1. Introduction
The laboratory conditions of sequential data from spectroscopy experiments are often chosen in such a way that the processes can be assumed to be autonomous. In autonomous processes, only the initial states of the entire system determine its future. The Markov property, the lack of a memory, relates to the microstates of the system in the processes examined, i.e., what happens in detail (metaphorically, behind the curtains) is Markovian. These details are often not searched for.
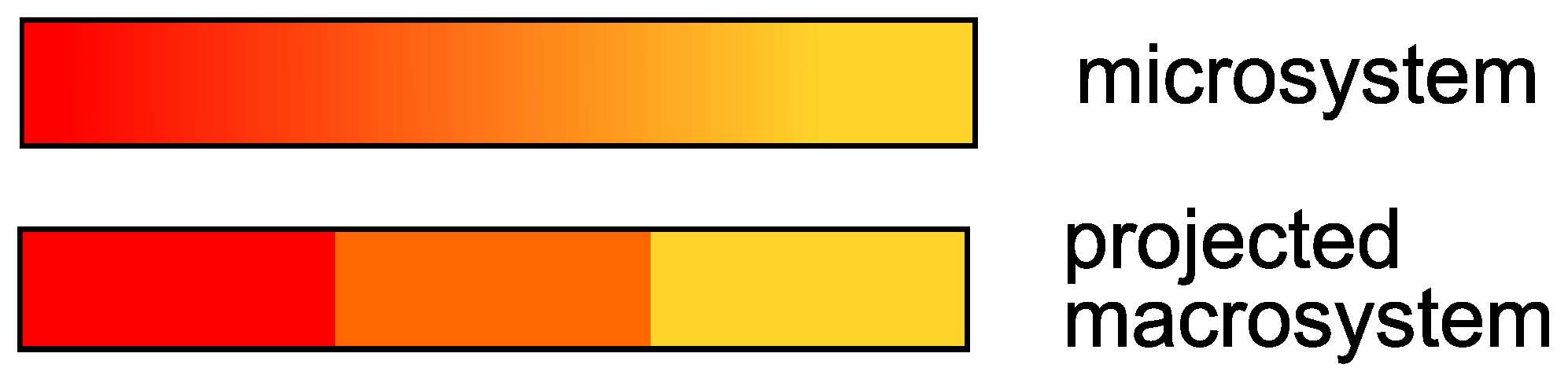
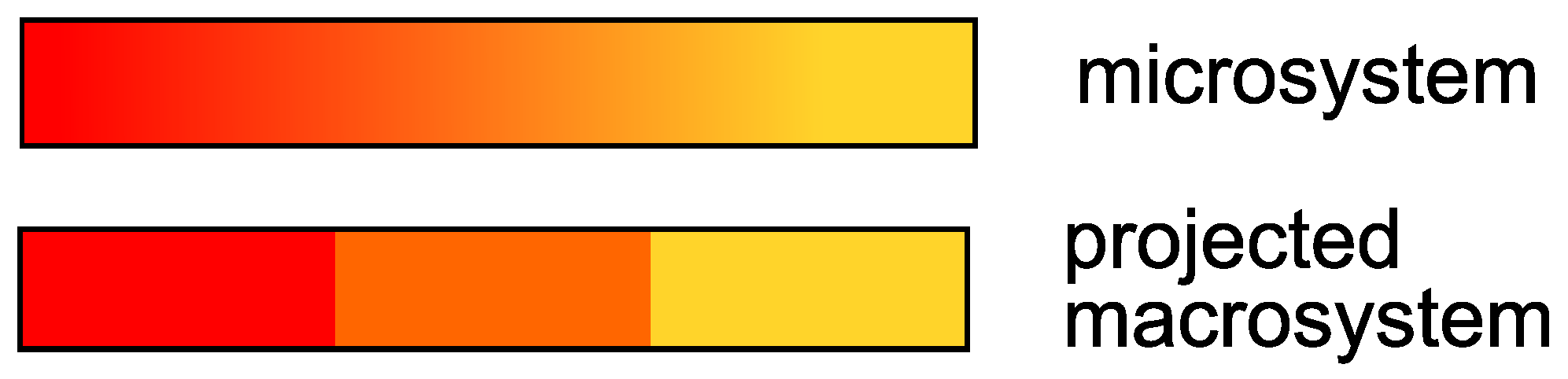
For instance, one could think of a toy-model reaction; in this reaction, a red solution gradually turns orange and then yellow with the time. By taking a picture of the solution with a certain time-step
, one observes the solution changing color. However, the exact nuance of the solution, e.g., the solution is orange-red, is not relevant for the experiment. Rather, one is interested only if the solution is red, orange or yellow. From the perspective of the solution, the overall system breaks down into three states; the Markov process, which takes places within the continuum of the colors that the solution can assume, is projected onto a three-dimensional space. A vector
h,
, indicates the proportion occupied by the colors red, orange and yellow in the overall system at each point in time of the measurement. This situation is displayed in
Figure 1.
In other words, the solution has a memory because its color cannot “switch” from red to yellow within the interval
; if the solution is red, and it was dark red before, in the next time step it will be more likely orange-red, not yellow. Meanwhile, the particles in the solution can jump from red to yellow or to any color, regardless of the past particle color (Markov). If the Markov property were also met on the level of the projected macrosystem (the solution can be red-orange-yellow), one could find, e.g., an ordinary differential equation (a Markov process) matrix for the evolution of the concentration vectors. A solution of this system for fixed time-step
could be represented by a matrix equation:
where the matrix
usually also depends on further quantities (e.g., temperature at which the reaction takes place), and
is the proportion of a state in the reaction. In the analysis of sequentially measured spectroscopic data,
h is associated to a spectral fingerprint, as explained later in this work.
The transition from a micro to a macrosystem can have a systematic source of error, because a projected Markov system loses its Markov property.
Mathematically, there are two possibilities to model this “non-Markovianity”:
- A
The process is assumed to have a memory. Not only the current (macro)state of the system, but also past steps of the process development have to be taken into account in order to derive a “forecast” for the future.
- B
The clustering of microstates into coarser macrostates is assumed to be “fuzzy”. Then Markoviantity can be preserved, but we lose control about what we will denote as a macrostate.
Chemically, this article develops option
B and presents a tool to compute the minimal past-time dependency of compounds in a reaction spectrum. A compound is a system’s state with an associated spectral fingerprint. The tool considers not only the compounds’ dependency on the past, which is called
memory, but also the dependency in the kinetic development between the compounds themselves. This memory effect has been investigated in the mathematical context (e.g., [
1,
2]) and it has been observed in chemistry, for instance in the context of protein folding [
3,
4].
The interpretation of the compounds’ kinetics is more meaningful if the development of each kinetic curve is as independent as possible to the development of the other kinetic curves. A correlated development of kinetics causes an overlap of the kinetic curves. This is because they represent distinct physical system states and they are not chemical components.
The tool presented in the following measures the memory of the reaction compounds by scoring the overlap of the kinetic curves, following the theory of [
5]. This analysis determines physical states corresponding to the basis vectors of an invariant subspace, as we will explain in the theory section. This way to compute the minimal memory effect is advantageous for spectral-data analysis, since it helps to determine the number of compounds in the reaction. If the number of compounds is too high, two or more compounds’ kinetic curves will overlap strongly since they are assigned to the same basis vector, or a linear combination of them.
Knowing the number of reaction compounds is important not only when analyzing the datasets, but also when performing experiments. In fact, the memory measure gives insights into the quality of the sample and the noise levels of the experimental setup.
The estimation of the minimal memory effect is possible with the SepFree NMF (separability-assumption free non-negative matrix factorization), a novel method based on the
non-negative matrix factorization without separability assumption framework [
6]. The non-negative matrix factorization without separability assumption is a matrix-decomposition method already used in the analysis of sequentially measured spectroscopic data. The structure of the datasets is the spectrum, in a certain wavenumber interval, measured for different times after the excitation of the sample. Furthermore, it is applicable also to time-resolved spectra; these investigate the evolution of photo-induced phenomena, as they measure the change of the optical signal (emission, absorption, Raman scattering) as a function of time (e.g., [
7,
8,
9]).
This work uses the SepFree NMF for the analysis of the sequentially measured spectra. The method examines the sequentially measured spectrum as a matrix
M to decompose into a product of matrix
H, describing concentration proportions of the compounds as function of time, and matrix
W, which describes the peak intensities of the compounds as function of wavenumber. In particular, the SepFree NMF models the evolution of the concentrations’ proportions of the compounds in
H as a memoryless process, called the Markovian process [
6].
A transition probability matrix matrix
governs the
-evolution of the columns of
H:
The main contribution of this work is to provide a measure for the overlap between the rows of the concentration–proportion matrix H.
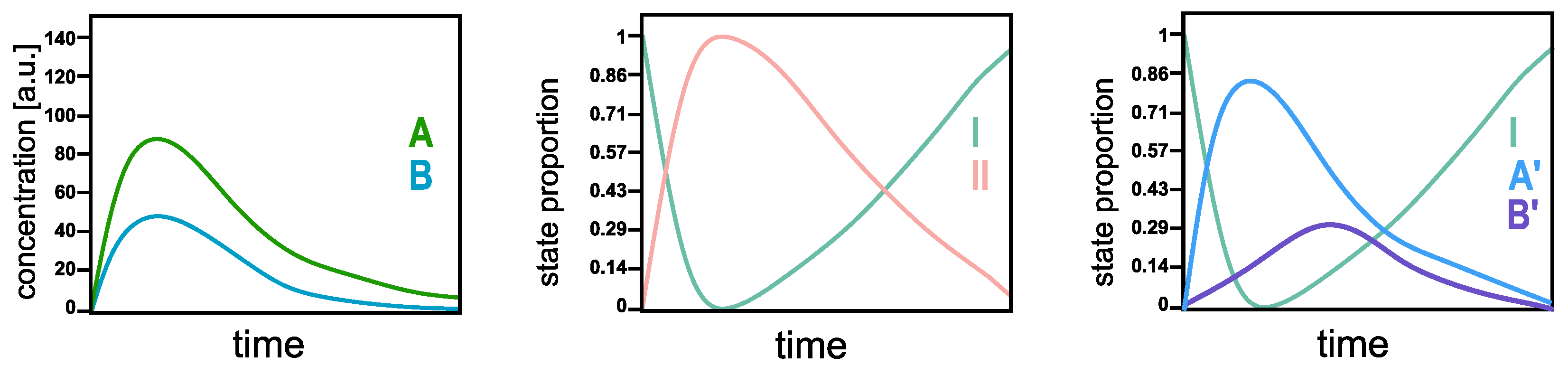
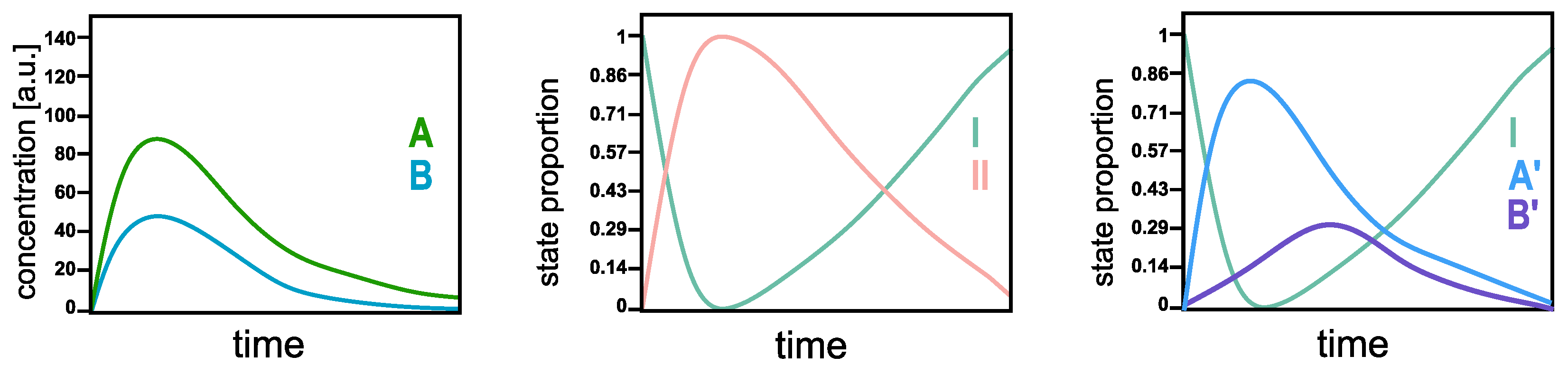
The SepFree NMF identifies the
physical states of the system, not necessarily the chemical ones. In order to understand the difference, think of a reaction in which all the molecules can react into two compounds, A and B, and then go back to the initial state. In a standard multivariate curve resolution analysis [
10], the concentration profile of the molecules in A and B evolves as in
Figure 2 left. These curves represent the concentrations of A and B in the system as a function of time; they are not between 1 and 0. By SepFree NMF of rank 2, the algorithm returns the amount of system that is either is state I or in state II. Their time development will look as in
Figure 2 middle. The first state I is the initial
physical state of the system at the beginning of the reaction; all the molecules are in this state at time
, so its proportion will be (almost) 1. During the reaction, a second state II arises. This state II represents the amount of the system that is not in the initial condition I, that is, the amount of system molecules that is in either A or B. So, being in A or B is the second
physical state of the system. By asking SepFree NMF for a decomposition into three physical compounds, the algorithm yields three states: the initial physical states, A’, and B’ (see
Figure 2 right). Although the curves in
Figure 2 right have a similar development to the concentration profiles in the left picture, their interpretation for the system is different. They represent the amount of the system that is in physical states A’ and B’, or in the initial state I, they are not concentration profiles.
This article is divided into three main parts. The first one explains the background of the SepFree NMF (
Section 2.1), and of the PCCA+ projection (
Section 2.2). The second part shows how we propose to compute the memory from the NMF results (
Section 3). In the third part, we present examples of sequentially measured Raman spectra: first for computer-generated data, then for an experiment measuring the Raman spectrum of the crystallization process of paracetamol in methanol (
Section 4).
4. Numerical Examples
These numerical examples illustrate how the minimal memory effect can be calculated from the SepFree NMF. The examined data are sequentially measured Raman spectra. One spectrum is computer generated (
Section 4.1), while the other one is an experimental spectrum (
Section 4.2).
The analyzed synthetic dataset is the same one proposed in [
14], by applying the algorithm in [
6] for the decomposition. For the experimental dataset, the algorithm is applied to a sequentially measured Raman spectrum of the crystallization process of paracetamol in methanol.
After the decomposition, every compound is labeled with a letter (A, B, C, etc.). These letters do not have any chemical meaning, but only serve as a naming convention for the data analysis routine. The labeling does not refer to the chemical meaning, and in each decomposition we can have a different order of the physical states. We will point it out in the analysis when such a situation is displayed in the figures.
Both the Python version of the algorithm to generate the synthetic data, as well as the algorithm in [
6] are available in the
supplementary materials. Furthermore, in the
supplementary material section, a tutorial explains how to use the hereby presented method for the analysis of a dataset. This version of the SepFree NMF algorithm was developed in [
18].
4.1. Synthetic Dataset
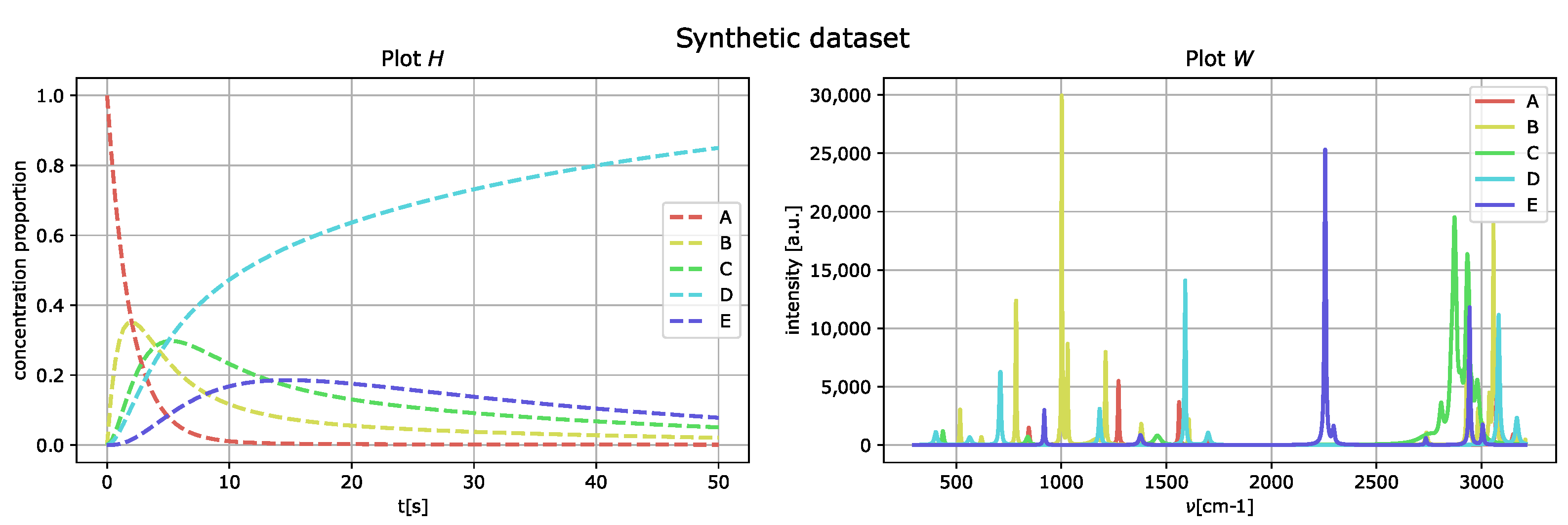
This first-order kinetics process describes the reaction of five compounds, called
A,
B,
C,
D, and
E. The rate matrix
Q that generates the kinetics is
The concentration proportions of the chemical moieties evolve in time following the rule, .
The generated peak intensities consist of Lorentzian curves in the matrix
W. The generated dataset is then given by
whereby no artificial noise is added, and the peaks of the matrix
W are well separated.
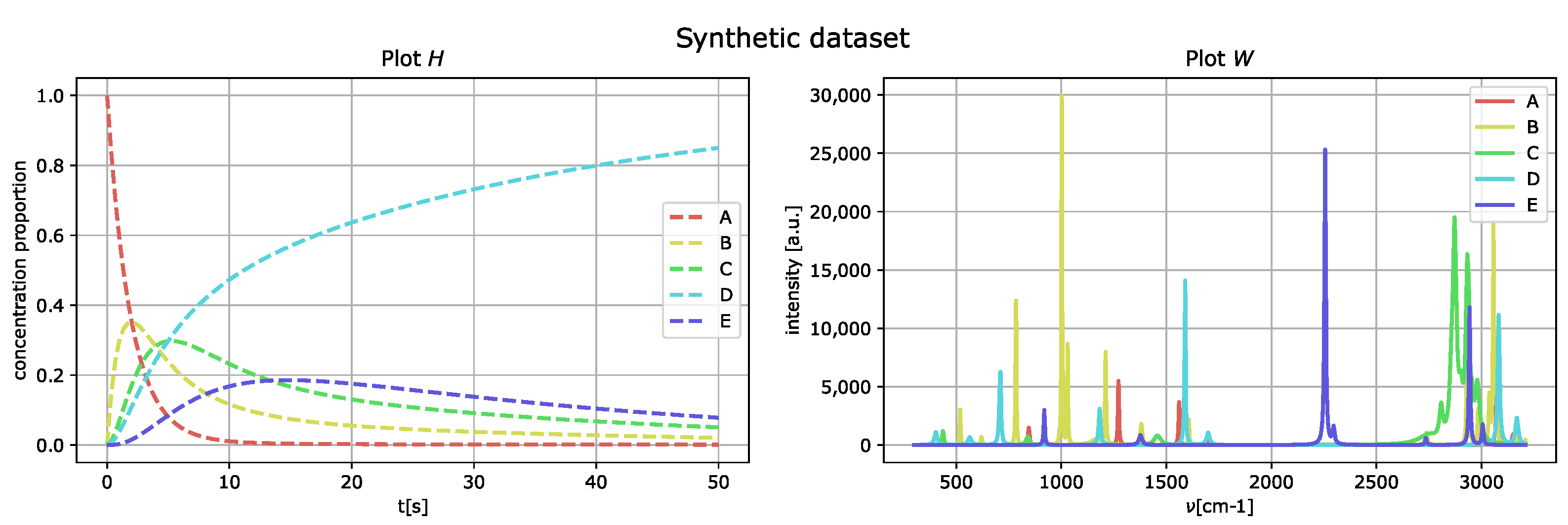
Figure 3 represents the generated spectral compounds as a function of the wavenumber
(right) and their concentration proportions as a function of delay time
t (left).
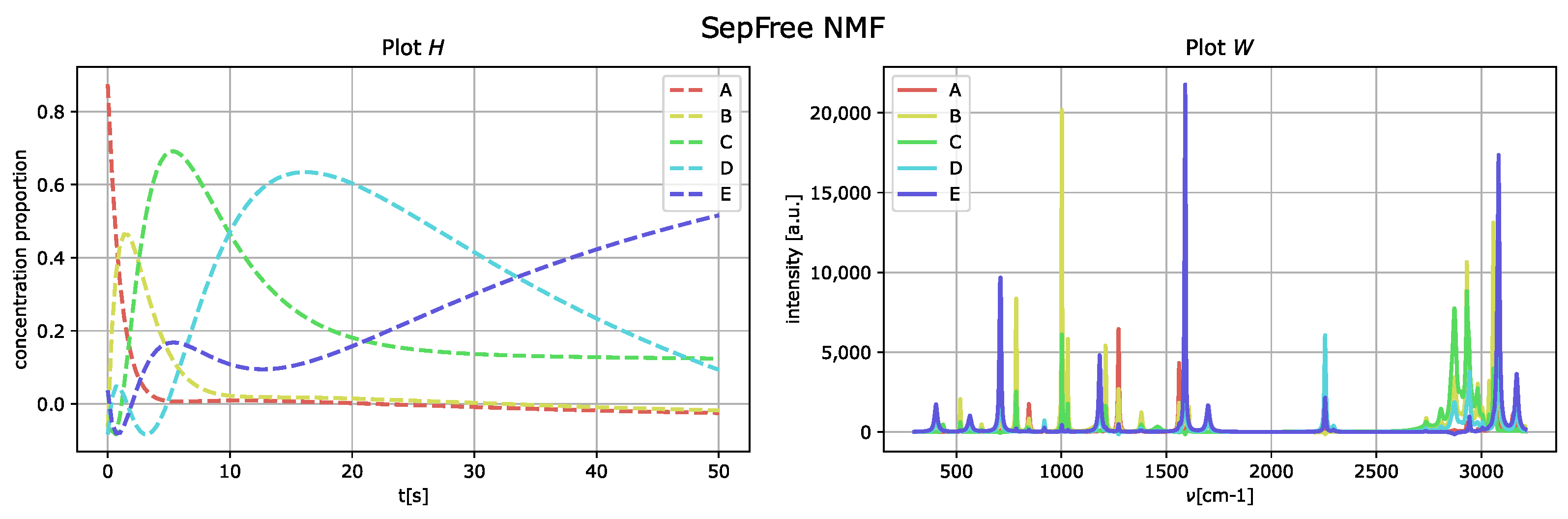
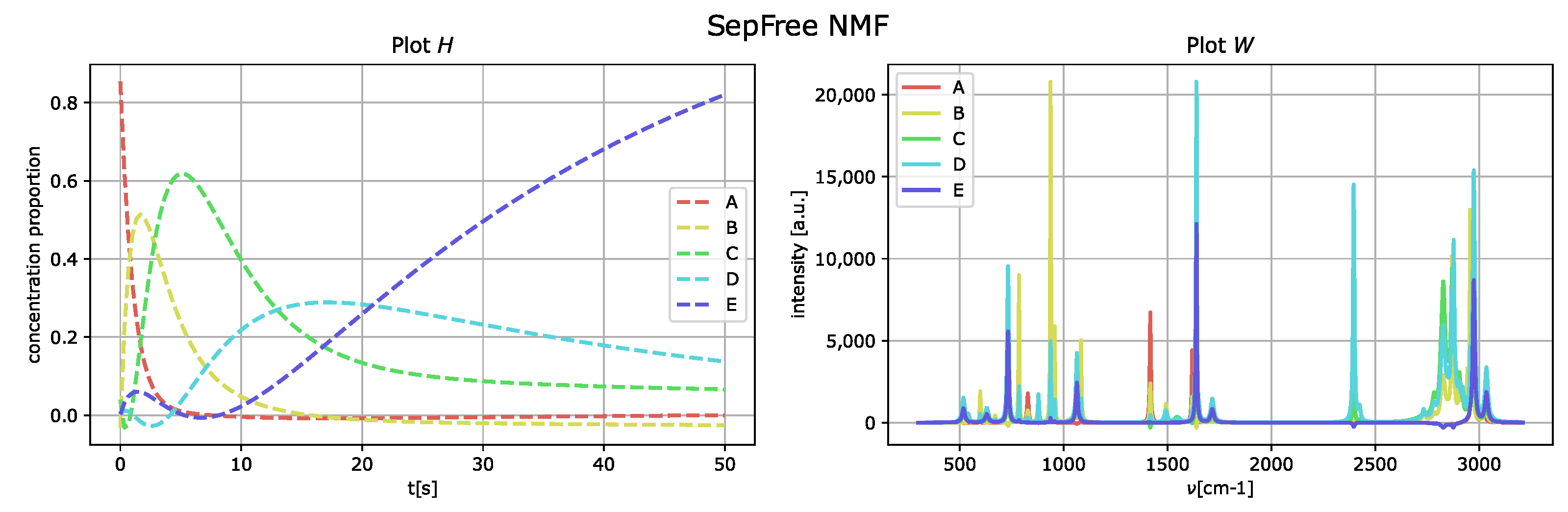
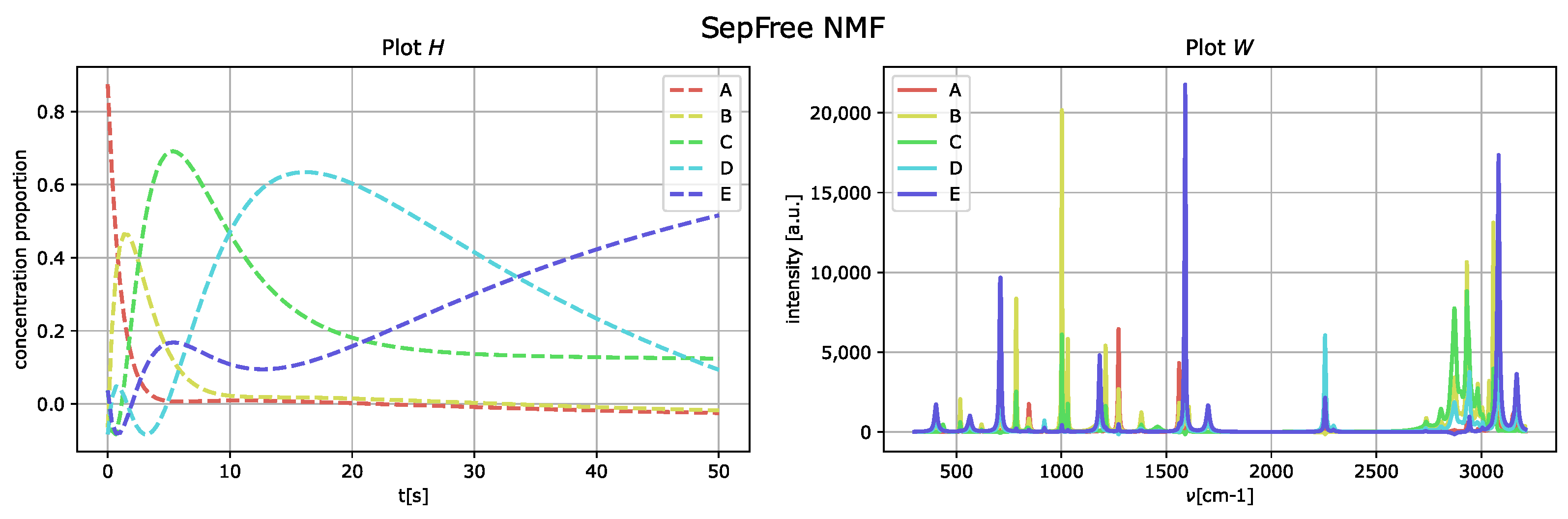
The outcome of the application of the algorithm in
Section 2.1 is presented in
Figure 4, and the parameters used for the optimization of the function
are
We set and to zero, since they are related to the computation of the Koopman transition probability matrix K. Thus, we do not need to bias the optimization of for K.
For determining the parameters for
, a heuristic method is used; see
Appendix B.
By comparing the spectral fingerprint of species
D and
E, in
Figure 4, to the spectral fingerprint of
D of the generated dataset,
Figure 3, we see that the species
E obtained corresponds to the component labeled as
D in
Figure 3.
The memory effect for this reconstruction is , which means that the memory effect for this decomposition is very strong. Additionally, the low value of the determinant is given by the fact that the reconstructed kinetic-matrix H contains some negative entries, even if they are close to zero.
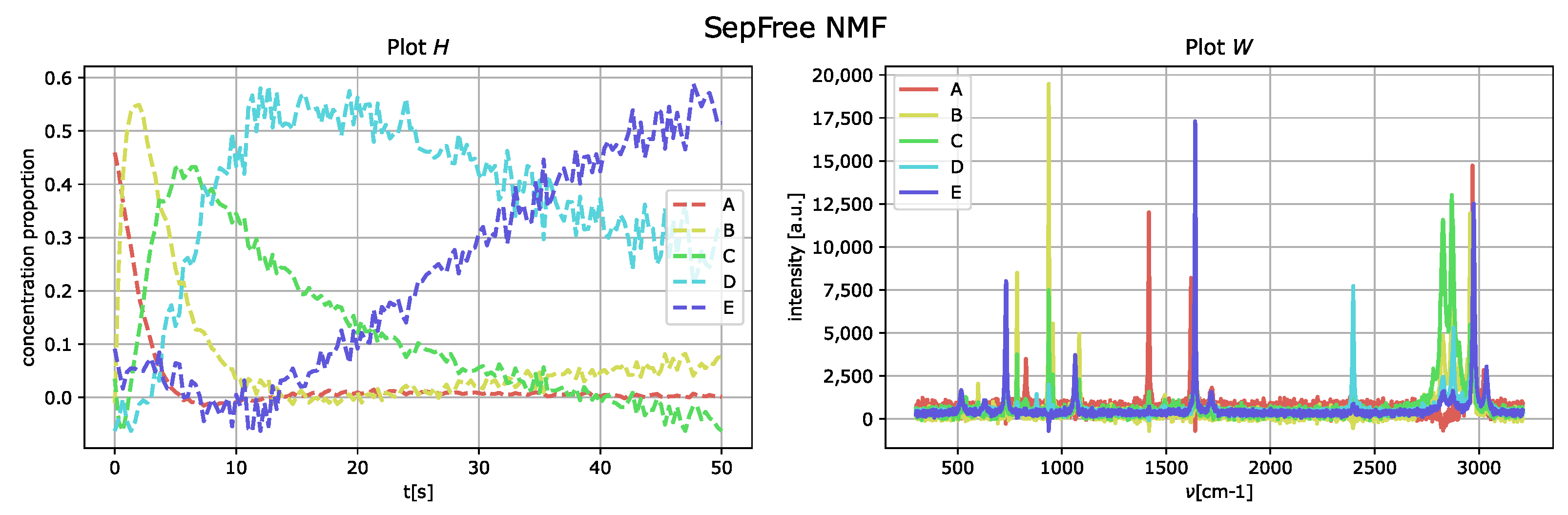
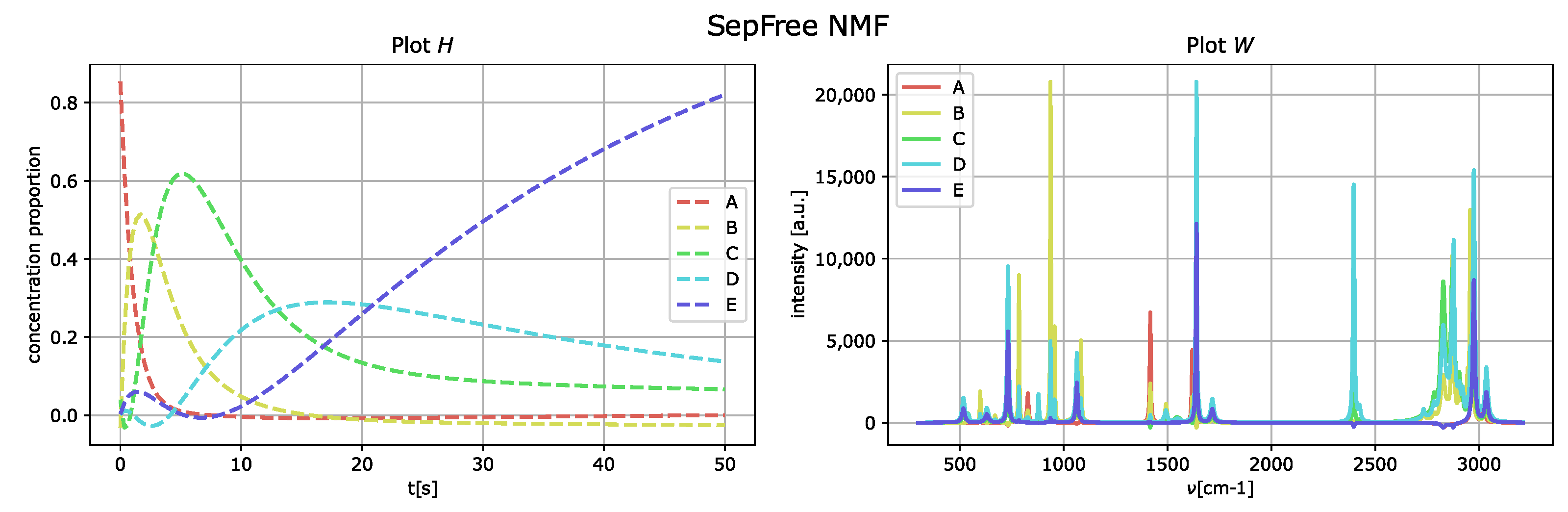
4.1.1. Synthetic Dataset with Increased Interference
To increase the interference level, the peaks of the matrix
W are moved next to each other. The results of the analysis with SepFree NMF are displayed in
Figure 5; the decomposition is computed for the same parameter set previously used. The minimal memory effect for increasing peak-interference of
is
, so the memory effect becomes twice bigger by adding interference to the data.
As in the obtained results for the data without noise nor interference, the component E corresponds to the one labeled as D of the generated dataset.
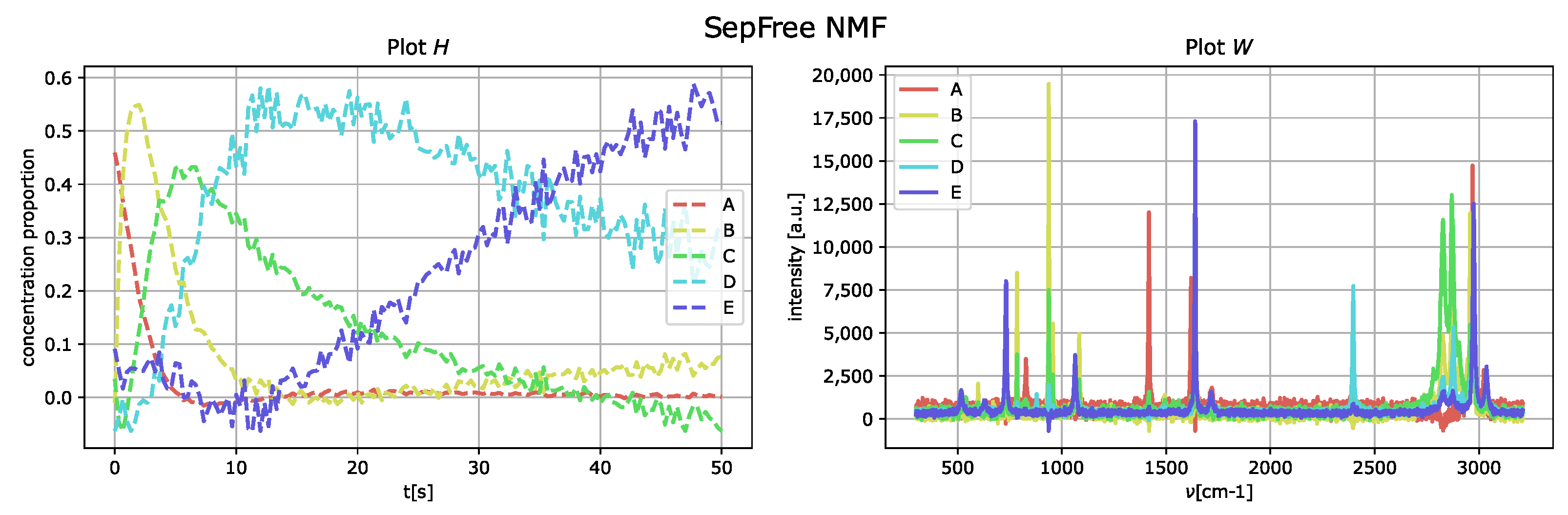
4.1.2. Synthetic Dataset with Noise and Increased Interference
In order to consider the effects of the (experimental) noise to the analysis and for the computation of the minimal memory effect, the normally distributed noise matrix
N,
is added to the data matrix
M so that
and the noise is scaled by a factor
, for this example
. The results of the decomposition are displayed in
Figure 6. As in the obtained results for the data without noise nor interference, the component
E corresponds to the species labeled as
D of the generated dataset. The minimal memory effect computed with the decomposition is 0.0005. The introduction of the noise makes a remarkable difference in the memory properties of the spectrum. In comparison to the dataset with well-separated peaks and no additional noise, the minimal memory effect is four times bigger.
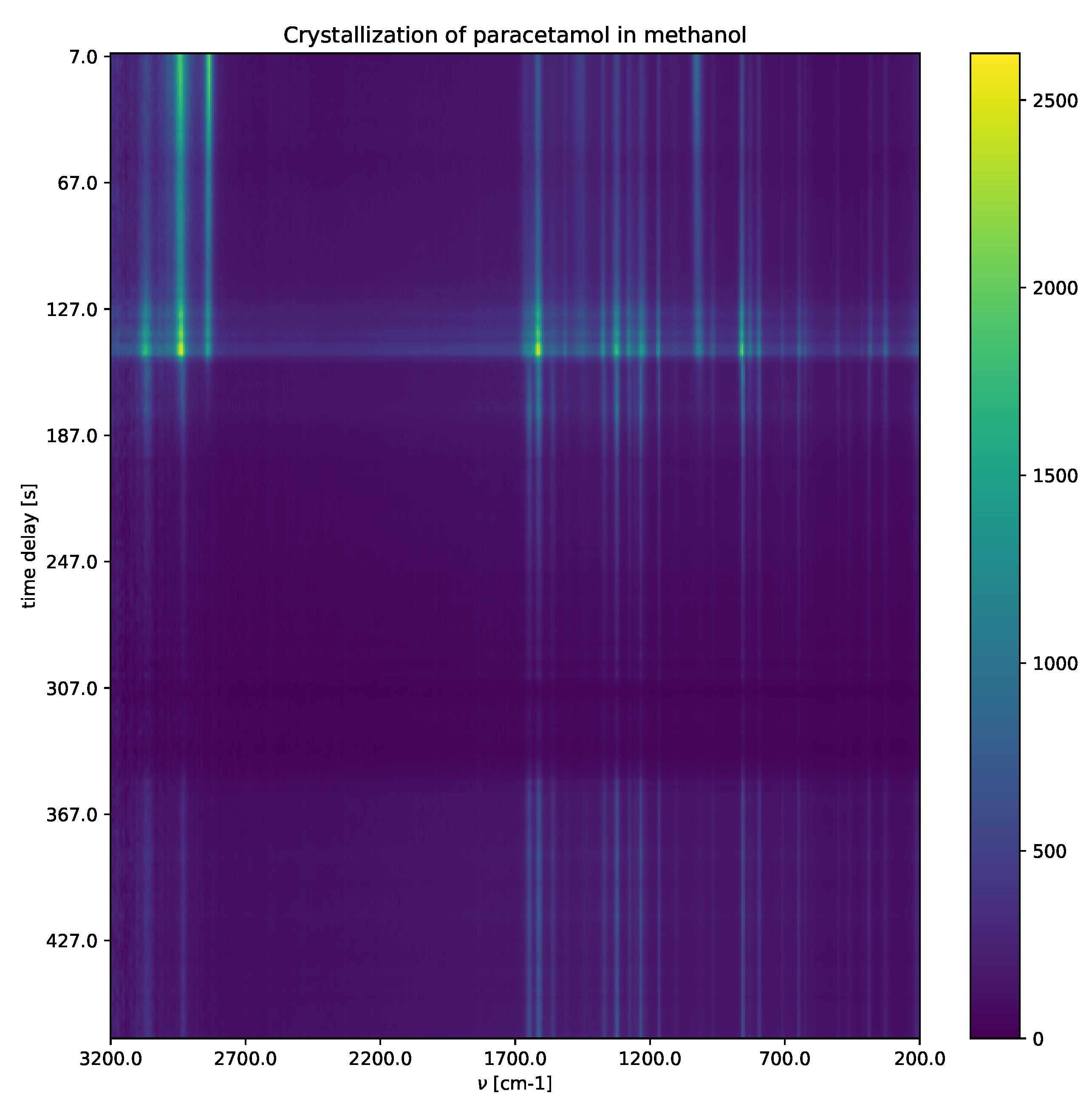
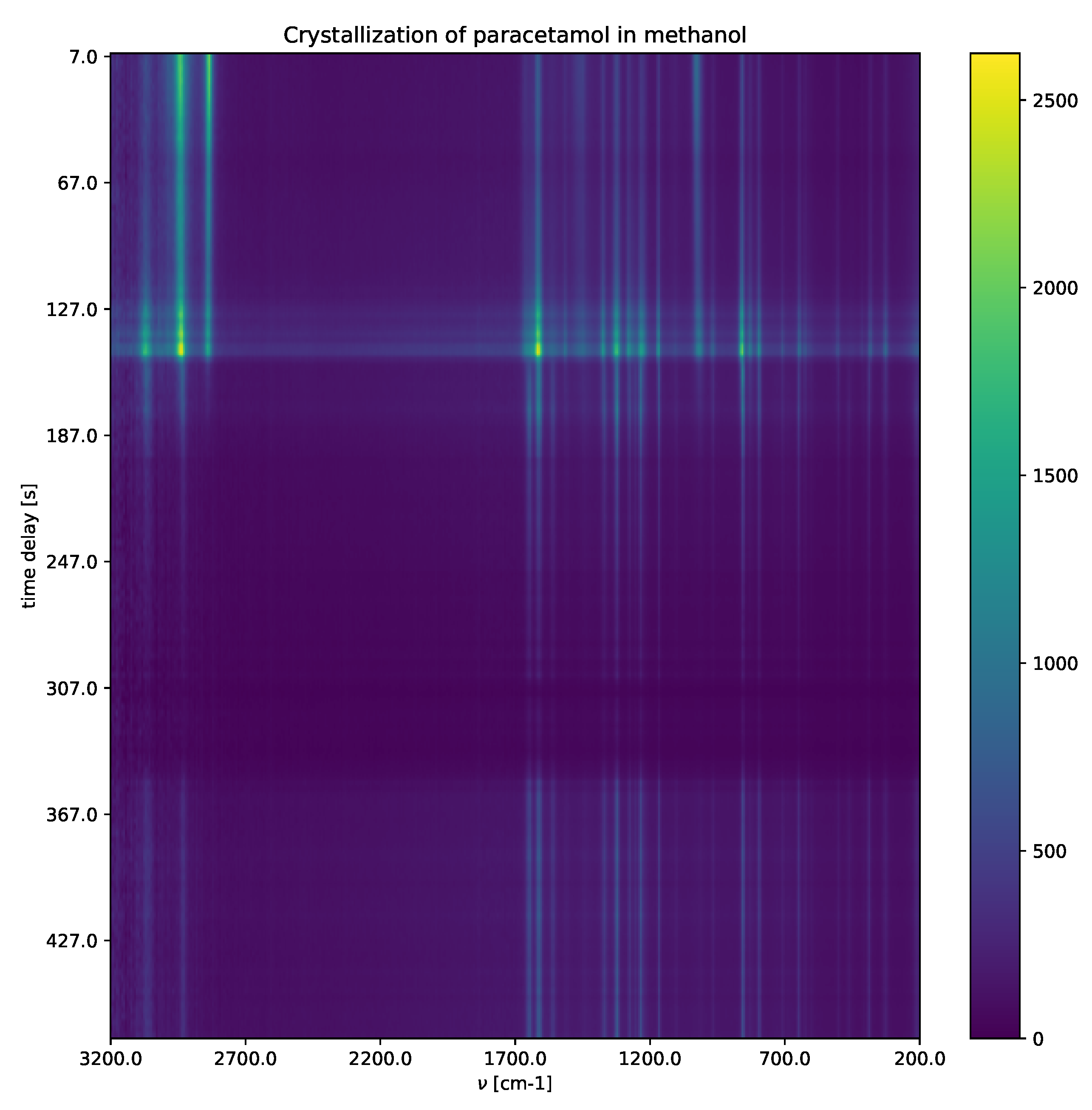
4.2. Raman Experiment-Crystallization of Paracetamol in Methanol
In this example, we show that the determinant of
, defined in (
12), is an effective measure for the physical meaningful number of compounds.
Crystallization of paracetamol in methanol (
Figure 7) shows a solvated paracetamol in methanol, a metastable intermediate phase, and a crystalline phase. For detailed explanations, including the experimental settings, see
Appendix A. Raman shifts characterize different paracetamol phases (solution phase, metastable intermediate phase and form II). After applying the SepFree NMF method, the role that the obtained compounds have in the reaction can be identified by the position of the peaks in matrix
W.
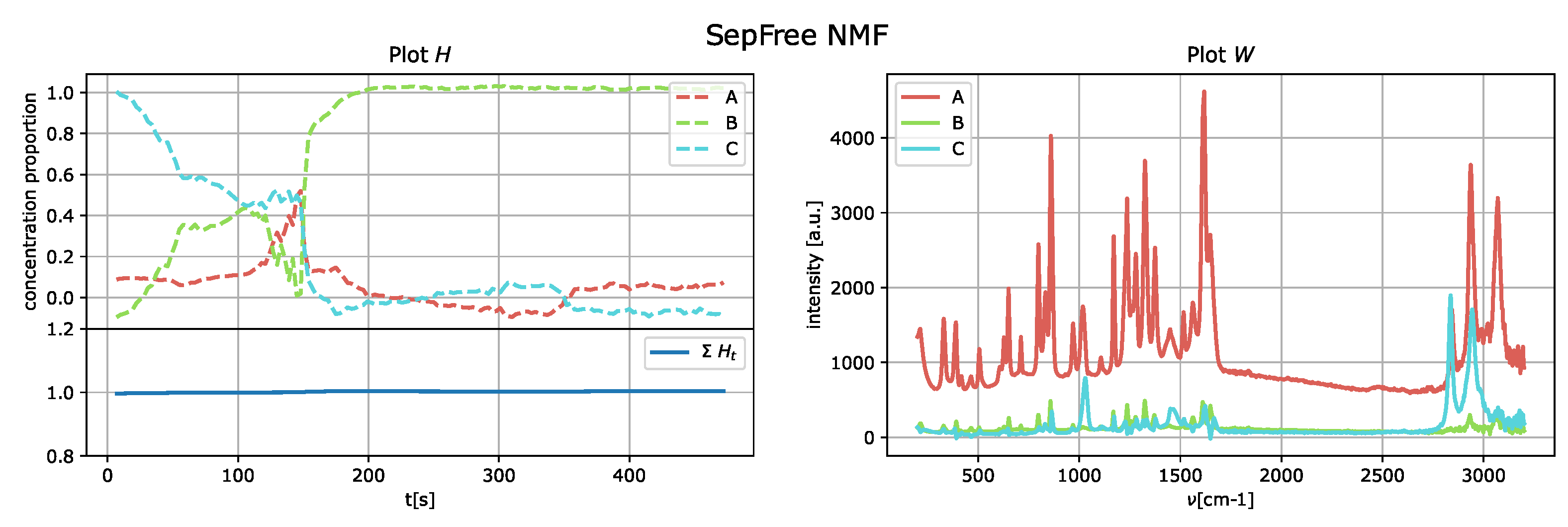
Firstly, the singular value decomposition of the sequentially measured unprocessed spectral data taken as matrix suggests that the number of compounds can probably be . Thus, SepFree NMF is tested then for these r values. The parameters used for the objective function are , , , , .
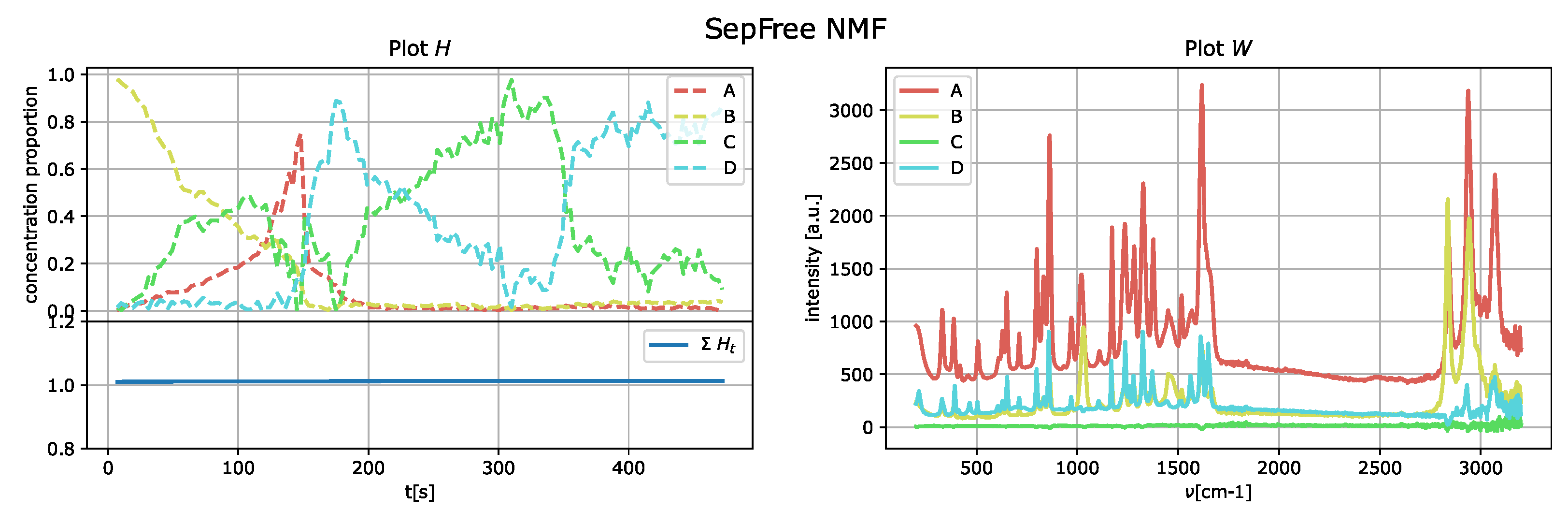
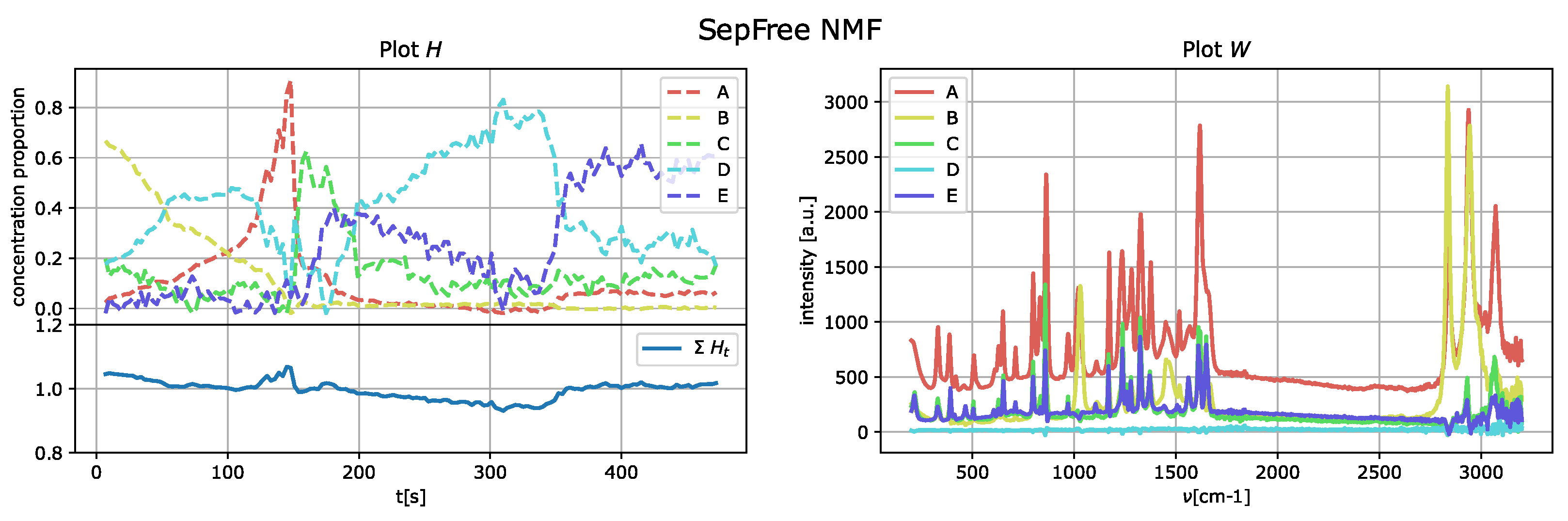
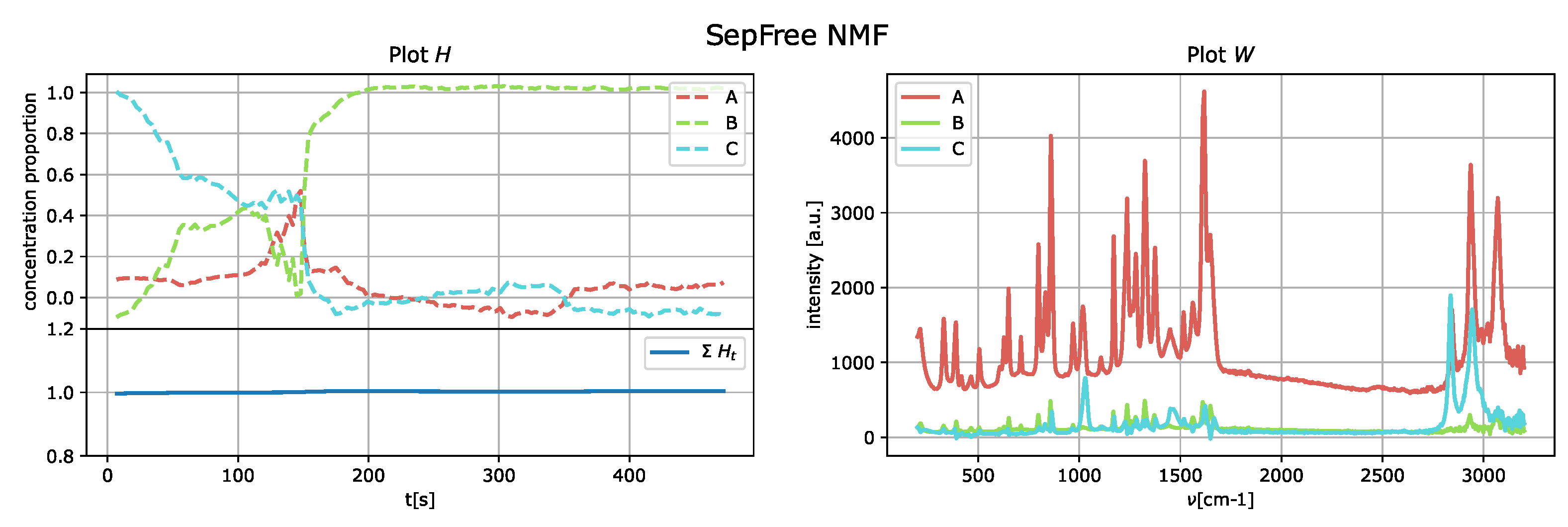
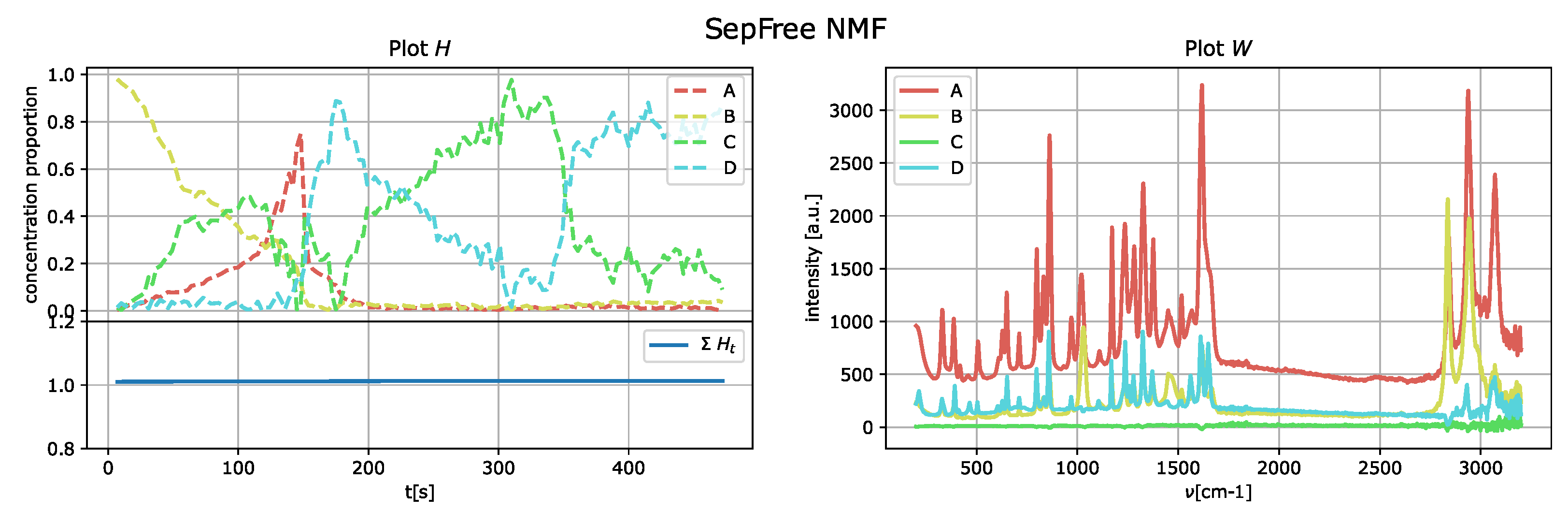
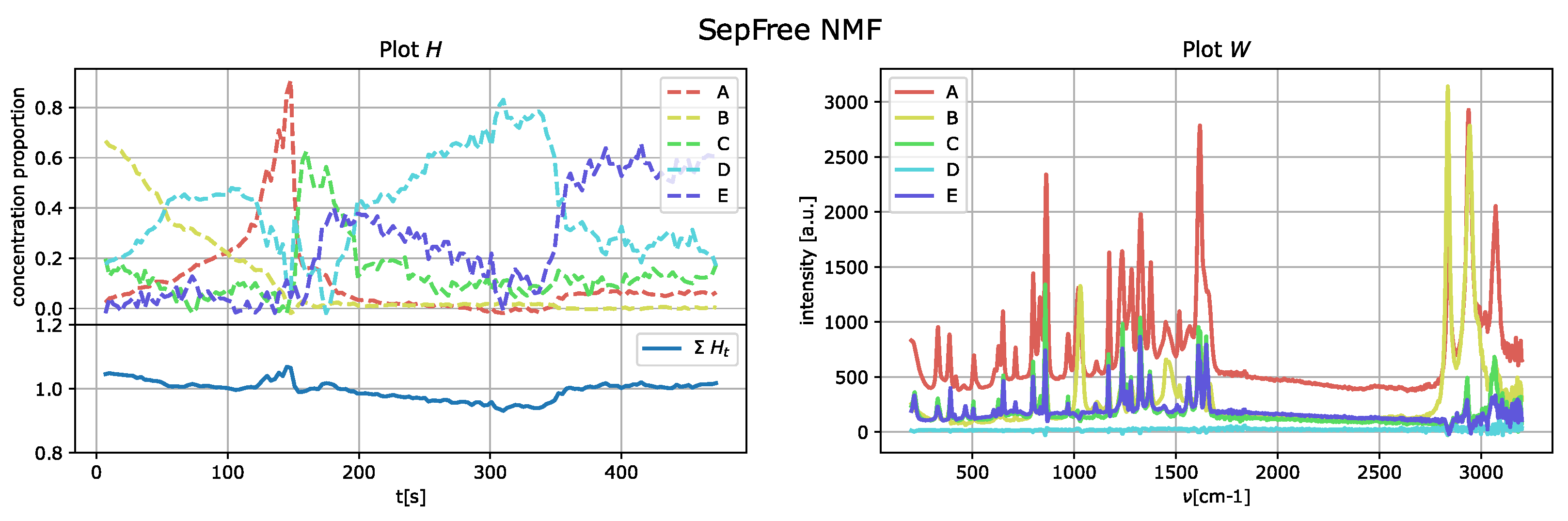
The results of the application of the SepFree NMF are shown in
Figure 8 for
, in
Figure 9 for
, and in
Figure 10 for
. In each figure, the left plot shows the row of the matrix
H as function of the delay time
t; the right plot displays the peak intensities of
W as function of the Raman shift (cm
−1). The sum of the relative concentrations
is also plotted on the left below, since the sum of the compounds can change. This change is due to variations of state occurring in the reaction, which modify the relative concentrations of the paracetamol moieties.
The decomposition with
and
compounds show kinetic curves in which the state’s proportion rises, decreases and rises again, as it can be seen in
Figure 9 and
Figure 10. This pattern cannot explain the state of a compound in a crystallization process. In contrast, the decomposition in
Figure 8 for
describes a sequential kinetic process, as expected for a crystallization.
Can we estimate the number of reaction compounds only by looking at ?
This question can be answered with
yes by
Table 1. In this table, the values of
for
are given. It can be noticed that for
the determinant has the highest value, in accordance with the above considerations on the kinetics.
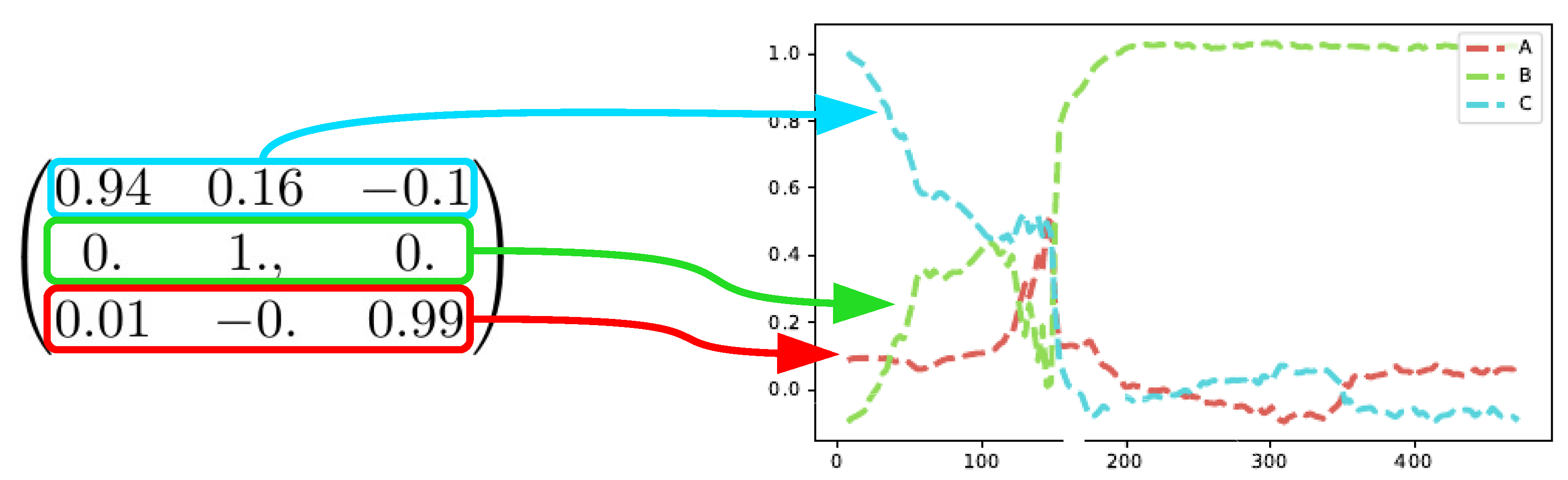
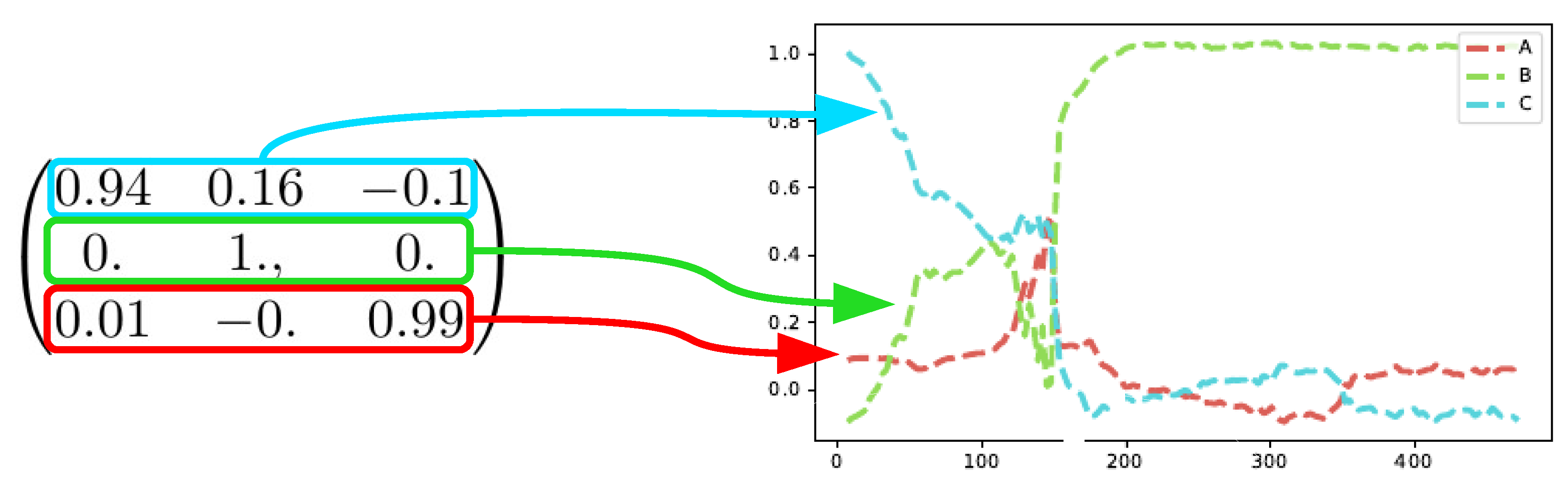
The SepFree NMF yields also a transition matrix
for the process [
6]. Examining the transition matrix allows to understand the kinetic model underlying the reaction mechanism [
18]. The crystallization process of this dataset is a sequential process.
The matrix
resulting from the decomposition
shows also a sequential process
and it shows that
B is an absorbing state of the Markovian process. This means that when the process reaches
B, it does not leave that condition anymore, as also shown in
Figure 11. The first compound for the analysis of the matrix
is
C since
C is the most abundant at time
, see
Figure 8.
In the
supplementary materials, we provide a similar analysis for the decomposition of ranks 4 and 5, showing that the kinetic model described is not sequential.
Recognizing the chemical moieties from the Raman shifts is advantageous for validating the number of compounds estimated with the minimal memory effect, as shown in the following analysis. To show that the results given by the decomposition of rank 3 are indeed in agreement with the experiment, we plot each peak intensity of
W (
Figure 8) with the peaks of form II and metastable amorphous form II. The values of the Raman peaks are reported in the
supplementary materials.
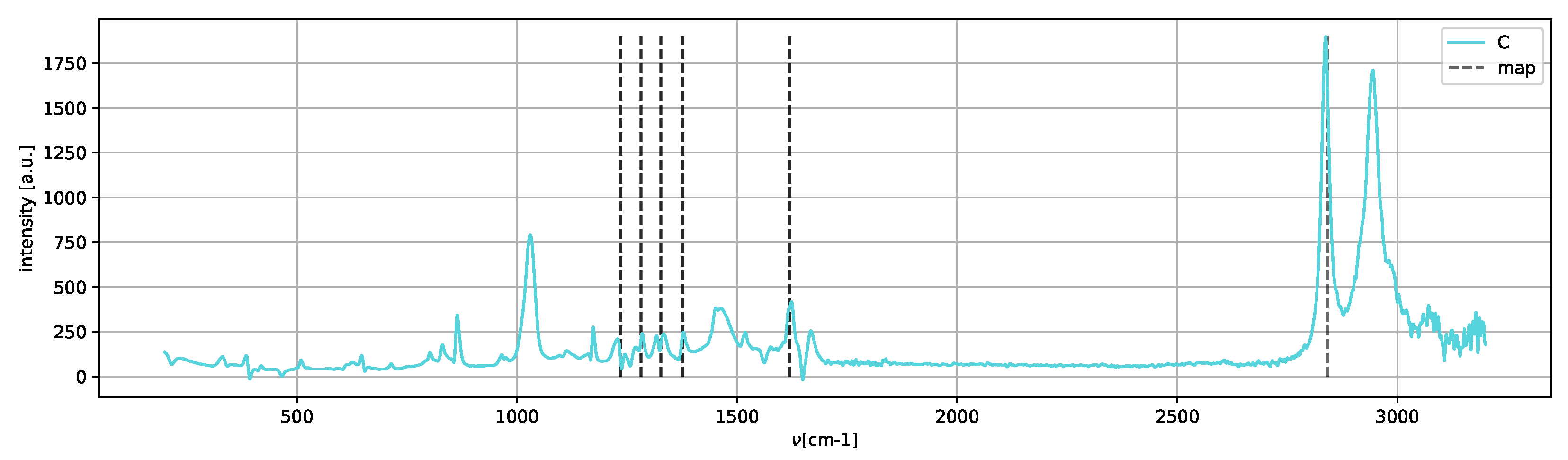
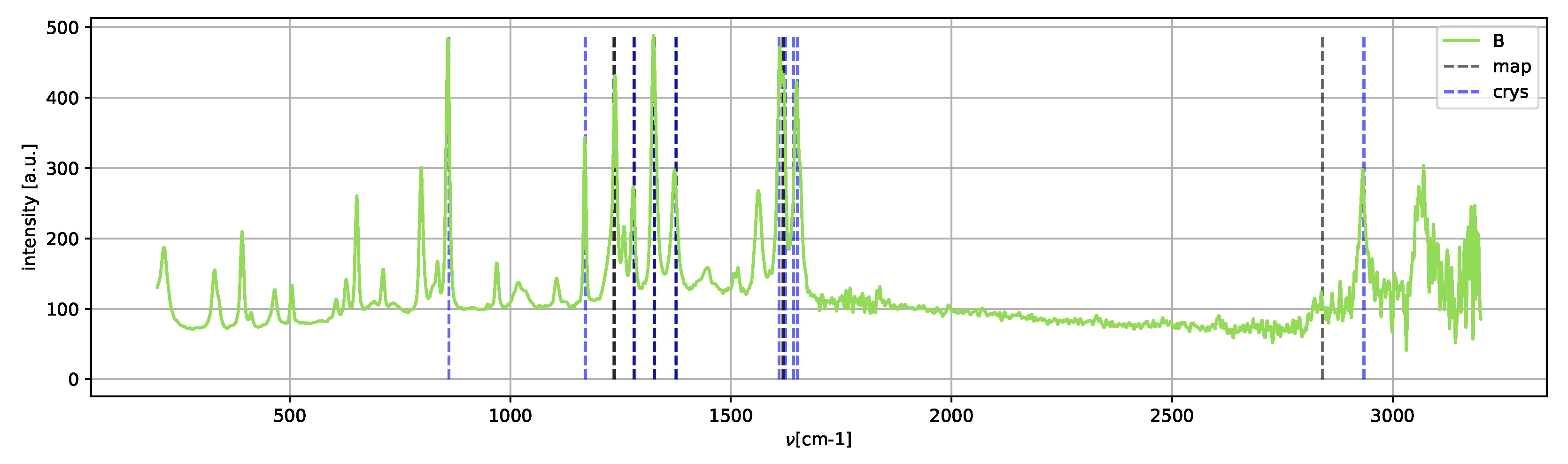
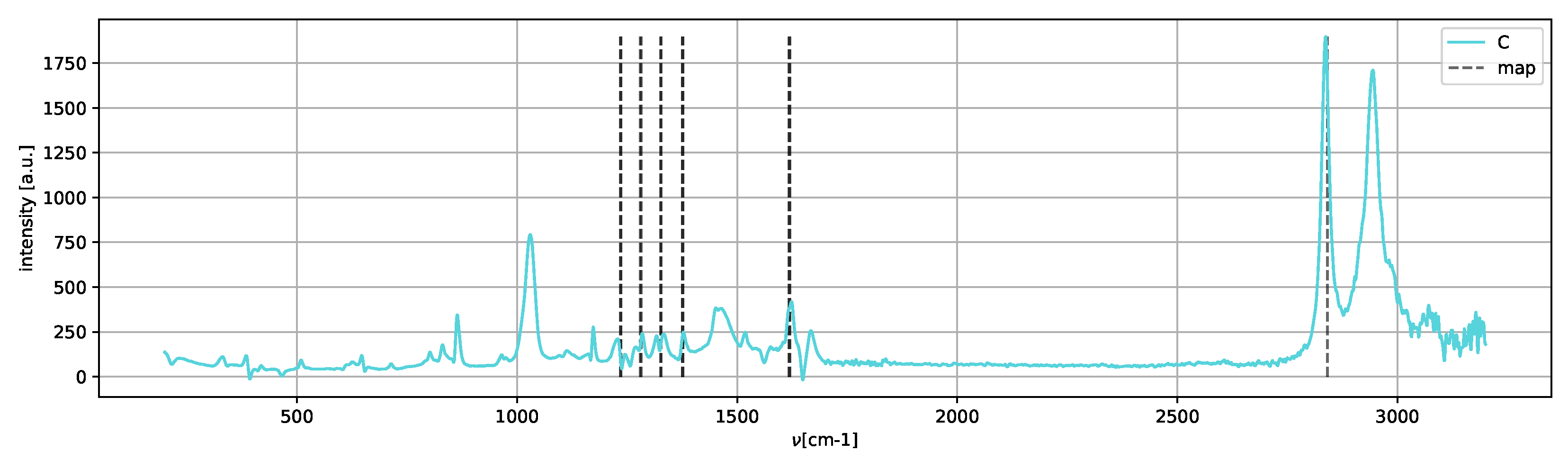
Spectrum
C (
Figure 12) shows only the peaks of the metastable amorphous phase and two clear peaks in the 2800–3000 cm
region, which can be assigned to methanol [
25].
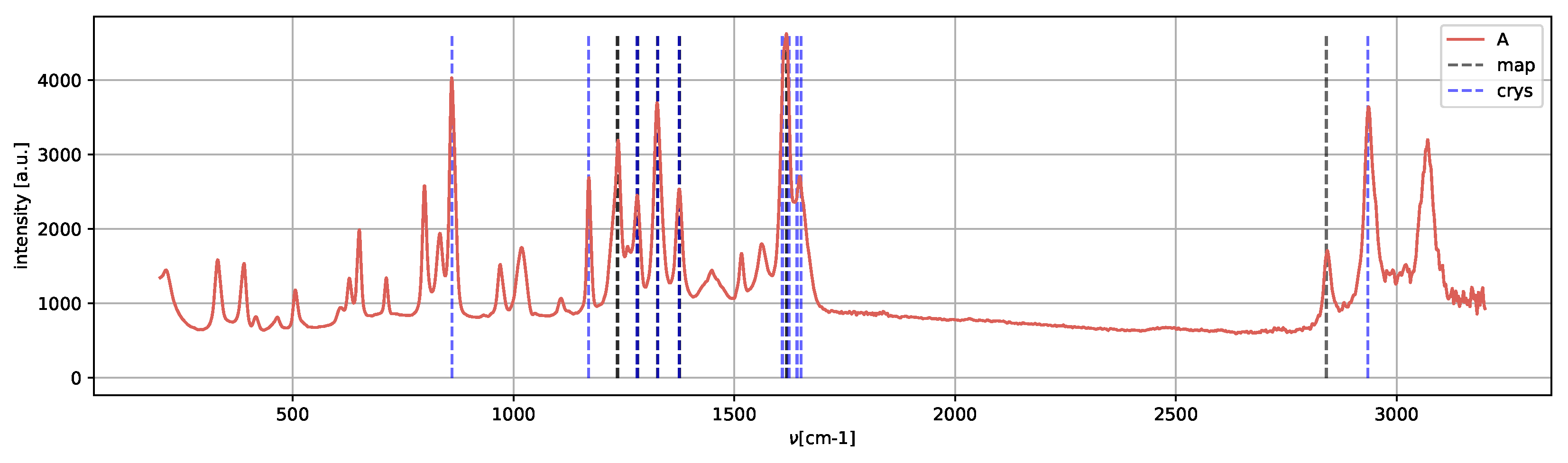
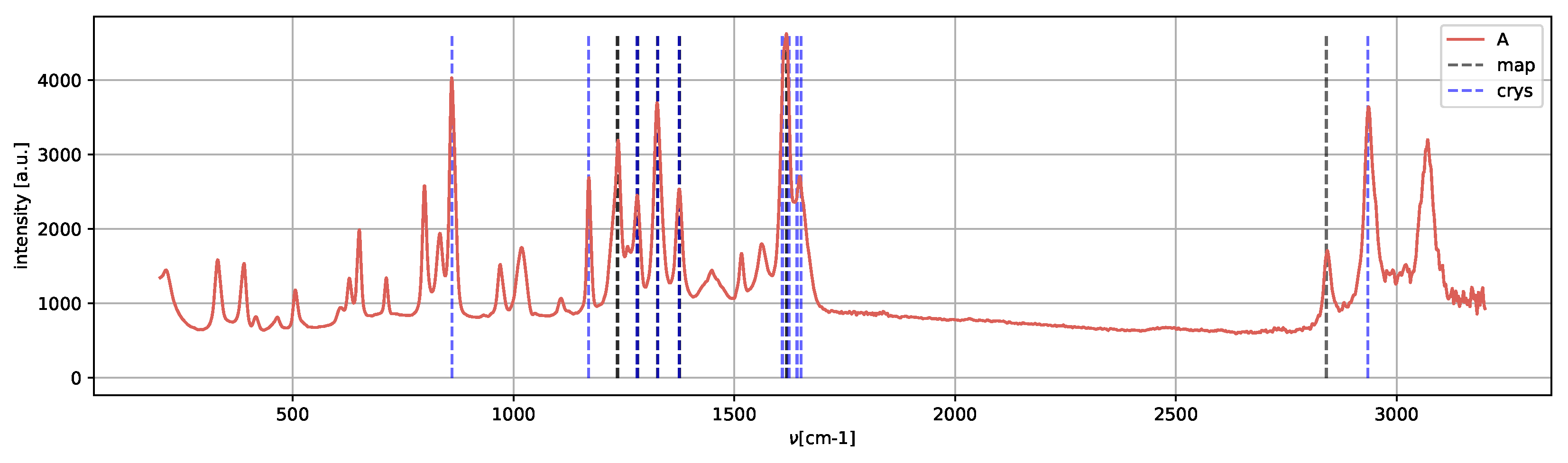
Spectrum
A (
Figure 13) presents the peaks characteristic of the metastable amorphous phase and solvent, as methanol Raman shifts are still present, in agreement with [
25].
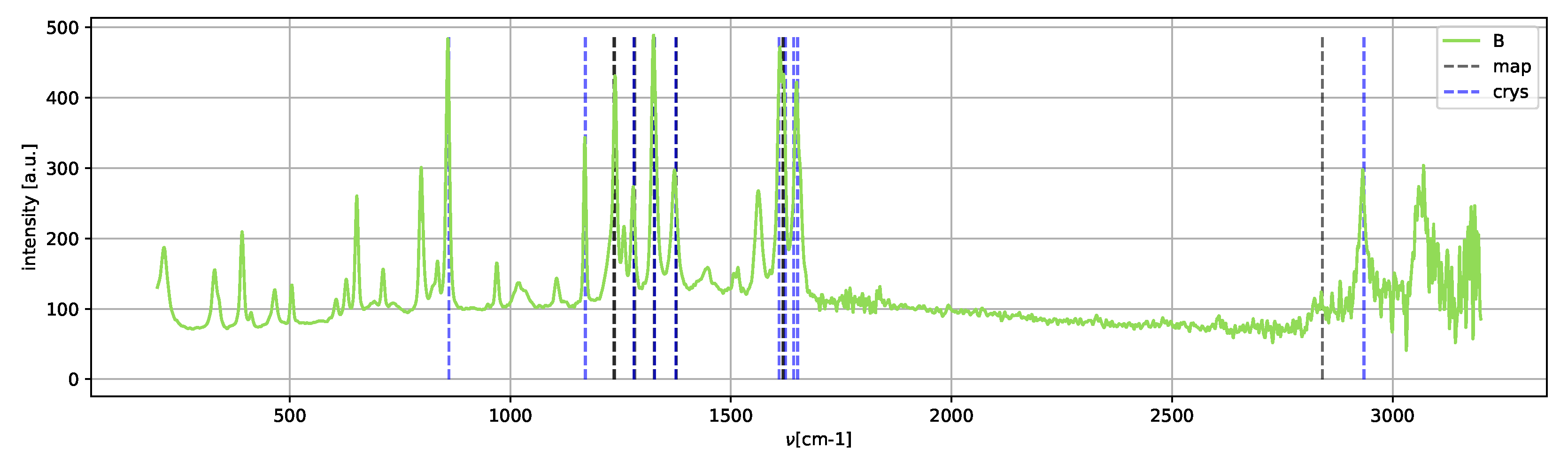
Spectrum
B (
Figure 14) shows the peaks of the crystallized form II; neither Raman shifts of the metastable amorphous phase nor methanol shifts are clearly present.
In conclusion, the decomposition analysis reveals three stages of the reaction:
In this way, only by computing the minimal memory effect, we obtain a decomposition (a) that describes the correct kinetic process, (b) whose spectral intensities have a precise and clear interpretation.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}