across all tested configurations, the embedding generation training times demonstrated moderate variability, with the standard deviations consistently ranging from 20.1 to 24.4 s. The classification performance metrics for the shallow MLP models utilizing the generated Skip-Gram embeddings are summarized in Table 5.
Table 4.
Computational times for Skip-Gram embedding training.
Table 5.
Condensed performance metrics and training times (F1-score, accuracy, ROC-AUC) for MLP models–Skip-Gram.
Across all of the Skip-Gram experiments, the MLP models without SMOTE demonstrated a moderate classification performance. The accuracy values remained fairly stable across dimensions, with the 50-dimensional model achieving the highest at 71.5 ± 0.8% and the others ranging from 71.1 ± 1.3% to 71.5 ± 0.4%. The F1-score for the minority class remained relatively low (61.1–62.0%), while the majority class scored higher (77.0–77.5%), resulting in macro F1-scores between 69.1 ± 1.6% and 69.6 ± 0.6%. The ROC-AUC values fell within a range of 76.7–78.0%, and the training durations ranged from 57.1 s to 60.2 s. With SMOTE applied, an improvement in the minority-class F1-score was observed across all dimensions, reaching 65.0–65.8%, which led to higher macro F1-scores (70.2–71.4%) and ROC-AUC values (77.1–78.6%). The F1-score for the majority class declined in some cases, reflecting improved balance. The training times also increased, ranging from 65.2 s to 70.6 s. The optimal configuration was the MLP model with 50-dimensional Skip-Gram embeddings and SMOTE, yielding the best accuracy (72.5 ± 0.9%), macro F1-score (71.4 ± 1.0%), and ROC-AUC (78.6%). Figure 4 displays the ROC-AUC curve for the Skip-Gram MLP model.
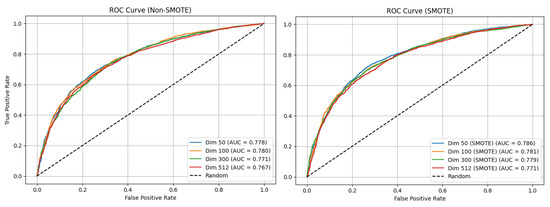
Figure 4.
The ROC-AUC curves for the original Skip-Gram MLP model. The left plot shows the performance without resampling, while the right plot shows the performance with SMOTE applied to the training data.
6.3. ChemBERTa Embeddings
The performance of the ChemBERTa model is summarized in Table 6. Across both configurations, the ChemBERTa embeddings demonstrated strong classification capabilities. Prior to resampling, the model achieved an accuracy of 79.2 ± 0.5% and a macro F1-score of 77.8 ± 0.7%, reflecting a balanced performance across classes. After applying SMOTE to addressing the class imbalance, the model’s accuracy improved to 80.5 ± 1.0%, with a notable increase in the macro F1-score to 79.3 ± 1.1%. This improvement highlights the effectiveness of SMOTE in enhancing the minority-class representation without compromising the overall performance. However, this gain in predictive performance came at the cost of an increased training time.
Table 6.
Test set performance for MLP with ChemBERTa (512 dimensions).
7. Discussion
Although ChemBERTa achieved the highest classification performance overall, it is important to consider the computational trade-offs. CBOW and Skip-Gram are significantly more lightweight in terms of model complexity and the cost of embedding training. For instance, the CBOW embeddings at 512 dimensions were trained in approximately 127.2 ± 9.5 s and Skip-Gram in 700.6 ± 23.0 s, whereas ChemBERTa leveraged a massive pretrained transformer whose pretraining cost was not reflected in the fine-tuning time alone. While ChemBERTa required about 70.1 seconds for shallow MLP training on 512-dimensional embeddings, this assumes access to a large-scale pretrained model, which may not be feasible in constrained or on-device scenarios.
Although multiple embedding dimensions (50, 100, 300, 512) were evaluated using a context window of , the resulting model performance remained essentially unchanged. This likely reflects the limited size and sparsity of the dataset, which restricts the practical benefit of increasing the embedding dimensionality. In settings with insufficient data, the additional parameters introduced by larger embeddings cannot be effectively leveraged, resulting in minimal gains. Consequently, lower-dimensional embeddings appear sufficient to represent the structural patterns required for this classification task. Moreover, faster embedding generation enables the rapid screening of large molecular libraries in early-stage drug discovery workflows. In such scenarios, high-throughput virtual screening benefits from computationally efficient models, especially when resources are limited or when rapid iteration is needed to explore a broader chemical space.
Performance-wise, ChemBERTa with SMOTE delivered the strongest results at 512 dimensions, achieving 80.5 ± 1.0% accuracy, a 79.3 ± 1.1% macro F1-score, and an 84.5% ROC-AUC. However, both the CBOW and Skip-Gram models showed a strong performance at lower dimensions. For example, Skip-Gram with 50-dimensional embeddings and SMOTE reached 72.5 ± 0.9% accuracy and a 71.4 ± 1.0% macro F1-score, and CBOW (50 d + SMOTE) achieved 71.9 ± 1.1% accuracy and a 70.9 ± 1.3% macro F1-score, with a minimal training time (e.g., 62.9 s for CBOW). This suggests that while transformer-based models like ChemBERTa offer a top-tier performance, simpler embedding methods like CBOW and Skip-Gram can still be highly effective, especially when optimized at lower dimensions, providing a good balance of performance and efficiency for real-world applications. The downstream task in all experiments is to predict the market approvability label of a given compound, serving as a practical benchmark for evaluating molecular representations. Overall, our experiments show that while ChemBERTa achieves the highest predictive performance, Word2Vec-based models the reduce training time by up to 10% and the embedding memory usage by over 90%, making them substantially more efficient for resource-constrained scenarios. Ultimately, the goal of these lightweight models is to provide scalable molecular representations that can accelerate early-stage drug screening, prioritization, and compound triage in both academic and industrial settings.
7.1. A Comparison with Prior Work
Compared to existing molecular representation learning methods such as SMILES2Vec [3], Mol2Vec [4], and ChemBERTa [5], our approach offers several practical advantages. First, it operates entirely in an unsupervised manner, decoupling embedding learning from the downstream task. This makes the embeddings reusable across multiple tasks and avoids label dependence during training. Second, the lightweight nature of Word2Vec models ensures a low computational overhead, enabling fast training even on modest hardware, a critical factor for deployment in low-resource settings or early-stage drug discovery pipelines. While ChemBERTa and other transformer-based models have demonstrated a superior performance in large-scale property prediction tasks, they require extensive pretraining and infrastructure, which may not be feasible for smaller research teams or industry partners operating under budget constraints.
While transformer-based models like ChemBERTa achieve a strong performance in large-scale property predictions, they require extensive pretraining and infrastructure. More recently, ChemAP [7] introduced a teacher–student framework that distills multi-modal semantic knowledge into a structure-only model, achieving state-of-the-art results in drug approval predictions. However, such architectures add complexity and remain task-specific, whereas our method emphasizes general-purpose, efficient, and easily deployable embeddings.
7.2. Limitations and Future Work
Despite the promising results, this study has several limitations. First, the character-level tokenization strategy may not fully capture higher-order structural motifs that are chemically relevant, such as rings or functional groups. Fragment-based methods like Mol2Vec or graph-based encodings may offer richer contextual representations in such cases. Notably, recent work such as ChemAP [7] has demonstrated the effectiveness of combining SMILES-based embeddings (including ChemBERTa) with fragment-level and graph-derived structural features for predicting drug approval outcomes. While our study focuses on lightweight Word2Vec-style embeddings, ChemAP highlights the advantages of deeper, structurally enriched representations. This distinction underscores that our contribution lies in benchmarking shallow embeddings under resource-constrained settings, rather than proposing a structurally comprehensive framework.
Second, the current embedding models focus solely on the local context within a fixed window, potentially missing long-range dependencies across the molecular sequence. Future work could explore hierarchical or multi-scale embeddings that incorporate both local and global structural information. Instead of viewing SMILES purely as a linear structure, where the structural connectivity cannot be captured, graph-based approaches can be used to enhance molecular embeddings. Advanced techniques such as weighted random walks or Graph Neural Networks (GNNs) [9] can be used to model the chemical bonds and topology of the molecules in the structure. By incorporating edge weights that reflect the bond type, partial charge, or atomic properties, future work could construct graph-based embeddings that more faithfully represent chemically relevant interactions and reactivity potential. Additionally, a GNN architecture could allow for direct encoding of the molecular graph in a way that captures a more robust and chemically informative representation of a given compound [9].
Additionally, future work could explore pretraining the Word2Vec embeddings on larger chemical datasets such as ZINC or ChEMBL [10], which may help improve the representation quality and enable better transfer learning across molecular tasks. Hybrid models that combine lightweight Word2Vec embeddings with attention-based mechanisms or transformer layers could also be investigated for capturing both the local and global patterns within molecular structures. Furthermore, expanding the downstream evaluation to include other molecular property prediction tasks, such as solubility, toxicity, or bioactivity predictions, would provide a more comprehensive validation of the approach. Finally, integrating explainable AI (XAI) techniques [11] to interpret the model predictions and identify important substructures within SMILES strings would increase the interpretability and applicability of the proposed method for real-world drug discovery workflows.
Author Contributions
Conceptualization: S.H.; methodology: S.H.; software: S.H.; validation: P.K. and Z.K.; formal analysis: Z.K. and S.H.; investigation: S.H. and Z.K.; resources: S.H.; data curation: S.H.; writing—original draft preparation: Z.K. and S.H.; writing—review and editing: Y.L., S.H. and Z.K.; visualization: S.H., Z.K. and P.K.; supervision: Y.L.; project administration: Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NSERC Discovery Grant.
Data Availability Statement
The data and code used in this study are available at https://github.com/RoosterMonkey777/SmilesWord2VecMarketApprovability (accessed on 25 August 2025).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Word2Vec | Word to Vector |
| SMILES | Simplified Molecular Input Line Entry System |
| CBOW | Continuous Bag of Words |
| MLP | Multi-Layer Perceptron |
| SMOTE | Synthetic Minority Over-Sampling Technique |
| MLM | Masked Language Modeling |
| GNN | Graph Neural Network |
References
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. arXiv 2013, arXiv:1301.3781. [Google Scholar] [CrossRef]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.O.; Baker, N. SMILES2Vec: An Interpretable General-Purpose Deep Neural Network for Predicting Chemical Properties. arXiv 2017, arXiv:1712.02034. [Google Scholar]
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J. Chem. Inf. Model. 2018, 58, 27–35. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Cai, C.; Lin, H.; Wang, H.; Xu, Y.; Ouyang, Q.; Lai, L.; Pei, J. miDruglikeness: Subdivisional Drug-Likeness Prediction Models Using Active Ensemble Learning Strategies. Biomolecules 2023, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.; Lee, S.; Bang, D.; Piao, Y.; Kim, S. ChemAP: Predicting drug approval with chemical structures before clinical trial phase by leveraging multi-modal embedding space and knowledge distillation. Sci. Rep. 2024, 14, 23010. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15: Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Liu, B.; Gomes, J.; Zitnik, M.; Liang, P.; Pande, V.; Leskovec, J. Strategies for pre-training graph neural networks. In Proceedings of the International Conference on Learning Representations (ICLR), Addis Ababa, Ethiopia, 26–30 April 2020. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. Why should I trust you?: Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD), San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).