
A Bioinformatics Analysis of Ovarian Cancer Data Using Machine Learning


Abstract
1. Introduction
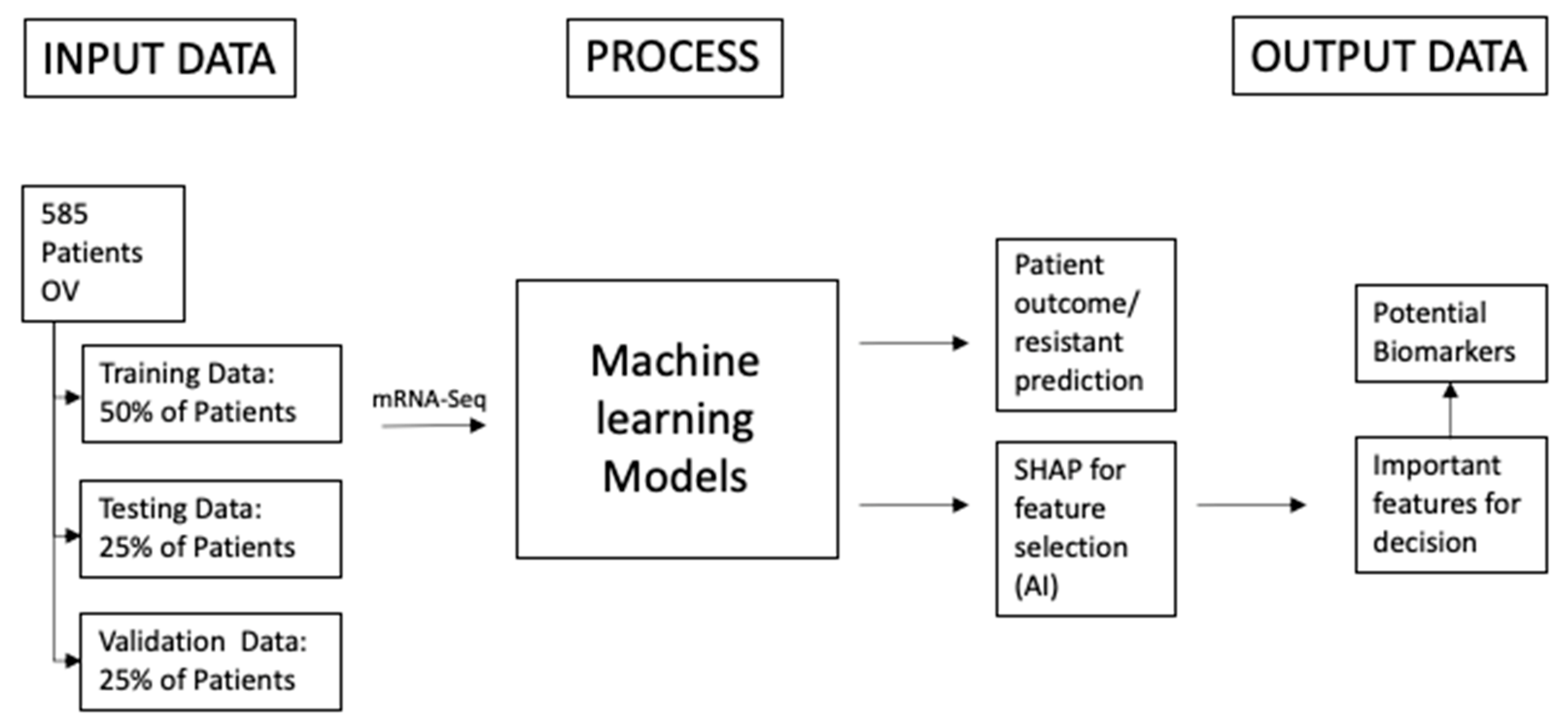
2. Materials and Methods
2.1. Materials
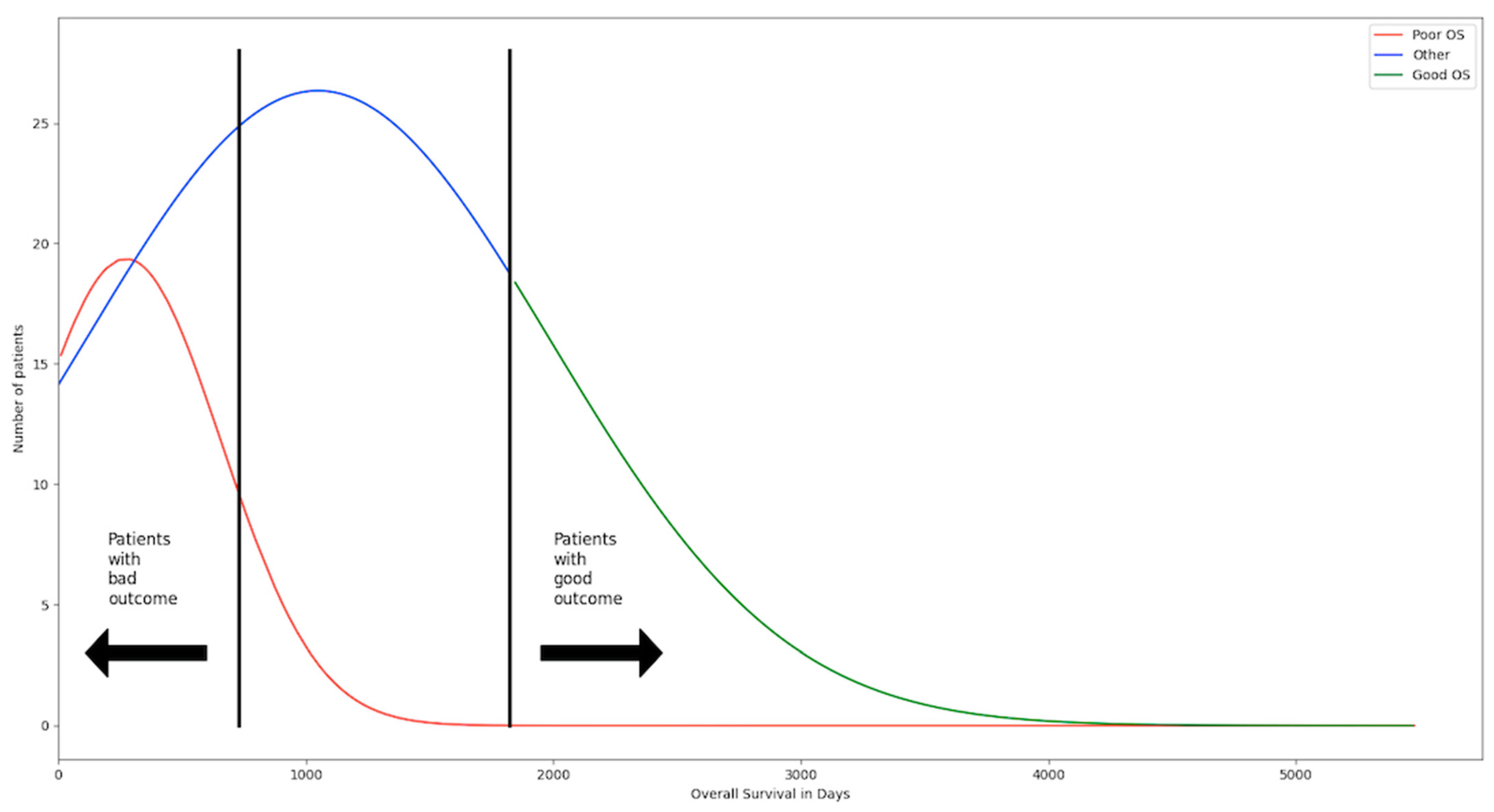
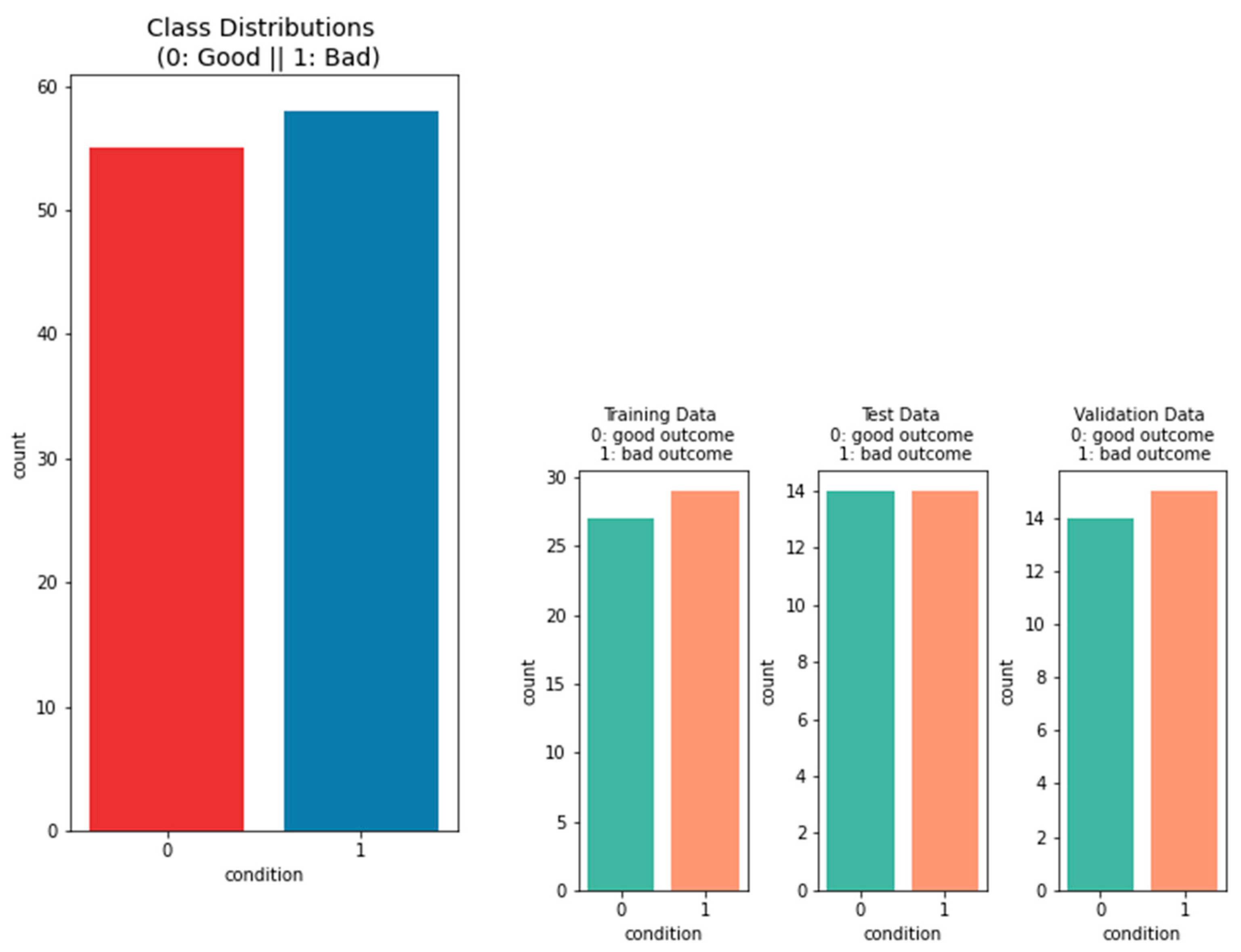
2.1.1. Data on the Outcome of the Patients
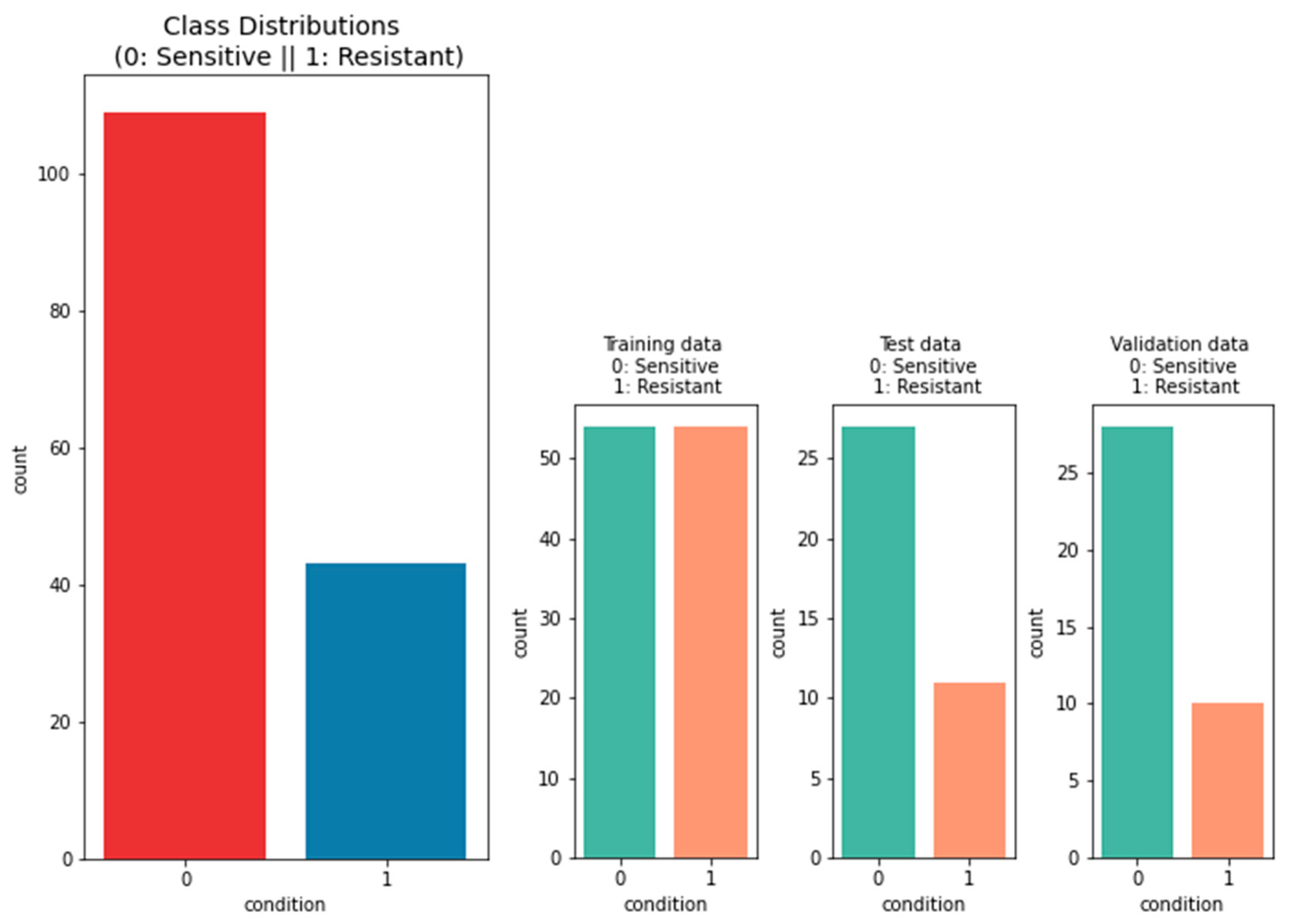
2.1.2. Data on the Platinum Response
2.2. Methods
2.2.1. Machine Learning Methods
2.2.2. SHAP
2.2.3. Bioinformatics Algorithms
2.2.4. Statistical Methods
3. Results
3.1. Performance of Machine-Learning Models
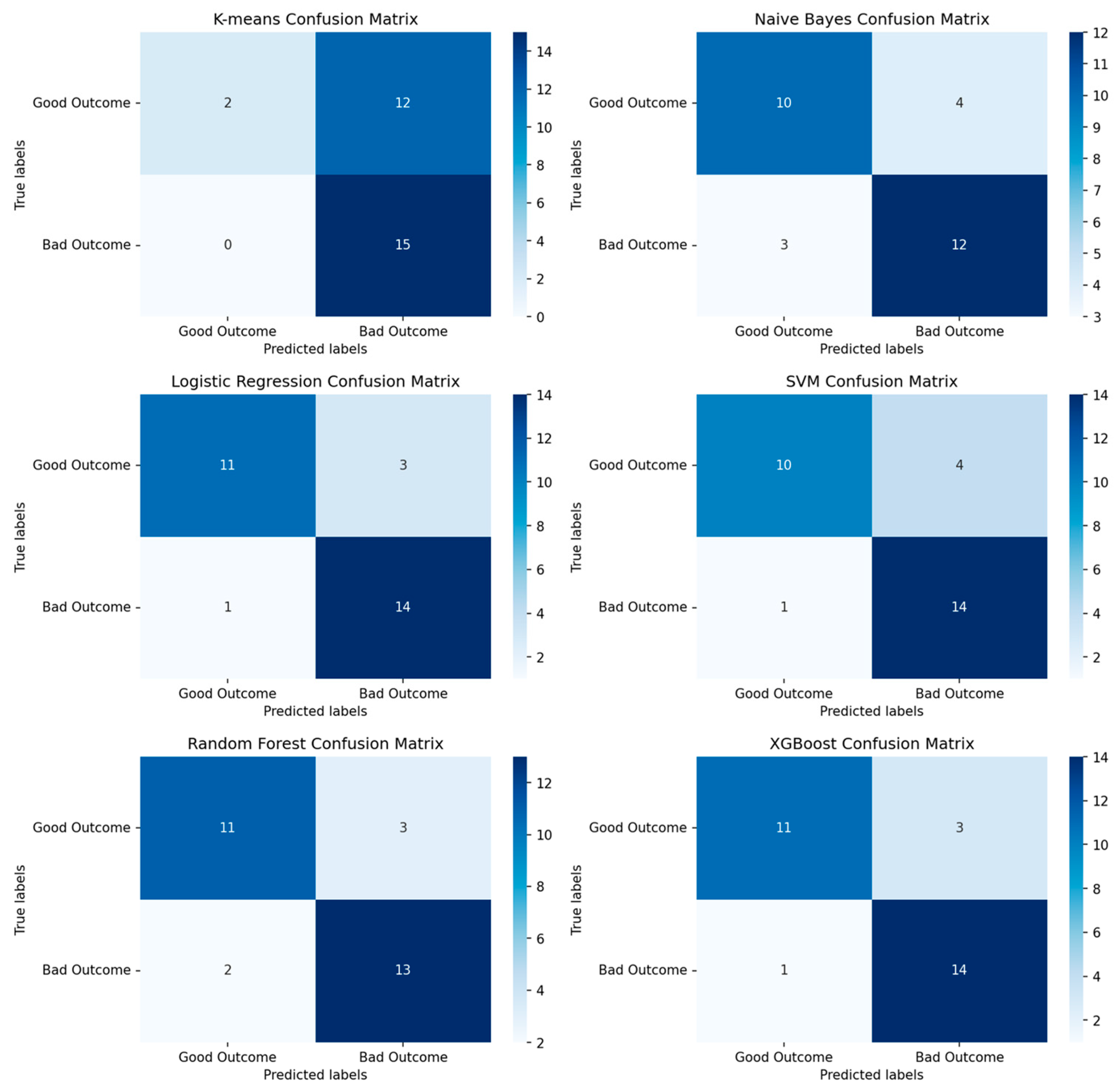
3.1.1. Performance of Machine-Learning Models for Patient Outcome
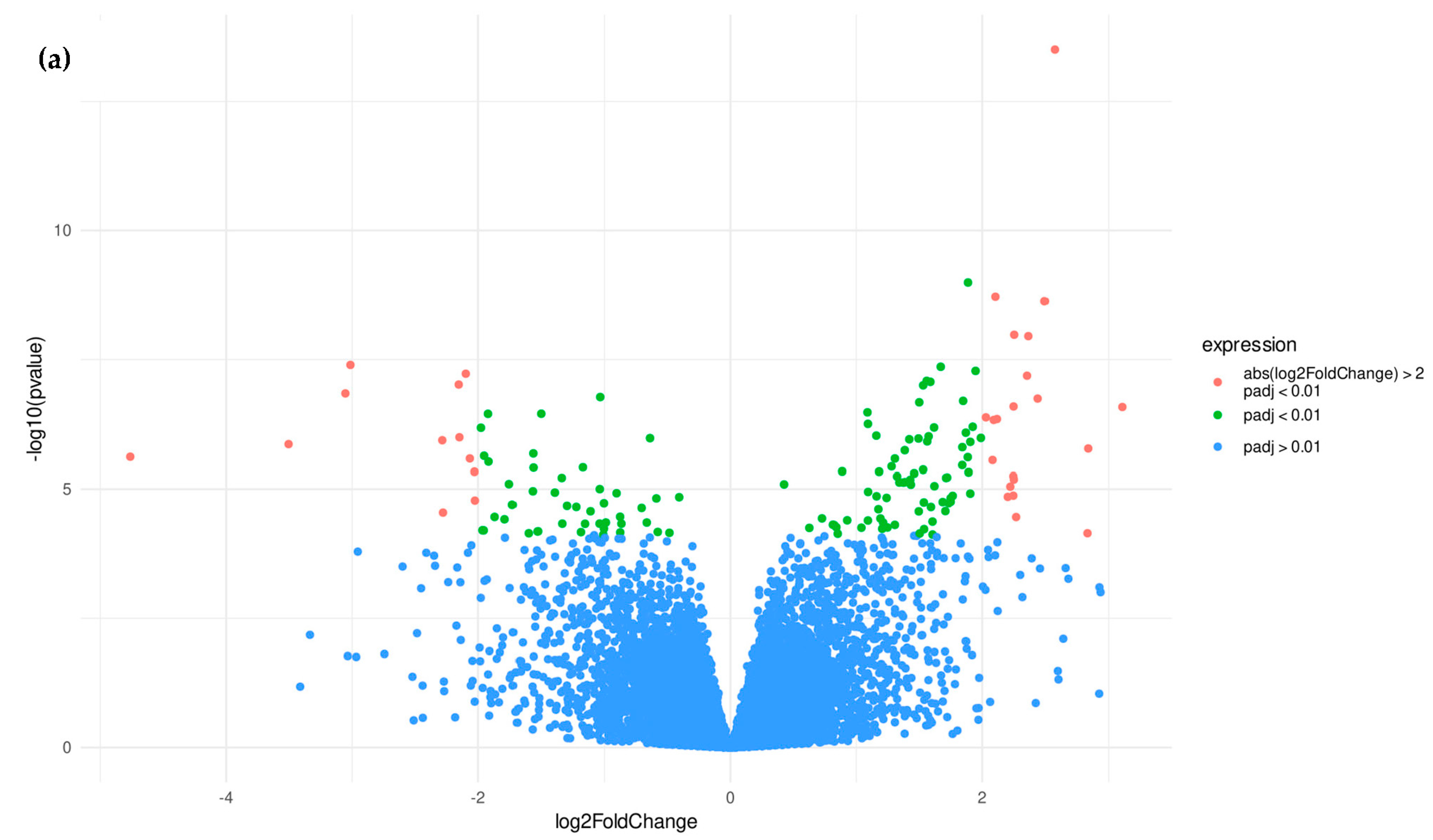
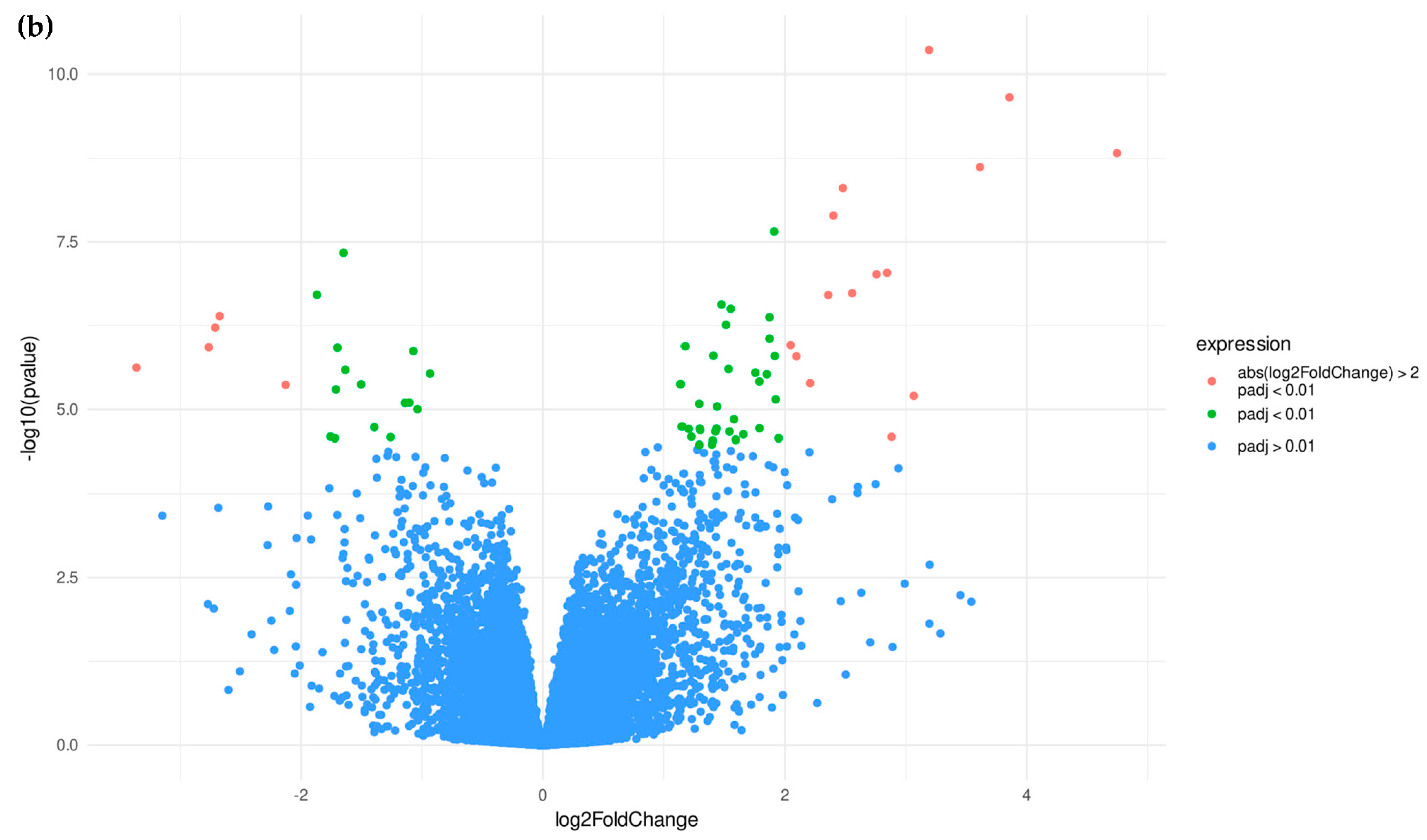
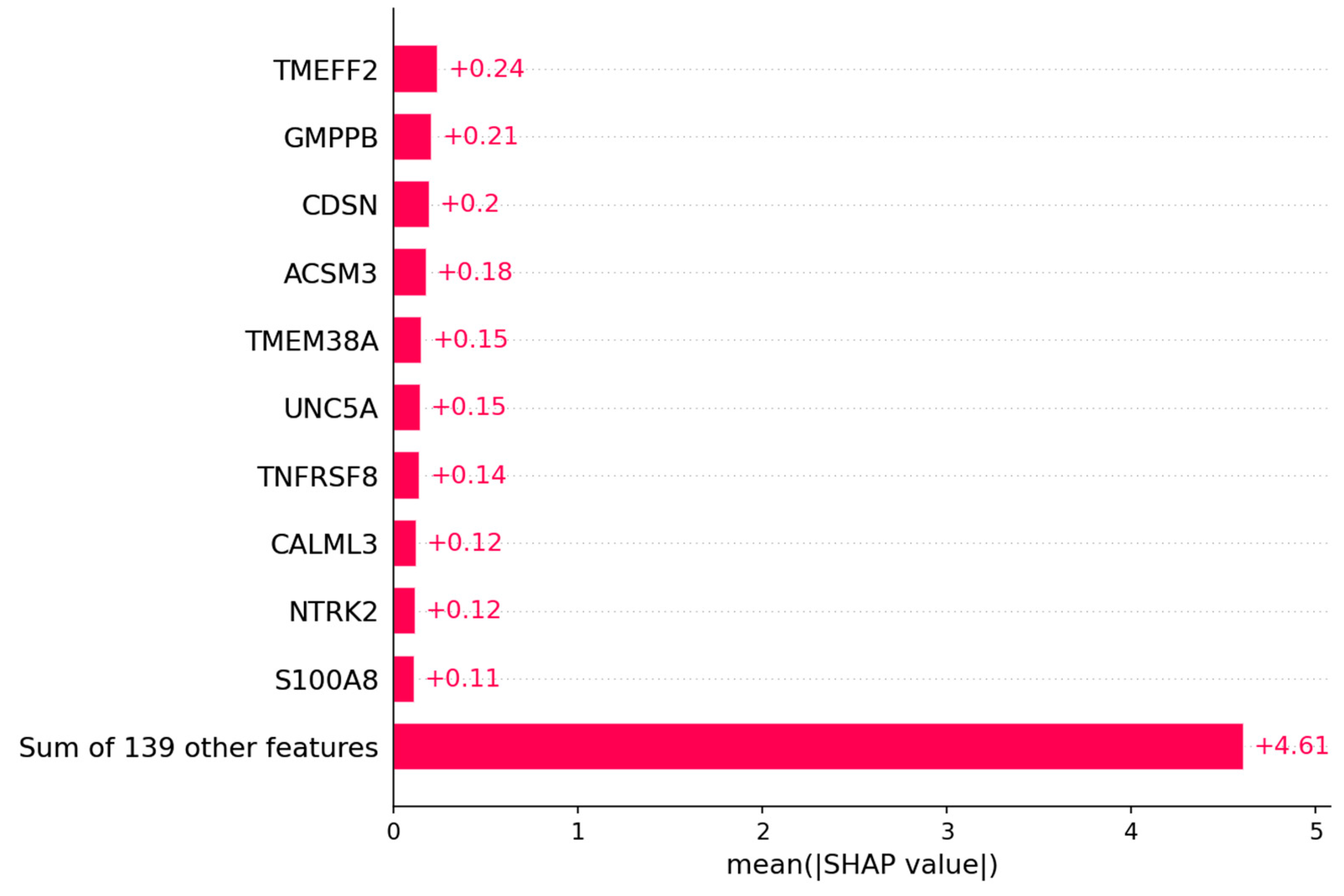
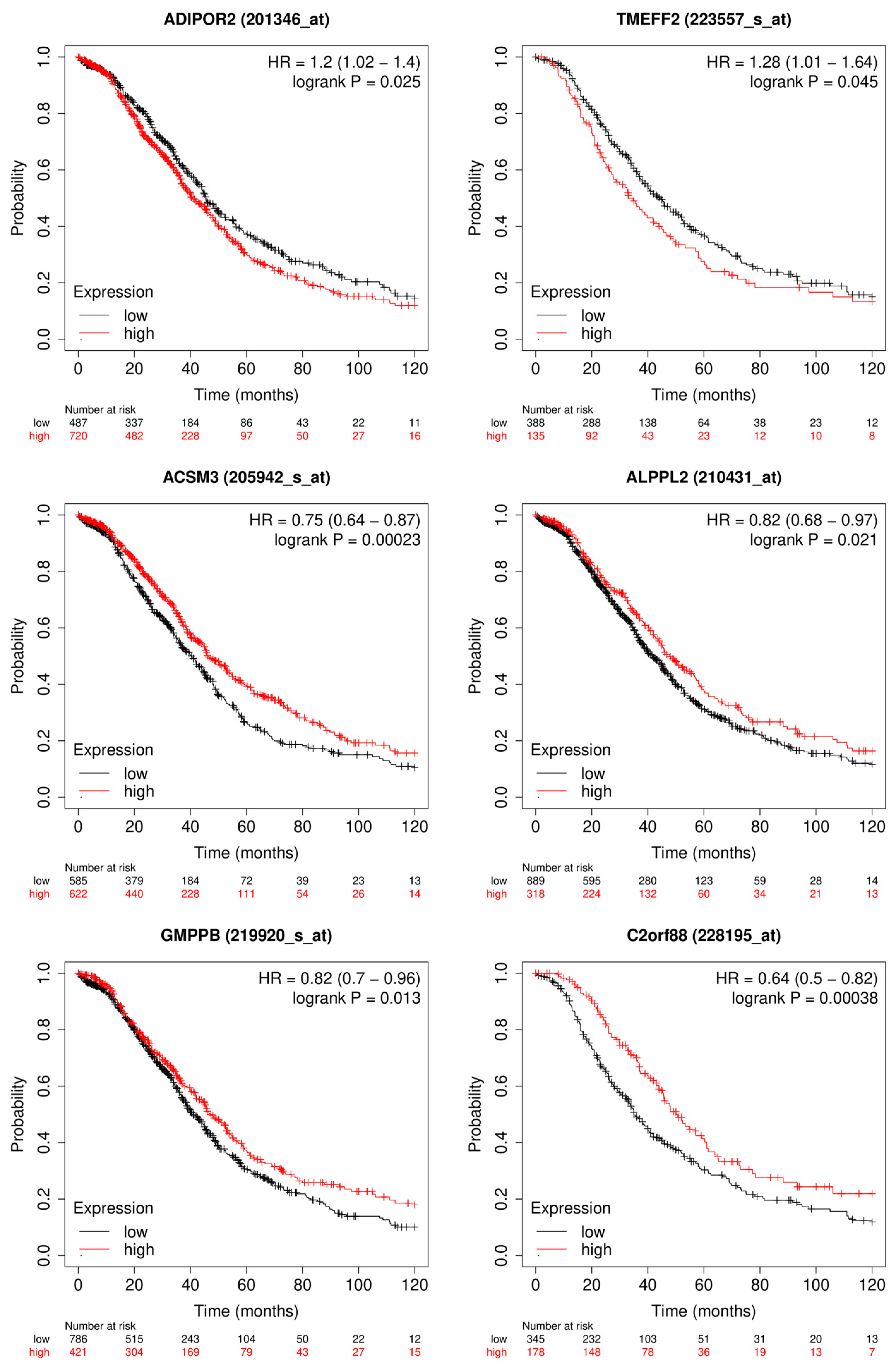
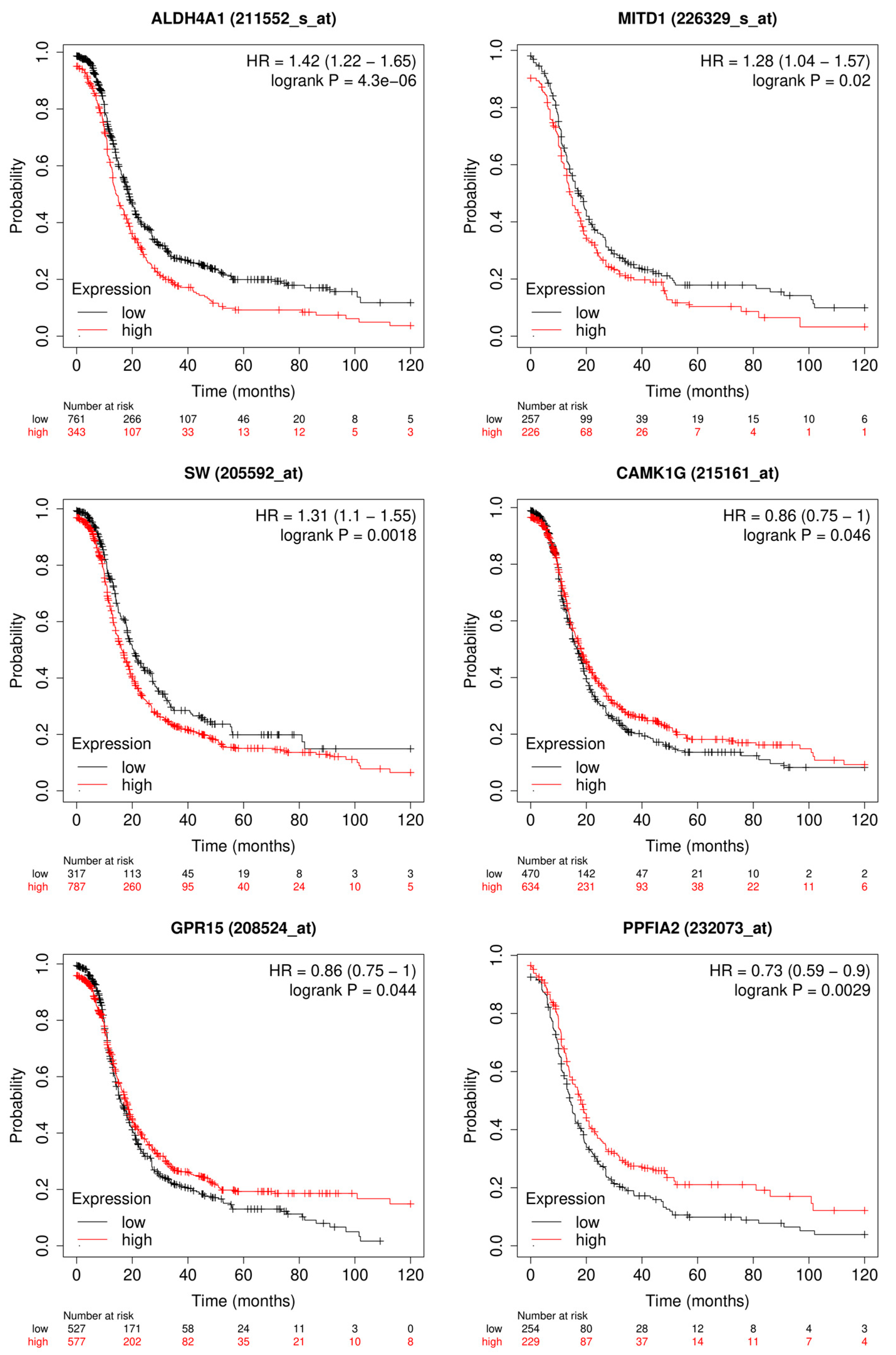
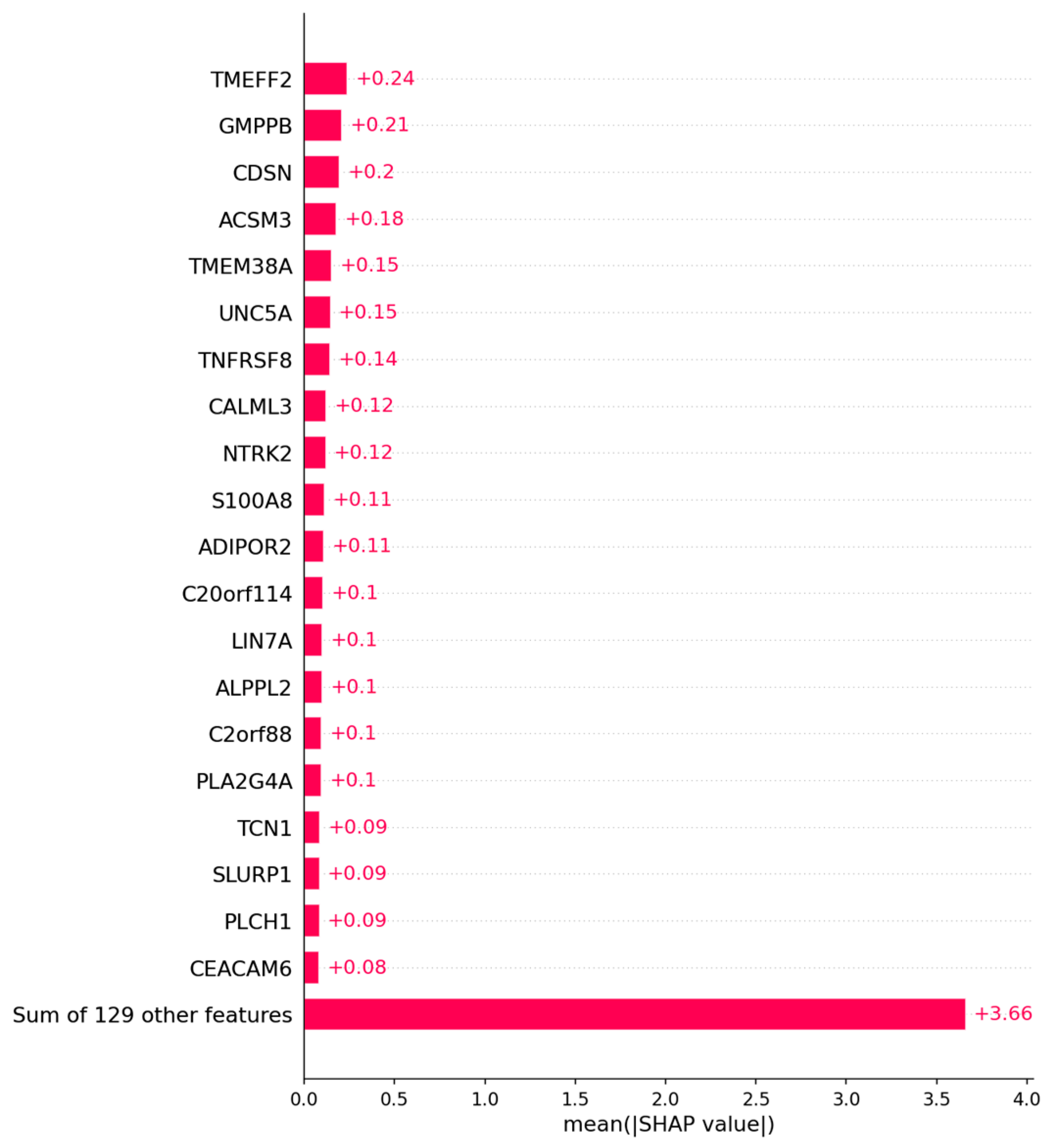
3.1.2. Candidate Genes
3.2. Platinum Resistance Prediction of Ovarian Cancer Patients
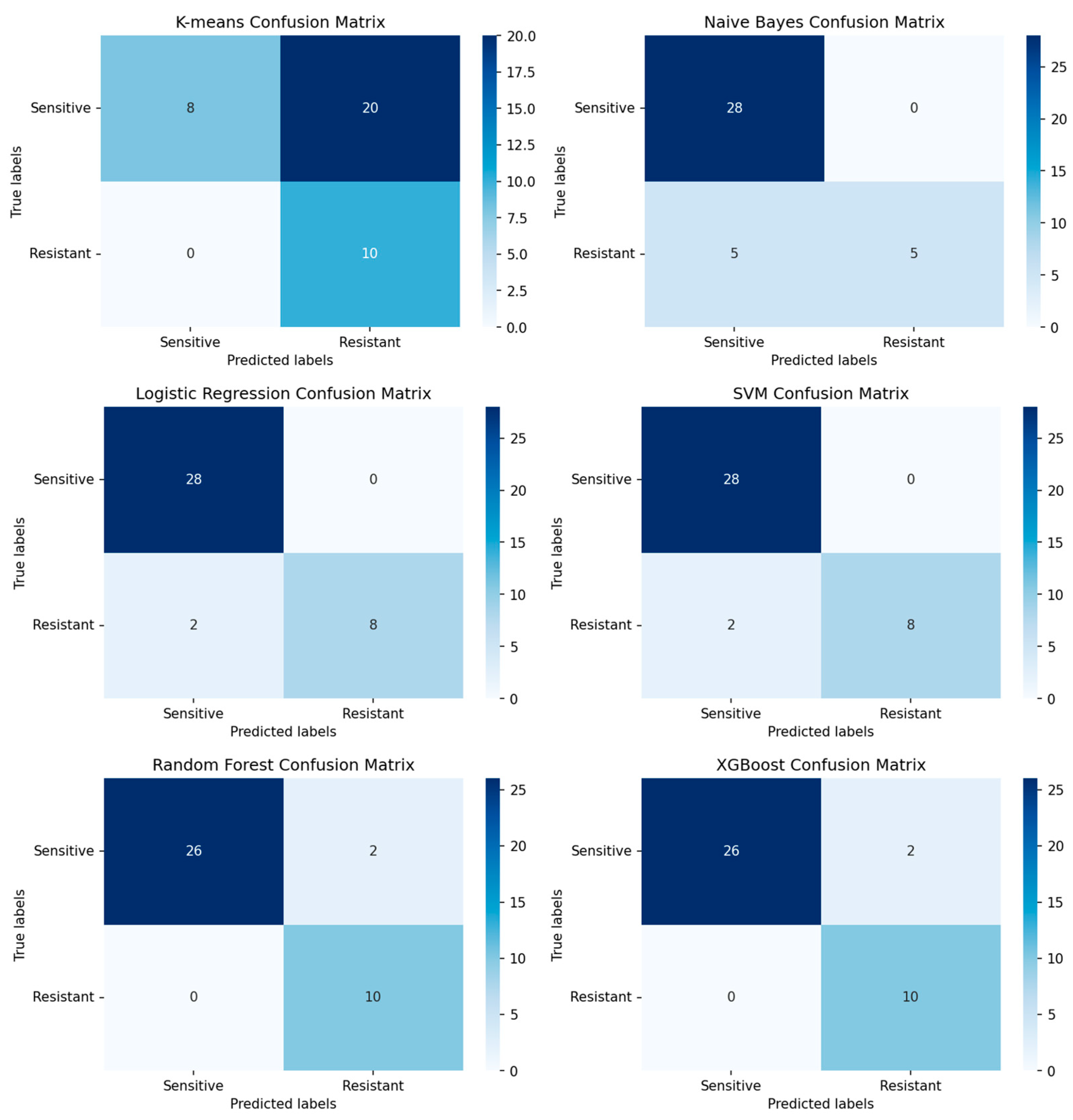
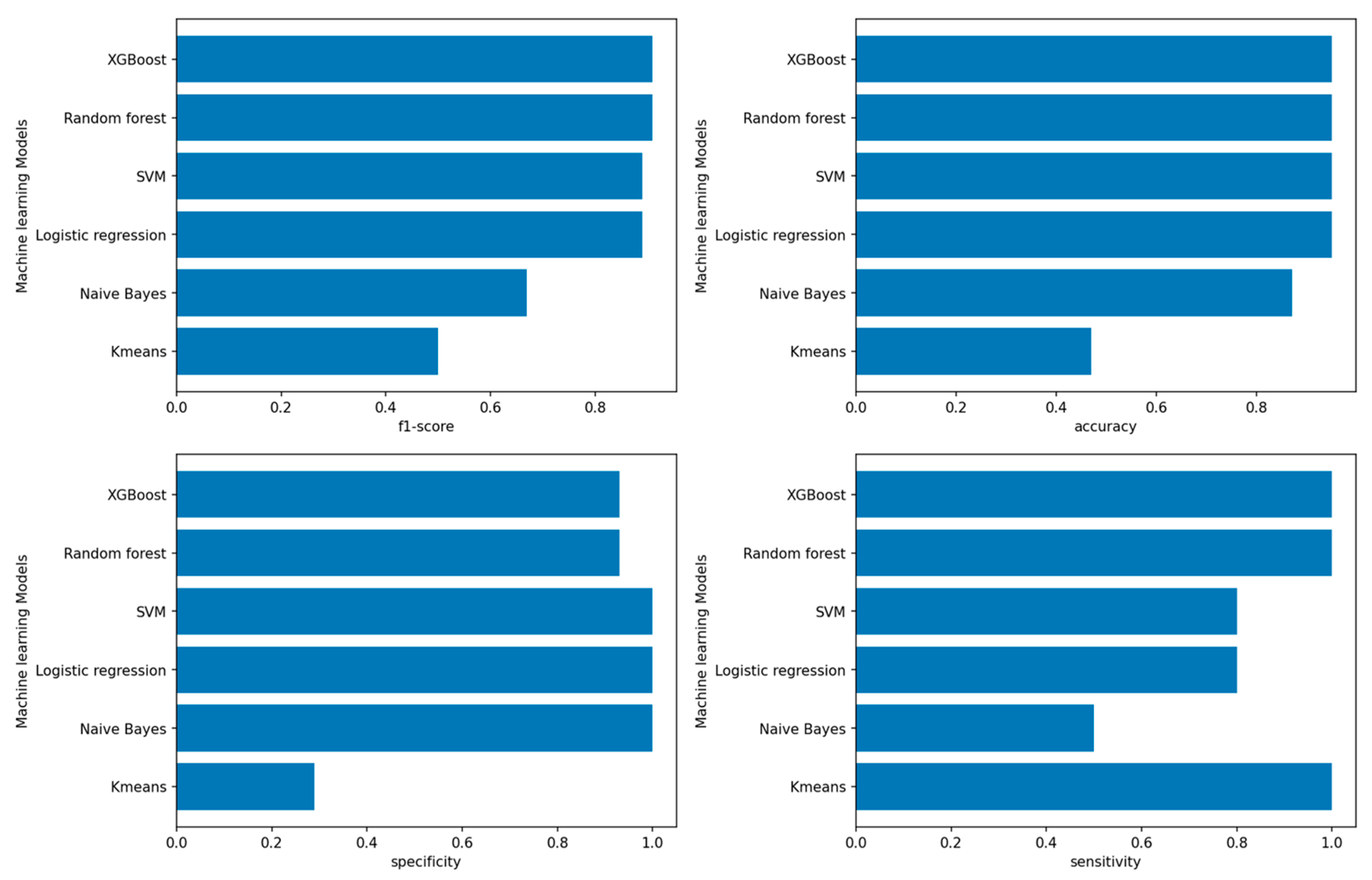
3.2.1. Performance of Machine-Learning Models for Platinum Response
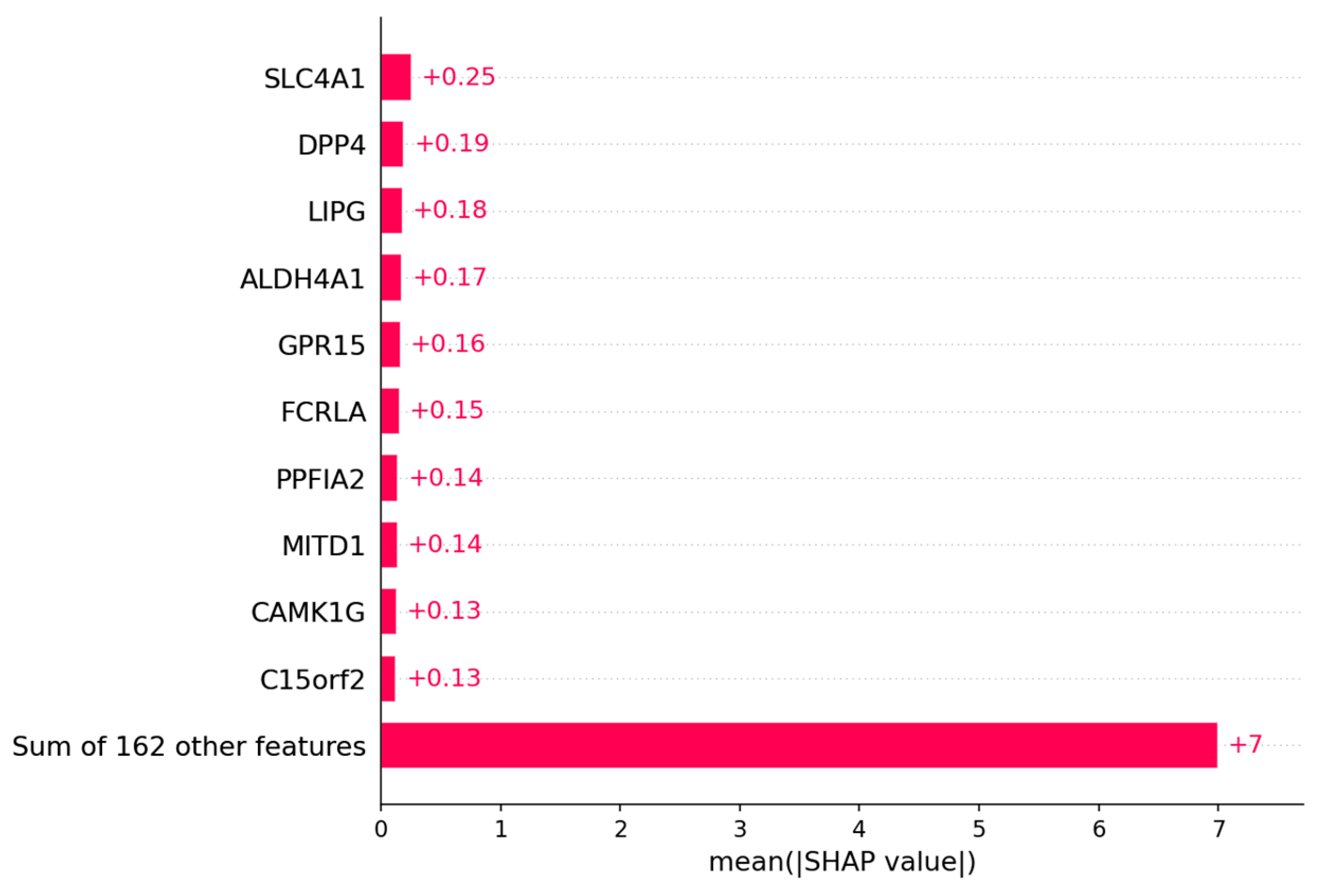
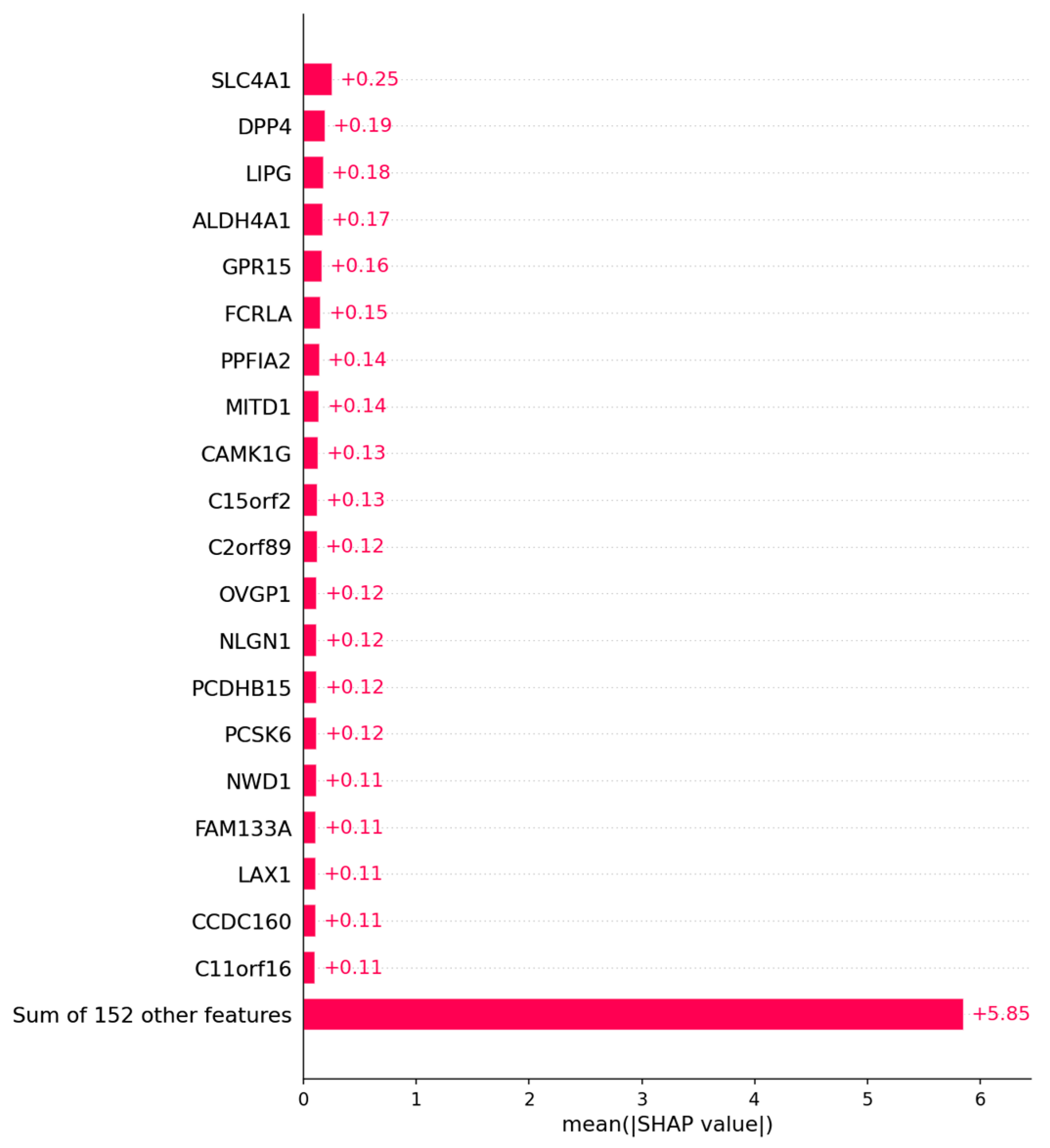
3.2.2. Candidate Genes
4. Discussion
4.1. Outcome Prediction for Patients with Ovarian Cancer
4.2. Prediction of Platinum Response Status
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | GO | Description | Hits |
|---|---|---|---|
| GO Biological Processes | GO:0042886 | amide transport | NTRK2|S100A8|SLC1A6 |
| Immunologic Signatures | M5353 | GSE37416 0H vs. 48H F TULARENSIS LVS NEUTROPHIL DN | TNFRSF8|CATSPERG|ADIPOR2 |
| GO Biological Processes | GO:0042060 | wound healing | S100A8|TMEFF2|ADIPOR2 |
| GO Biological Processes | GO:0099537 | trans-synaptic signaling | NTRK2|SLC1A6|LIN7A |
| GO Biological Processes | GO:0048514 | blood vessel morphogenesis | NTRK2|ANGPTL4|ADIPOR2 |
| GO Biological Processes | GO:0009611 | response to wounding | S100A8|TMEFF2|ADIPOR2 |
| GO Biological Processes | GO:0099536 | synaptic signaling | NTRK2|SLC1A6|LIN7A |
| GO Biological Processes | GO:0001568 | blood vessel development | NTRK2|ANGPTL4|ADIPOR2 |
| GO Biological Processes | GO:0046903 | secretion | NTRK2|S100A8|LIN7A |
| GO Biological Processes | GO:0001944 | vasculature development | NTRK2|ANGPTL4|ADIPOR2 |
| GO Biological Processes | GO:0043065 | positive regulation of apoptotic process | TNFRSF8|S100A8|TIGAR |
| GO Biological Processes | GO:0043068 | positive regulation of programmed cell death | TNFRSF8|S100A8|TIGAR |
| GO Biological Processes | GO:0010942 | positive regulation of cell death | TNFRSF8|S100A8|TIGAR |
| GO Biological Processes | GO:0030855 | epithelial cell differentiation | CDSN|CASP14|TIGAR |
| GO Biological Processes | GO:0035239 | tube morphogenesis | NTRK2|ANGPTL4|ADIPOR2 |
| Reactome Gene Sets | R-HSA-382551 | Transport of small molecules | APOC4|SLC1A6|ANGPTL4 |
| Category | GO | Description | Hits |
|---|---|---|---|
| GO Biological Processes | GO:0098742 | cell–cell adhesion via plasma–membrane adhesion molecules | NLGN1|PCDHB15| PCDHB7 |
| GO Biological Processes | GO:0098609 | cell–cell adhesion | NLGN1|PCDHB15| PCDHB7 |
References
- Ovarian Cancer Survival Rates|Ovarian Cancer Prognosis. Available online: https://www.cancer.org/cancer/ovarian-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 28 March 2023).
- Surgery for Recurrent Ovarian Cancer May Help Selected Patients-NCI. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2022/ovarian-cancer-return-surgery-desktop-iii (accessed on 28 March 2023).
- Flynn, M.J.; Ledermann, J.A. Ovarian Cancer Recurrence: Is the Definition of Platinum Resistance Modified by PARPi and Other Intervening Treatments? The Evolving Landscape in the Management of Platinum-Resistant Ovarian Cancer. Cancer Drug Resist. 2022, 5, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian Cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- How to Check for Ovarian Cancer|Ovarian Cancer Screening. Available online: https://www.cancer.org/cancer/ovarian-cancer/detection-diagnosis-staging/detection.html (accessed on 26 April 2023).
- Klein, M.E.; Dabbs, D.J.; Shuai, Y.; Brufsky, A.M.; Jankowitz, R.; Puhalla, S.L.; Bhargava, R. Prediction of the Oncotype DX Recurrence Score: Use of Pathology-Generated Equations Derived by Linear Regression Analysis. Mod. Pathol. 2013, 26, 658–664. [Google Scholar] [CrossRef]
- Kumar, L.; Greiner, R. Gene Expression Based Survival Prediction for Cancer Patients—A Topic Modeling Approach. PLoS ONE 2019, 14, e0224446. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.-Y.; Brain, E.; Causeret, S.; DeLorenzi, M.; et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Ghoniem, R.M.; Algarni, A.D.; Refky, B.; Ewees, A.A. Multi-Modal Evolutionary Deep Learning Model for Ovarian Cancer Diagnosis. Symmetry 2021, 13, 643. [Google Scholar] [CrossRef]
- Hartmann, L.C.; Lu, K.H.; Linette, G.P.; Cliby, W.A.; Kalli, K.R.; Gershenson, D.; Bast, R.C.; Stec, J.; Iartchouk, N.; Smith, D.I.; et al. Gene Expression Profiles Predict Early Relapse in Ovarian Cancer after Platinum-Paclitaxel Chemotherapy. Clin. Cancer Res. 2005, 11, 2149–2155. [Google Scholar] [CrossRef]
- Millstein, J.; Budden, T.; Goode, E.L.; Anglesio, M.S.; Talhouk, A.; Intermaggio, M.P.; Leong, H.S.; Chen, S.; Elatre, W.; Gilks, B.; et al. Prognostic Gene Expression Signature for High-Grade Serous Ovarian Cancer. Ann. Oncol. 2020, 31, 1240–1250. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Cannistra, S.A. Gene-Expression Profiling in Epithelial Ovarian Cancer. Nat. Rev. Clin. Oncol. 2008, 5, 577–587. [Google Scholar] [CrossRef]
- Welsh, J.B.; Zarrinkar, P.P.; Sapinoso, L.M.; Kern, S.G.; Behling, C.A.; Monk, B.J.; Lockhart, D.J.; Burger, R.A.; Hampton, G.M. Analysis of Gene Expression Profiles in Normal and Neoplastic Ovarian Tissue Samples Identifies Candidate Molecular Markers of Epithelial Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1176–1181. [Google Scholar] [CrossRef]
- Spentzos, D.; Levine, D.A.; Ramoni, M.F.; Joseph, M.; Gu, X.; Boyd, J.; Libermann, T.A.; Cannistra, S.A. Gene Expression Signature with Independent Prognostic Significance in Epithelial Ovarian Cancer. J. Clin. Oncol. 2004, 22, 4700–4710. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Kaur, S.; Volinia, S.; Greshock, J.; Lassus, H.; Hasegawa, K.; Liang, S.; Leminen, A.; Deng, S.; Smith, L.; et al. MicroRNA Microarray Identifies Let-7i as a Novel Biomarker and Therapeutic Target in Human Epithelial Ovarian Cancer. Cancer Res. 2008, 68, 10307–10314. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically Relevant Gene Signatures of High-Grade Serous Ovarian Carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ota, T.; Shridhar, V.; Chien, J.; Wu, B.; Kuang, R. Network-Based Survival Analysis Reveals Subnetwork Signatures for Predicting Outcomes of Ovarian Cancer Treatment. PLoS Comput. Biol. 2013, 9, e1002975. [Google Scholar] [CrossRef]
- Lundberg, S.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. arXiv 2017. [Google Scholar] [CrossRef]
- Nasimian, A.; Ahmed, M.; Hedenfalk, I.; Kazi, J.U. A Deep Tabular Data Learning Model Predicting Cisplatin Sensitivity Identifies BCL2L1 Dependency in Cancer. Comput. Struct. Biotechnol. J. 2023, 21, 956–964. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- CBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/study/clinicalData?id=ov_tcga_pan_can_atlas_2018 (accessed on 4 June 2023).
- Steinhaus, H. Bulletin de L’Académie Polonaise Des Sciences: Série des sciences mathématiques, astronomiques, et physiques. Państowowe Wydawn 1956, 4, 801–804. [Google Scholar]
- Lloyd, S. Least Squares Quantization in PCM. IEEE Trans. Inform. Theory 1982, 28, 129–137. [Google Scholar] [CrossRef]
- Bayes, T.; Price, R. LII. An Essay towards Solving a Problem in the Doctrine of Chances. By the Late Rev. Mr. Bayes, F. R. S. Communicated by Mr. Price, in a Letter to John Canton, A. M. F. R. S. Philos. Trans. R. Soc. Lond. 1763, 53, 370–418. [Google Scholar] [CrossRef]
- Garnier, J.-G.; Quetelet, A. Correspondance Mathématique et Physique; Hayez, M., Imprimeur; Harvard University: Cambridge, MA, USA, 1838; Volume 10. [Google Scholar]
- Verhulst, P.-F. Recherches Mathématiques sur la loi D’accroissement de la Population; Nouveaux Mémoires de l’Académie Royale des Sciences et Belles-Lettres de Bruxelles, Harvard University: Cambridge, MA, USA, 1845; pp. 14–54. [Google Scholar]
- Vapnik, V.N.; Lerner, A.Y. Recognition of Patterns with help of Generalized Portraits. Recognit. Patterns Help. Gen. Portraits 1963, 24, 774–780. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Ho, T.K. Random Decision Forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13 August 2016; pp. 785–794. [Google Scholar]
- Shapley, L.S. Stochastic Games*. Proc. Natl. Acad. Sci. USA 1953, 39, 1095–1100. [Google Scholar] [CrossRef]
- Kuo, C. Explain Your Model with the SHAP Values. Medium. 2019. Available online: https://medium.com/dataman-in-ai/explain-your-model-with-the-shap-values-bc36aac4de3d (accessed on 31 May 2023).
- Piper, M.M.; Khetani, R.; Gene-Level, M. Differential Expression Analysis with DESeq2. Available online: https://hbctraining.github.io/DGE_workshop/lessons/04_DGE_DESeq2_analysis.html (accessed on 31 May 2023).
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority over-Sampling Technique. J. Artif. Int. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Korstanje, J. SMOTE. Available online: https://towardsdatascience.com/smote-fdce2f605729 (accessed on 31 May 2023).
- Pearson, K. LIII. On Lines and Planes of Closest Fit to Systems of Points in Space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- Hotelling, H. Analysis of a Complex of Statistical Variables into Principal Components. J. Educ. Psychol. 1933, 24, 417–441. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Gao, L.; Nie, X.; Zheng, M.; Li, X.; Guo, Q.; Liu, J.; Liu, Q.; Hao, Y.; Lin, B. TMEFF2 Is a Novel Prognosis Signature and Target for Endometrial Carcinoma. Life Sci. 2020, 243, 116910. [Google Scholar] [CrossRef]
- Alabiad, M.A.; Harb, O.A.; Hefzi, N.; Ahmed, R.Z.; Osman, G.; Shalaby, A.M.; Alnemr, A.A.-A.; Saraya, Y.S. Prognostic and Clinicopathological Significance of TMEFF2, SMOC-2, and SOX17 Expression in Endometrial Carcinoma. Exp. Mol. Pathol. 2021, 122, 104670. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Ocon-Grove, O.M.; Hadley, J.A.; Giles, J.R.; Johnson, P.A.; Ramachandran, R. Expression of Adiponectin and Its Receptors Is Altered in Epithelial Ovarian Tumors and Ascites-Derived Ovarian Cancer Cell Lines. Int. J. Gynecol. Cancer 2015, 25. [Google Scholar] [CrossRef] [PubMed]
- Rider, J.R.; Fiorentino, M.; Kelly, R.; Gerke, T.; Jordahl, K.; Sinnott, J.A.; Giovannucci, E.L.; Loda, M.; Mucci, L.A.; Finn, S. Tumor Expression of Adiponectin Receptor 2 and Lethal Prostate Cancer. Carcinogenesis 2015, 36, 639–647. [Google Scholar] [CrossRef]
- Yan, L.; He, Z.; Li, W.; Liu, N.; Gao, S. The Overexpression of Acyl-CoA Medium-Chain Synthetase-3 (ACSM3) Suppresses the Ovarian Cancer Progression via the Inhibition of Integrin Β1/AKT Signaling Pathway. Front. Oncol. 2021, 11, 644840. [Google Scholar] [CrossRef]
- Yang, X.; Wu, G.; Zhang, Q.; Chen, X.; Li, J.; Han, Q.; Yang, L.; Wang, C.; Huang, M.; Li, Y.; et al. ACSM3 Suppresses the Pathogenesis of High-Grade Serous Ovarian Carcinoma via Promoting AMPK Activity. Cell Oncol. 2022, 45, 151–161. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, X.; Bidlingmaier, S.; Behrens, C.R.; Lee, N.-K.; Liu, B. ALPPL2 Is a Highly Specific and Targetable Tumor Cell Surface Antigen. Cancer Res. 2020, 80, 4552–4564. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Feng, G.; Meng, H.; Nie, S.; Sun, R.; Yang, J.; Cheng, W. Nine Glycolysis-Related Gene Signature Predicting the Survival of Patients with Endometrial Adenocarcinoma. Cancer Cell Int. 2020, 20, 183. [Google Scholar] [CrossRef]
- Bi, J.; Bi, F.; Pan, X.; Yang, Q. Establishment of a Novel Glycolysis-Related Prognostic Gene Signature for Ovarian Cancer and Its Relationships with Immune Infiltration of the Tumor Microenvironment. J. Transl. Med. 2021, 19, 382. [Google Scholar] [CrossRef] [PubMed]
- C2orf88 Chromosome 2 Open Reading Frame 88 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/84281#summary (accessed on 26 April 2023).
- Vallacchi, V.; Vergani, E.; Camisaschi, C.; Deho, P.; Cabras, A.D.; Sensi, M.; De Cecco, L.; Bassani, N.; Ambrogi, F.; Carbone, A.; et al. Transcriptional Profiling of Melanoma Sentinel Nodes Identify Patients with Poor Outcome and Reveal an Association of CD30+ T Lymphocytes with Progression. Cancer Res. 2014, 74, 130–140. [Google Scholar] [CrossRef] [PubMed]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 Biology and Therapeutic Targeting: A Historical Perspective Providing Insight into Future Directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; De Souza, C.; Minn, K.; Chien, J. Genome-Scale CRISPR Knockout Screen Identifies TIGAR as a Modifier of PARP Inhibitor Sensitivity. Commun. Biol. 2019, 2, 335. [Google Scholar] [CrossRef]
- Bixel, K.; Hays, J.L. Olaparib in the Management of Ovarian Cancer. Pharmgenomics Pers. Med. 2015, 8, 127–135. [Google Scholar] [CrossRef]
- Qin, L.; Li, T.; Liu, Y. High SLC4A11 Expression Is an Independent Predictor for Poor Overall Survival in Grade 3/4 Serous Ovarian Cancer. PLoS ONE 2017, 12, e0187385. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Lu, R.; Song, Y.-N.; Zhu, J.-Y.; Xia, W.; Zhang, M.; Shao, Z.-Y.; Huang, Y.; Zhou, Y.; Zhang, H.; et al. Knockdown of Anion Exchanger 2 Suppressed the Growth of Ovarian Cancer Cells via MTOR/P70S6K1 Signaling. Sci. Rep. 2017, 7, 6362. [Google Scholar] [CrossRef]
- Parks, S.K.; Chiche, J.; Pouysségur, J. Disrupting Proton Dynamics and Energy Metabolism for Cancer Therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef]
- Damaghi, M.; Wojtkowiak, J.; Gillies, R. PH Sensing and Regulation in Cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde Dehydrogenase 1A1 in Stem Cells and Cancer. Oncotarget 2016, 7, 11018. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Korkaya, H.; Dontu, G.; Birnbaum, D.; Wicha, M.S.; Charafe-Jauffret, E. The cancer stem cell: The breast cancer driver. Med. Sci. 2007, 23, 1133–1139. [Google Scholar] [CrossRef]
- Dong, S.; Hou, D.; Peng, Y.; Chen, X.; Li, H.; Wang, H. Pan-Cancer Analysis of the Prognostic and Immunotherapeutic Value of MITD1. Cells 2022, 11, 3308. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chang, J.; Renvoisé, B.; Tipirneni, A.; Yang, S.; Blackstone, C. MITD1 Is Recruited to Midbodies by ESCRT-III and Participates in Cytokinesis. Mol. Biol. Cell 2012, 23, 4347–4361. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, J.S.; Skelding, K.A. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Xiong, Y.; Li, C.-D.; Xu, Q.; Shen, L.; Chandra Kaushik, A.; Wei, D.-Q. An Integrated Pan-Cancer Analysis and Structure-Based Virtual Screening of GPR15. Int. J. Mol. Sci. 2019, 20, 6226. [Google Scholar] [CrossRef]
- PPFIA2 PTPRF Interacting Protein Alpha 2 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/8499#summary (accessed on 27 April 2023).
- Pergolizzi, M.; Bizzozero, L.; Maione, F.; Maldi, E.; Isella, C.; Macagno, M.; Mariella, E.; Bardelli, A.; Medico, E.; Marchiò, C.; et al. The Neuronal Protein Neuroligin 1 Promotes Colorectal Cancer Progression by Modulating the APC/β-Catenin Pathway. J. Exp. Clin. Cancer Res. 2022, 41, 266. [Google Scholar] [CrossRef]
- Carrier, A.; Desjobert, C.; Lobjois, V.; Rigal, L.; Busato, F.; Tost, J.; Ensenyat-Mendez, M.; Marzese, D.M.; Pradines, A.; Favre, G.; et al. Epigenetically Regulated PCDHB15 Impairs Aggressiveness of Metastatic Melanoma Cells. Clin. Epigenetics 2022, 14, 156. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell Adhesion in Cancer: Beyond the Migration of Single Cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- Moh, M.C.; Shen, S. The Roles of Cell Adhesion Molecules in Tumor Suppression and Cell Migration: A New Paradox. Cell Adh Migr. 2009, 3, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.N. Editorial: Metabolism and Cell Adhesion in Cancer. Front. Cell Dev. Biol. 2022, 10, 871471. [Google Scholar] [CrossRef] [PubMed]
- Garay, T.; Juhász, É.; Molnár, E.; Eisenbauer, M.; Czirók, A.; Dekan, B.; László, V.; Hoda, M.A.; Döme, B.; Tímár, J.; et al. Cell Migration or Cytokinesis and Proliferation? – Revisiting the “Go or Grow” Hypothesis in Cancer Cells in Vitro. Exp. Cell Res. 2013, 319, 3094–3103. [Google Scholar] [CrossRef] [PubMed]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilling, V.; Beyerlein, P.; Chien, J. A Bioinformatics Analysis of Ovarian Cancer Data Using Machine Learning. Algorithms 2023, 16, 330. https://doi.org/10.3390/a16070330
Schilling V, Beyerlein P, Chien J. A Bioinformatics Analysis of Ovarian Cancer Data Using Machine Learning. Algorithms. 2023; 16(7):330. https://doi.org/10.3390/a16070330
Chicago/Turabian StyleSchilling, Vincent, Peter Beyerlein, and Jeremy Chien. 2023. "A Bioinformatics Analysis of Ovarian Cancer Data Using Machine Learning" Algorithms 16, no. 7: 330. https://doi.org/10.3390/a16070330
APA StyleSchilling, V., Beyerlein, P., & Chien, J. (2023). A Bioinformatics Analysis of Ovarian Cancer Data Using Machine Learning. Algorithms, 16(7), 330. https://doi.org/10.3390/a16070330