1. Introduction
Ribosomes perform protein synthesis from mRNA templates by a highly regulated process called translation. Translation control plays a key role in the regulation of gene expression, both in physiological and pathological conditions [
1].
The advent of high–throughput methods to measure the levels of gene expression has revealed the implications of multiple factors that might impact the rate at which an mRNA is translated. In recent years, the Ribosome profiling technique (Ribo–seq) has emerged as a powerful method for globally monitoring the translation process in vivo at single nucleotide resolution [
2]. The application of this method to a different number of organisms subjected to different conditions, from the deprivation of nutrients in bacterial cells to the development of cancer in human cells, has allowed to investigate fundamental aspect of cell biology [
3].
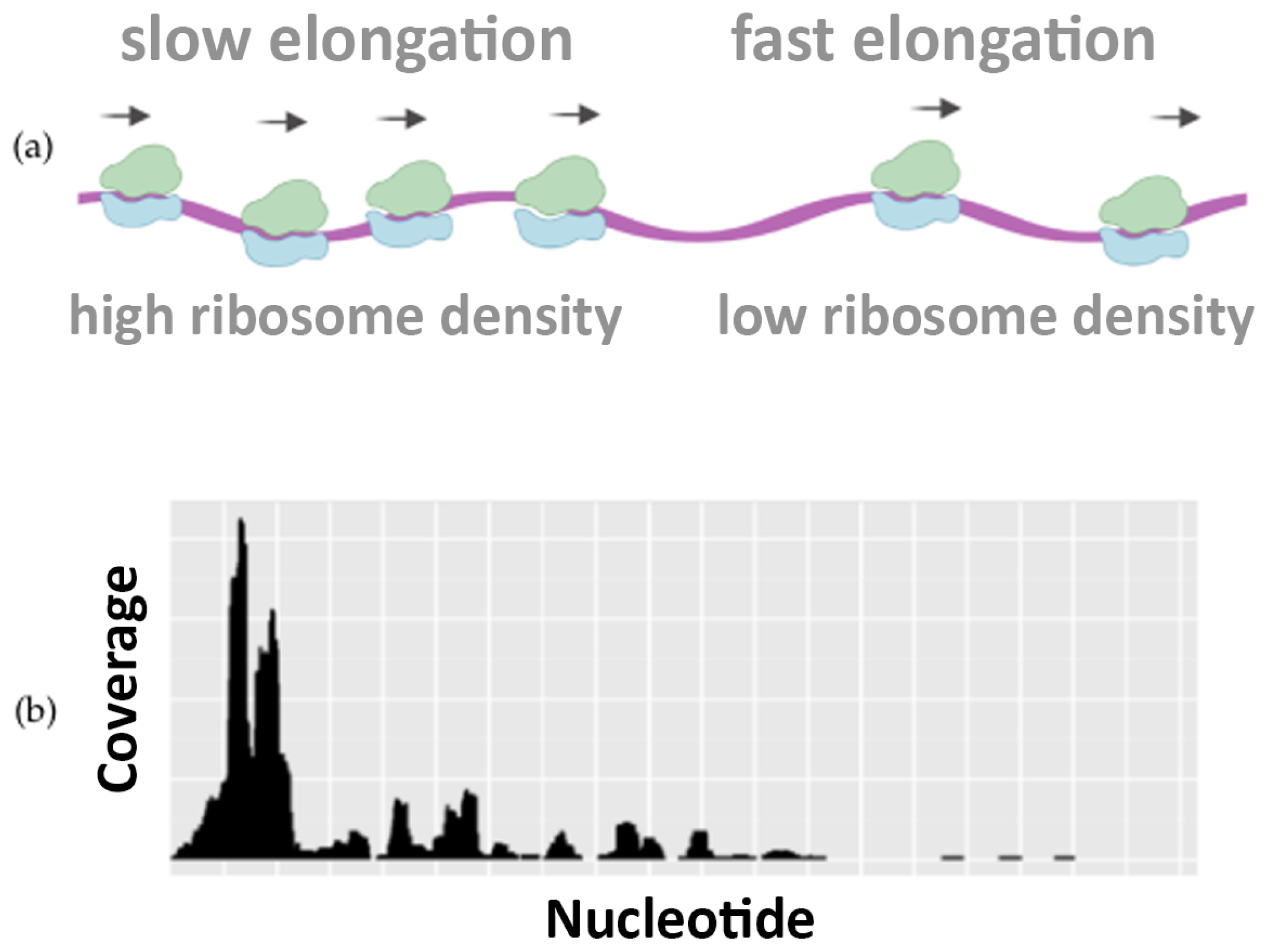
Interestingly, the nucleotide–level resolution of Ribo–seq experiments reveals the density of the ribosomes at each position along the mRNA template. Local differences in the density of Ribosome Protected Fragments (RPFs) along the Open Reading Frame (ORF) reflect differences in the speed of translation and elongation, determining regions where the translation is slower or faster.
Figure 1 illustrates how the translation speed is not uniform, highlighting the differences in ribosome occupancy. This piece of information is well visible in Ribo–seq profiling data and can be used to infer how the codon usage, the protein sequences, and other features can regulate the speed of translation [
4].
Unfortunately, the reproducibility of Ribo–seq experiments can be affected by multiple variables due to the complexity of the experimental protocol and the lack of standardization in computational data analysis [
5].
Thereofore, our work aims to overcome the aforementioned limitations by introducing a new statistical approach, designed to extract a set of highly reproducible profiles.
In particular, inspired by the seminal work proposed in [
6], we perform a novel analysis procedure for Ribo–seq data that allows to identify the reproducible Ribo–seq profiles emerging from the comparison of independent Ribo–seq experiments performed in different laboratories under the same conditions. These significantly reproducible profiles are then collected into a library of consensus sequences, in which sub-regions characterized by different translation speeds can be isolated. The aforementioned procedure has been applied to
E. coli sequences (
Escherichia Coli (E. coli) is a bacterium that lives in the lower intestine of warm-blooded animals and has a genome composed by approximately 4,600,000 base pairs. E. coli contains a total of 4288 genes, with coding sequences which are long, on average, 950 base pairs and separated, on average, from 118 bases. Considering the protein counterpart, in E. coli, the average length of a coding region is 316.8 codons, whereas less than 1.8% of the genes are shorter than 60 codons.), resulting in 40 highly reproducible profiles.
Differently from [
6], based on the collected data, a statistical analysis has been carried out that gave new insights on the dynamics of the ribosome translation, showing a statistically significant difference in the nucleotide composition between sub-sequences characterized by different translation speeds. Moreover, to validate the procedure, the selected highly reproducible profiles have been analyzed through state-of-the-art Machine Learning (ML) models, accurately classifying subsequences according to their speed of translation (slow or fast).We have made our source code public available (
https://github.com/pandrein/Ribo-Seq-analysis, accessed on 29 June 2022). Furthermore, these experiments allowed to discover that the translation speed is modulated both by the nucleotide composition of the sequences and by the order in which they appear within each sequence.
The rest of the paper is organized as follows:
Section 2 collects works from the literature dealing with related topics;
Section 3 describes the ribosome profiling data extraction and preprocessing, together with their analyses based on both statistical and ML methods;
Section 4 summarizes the results, discussing their meaning and their biological interpretation. Finally,
Section 5 draws some conclusions and derives future perspectives.
2. Related works
The Ribosome profiling approach offers a promising method for developing unbiased translation models from data, but the quantitative analysis of ribosome profiling data is challenging, because of high measurement variance and the inability to distinguish the ribosome rate of translation. The Ribosome profiling strategy based on deep sequencing of ribosome–protected mRNA fragments enables genome–wide investigation of translation at the codon and sub–codon resolution [
7].
In recent years, techniques based on machine learning have been employed with increasing frequency and intensity in many different fields, ranging from computer vision [
8,
9,
10,
11] to natural language processing [
12,
13,
14] and bioinformatics [
15,
16]. The popularity of these approaches stems from their success in the automatic inference of complex functions directly from the data. In particular, both statistical and ML approaches have been successfully applied to non–biological
sequential data classification tasks [
17,
18,
19], in which each sequence is associated with a class label and the classification is performed on the whole sequence. Within bioinformatics, examples of ML applications include the prediction of splicing patterns and protein secondary structures, protein–protein interface prediction, protein subcellular localization, drug side-effect prediction, and DNA/RNA motif mining, to name just a few [
20,
21,
22,
23]. Moreover, ML and deep learning approaches have been used to process Ribo–seq data for gene annotation in prokaryotes [
24], to predict ribosome stalling [
25] and for micropeptide identification [
26]. In particular, in [
27], a deep learning based approach, called RiboMIMO, was proposed, based on a multi-input and multi–output framework, for modeling the ribosome density distributions of full-length mRNA Coding Sequence (CDS) regions. Through considering the underlying correlations in translation efficiency among neighboring and remote codons, and extracting hidden features from the input full-length coding sequence, RiboMIMO can accurately predict the ribosome density distributions along with the whole mRNA CDS regions, a problem strictly correlated with the one we intend to face in this paper. Indeed, we propose a machine learning-based approach to validate the extraction procedure of highly reproducible profiles, by classifying the translation speed of the extracted regions, which deeply depends on the ribosome density along mRNA.
3. Materials and Methods
In this work, a new software, written in Python, has been developed, which reproduces the procedure described in [
6]. In this section, the method used to obtain a set of reproducible Ribo–seq profiles and their analysis through statistical and ML methods are presented. In particular, in
Section 3.1, the procedure employed to extract the profiles is described, while in
Section 3.3, a statistic analysis of the nucleotide composition is presented. Finally, in
Section 3.4, two different ML approaches are proposed to analyze the data and asses their quality.
3.1. Ribosome Profiling Data Extraction
The Ribosomal profiling technique (Ribo–seq) is currently the most effective tool to study the protein synthesis process in vivo. The advantage of this method, over other approaches, lies in its ability to monitor translation by precisely mapping the position and number of ribosomes on an mRNA transcript. Ribo–seq involves the extraction of mRNA molecules associated with ribosomes undergoing active translation and a digestion phase, during which the RNase enzyme processes all the RNA molecules, with the exception of the “protected” parts, to which ribosomes are attached. This step is followed by rRNA depletion and preparation of the sequencing library as in an RNA–seq approach. However, since reads are obtained relating only to actively transcribed mRNA molecules, Ribo–seq better reflects the translation rate than mRNA abundance alone, although it requires particularly laborious preparation and is only applicable to species that have a reference genome available. The strategy employed in our work consists in the identification of high resolution Ribo–seq profiles through the systematic comparison of Ribo–seq datasets referring to experiments performed independently in different laboratories and in different time periods.
In particular, the approach is composed by the following phases:
Preprocessing—the ORF–specific ribosome profiling data from multiple datasets are collected and then processed by a bioinformatic pipeline;
Signal digitalization—Ribo–seq profiles are digitalized by associating to each nucleotide a slow or fast label;
Comparison of digital profiles—Digital profiles are used to quantify similarities and differences between Ribo–seq profiles of different datasets referring to the same ORF.
Identification of significantly reproducible Ribo–seq profiles—A set of highly reproducible profiles is obtained and, among them, reproducible sub-sequences are identified.
3.1.1. Preprocessing of Ribosome Profiling Data
To illustrate the statistical procedure employed in this work, we analyse a set of
E. coli Ribo–seq profiles. For this purpose, the data stored in the Gene Expression Omnibus [
28] repository were used. Specifically, our analysis concerns a systematic comparison of Ribo–seq profiles, each belonging to a different GEO series (A
series is a collection of datasets that include at least one group of data—
sample—from Ribo–seq experiments performed on
E. coli in various conditions according to the most used experimental protocol.), referring to experiments performed culturing wild–type E. coli strains under control conditions. In particular, our analysis regarded a subset of eight samples (labelled from Dataset 1 to Dataset 8) obtained through experiments characterised by K–12 MG1655 genotype and cultured in a MOPS–based medium.
Table 1 reports the GEO Series ID and GEO sample ID of raw Ribo–seq dataset used in this experiment.
To reconstruct the Ribo–seq profiles starting from raw Ribo–seq data, represented by the FASTA format (The FASTA format is a text–based format for representing nucleotide sequences in which base pairs are indicated using single-letter codes [A,C,G,T] where A = Adenosine, C = Cytosine, G = Guanine, T = Thymidine), the reads are mapped against the whole set of coding sequences in
E. coli, taken from the EnsemblBacteria database [
37]. Then, we extracted and counted the number of reads mapping to each gene from the SAM alignment file (The Sequence Alignment Map (SAM) is a text–based format originally used for storing biological sequences aligned to a reference sequence.) using BEDTools [
38]. The genomic coordinates are stored in a BED file (The BED file is a tab-delimited text file used to store genomic regions where each feature is described by chromosome, start, end, name, score, and strand.) to build the Ribo–seq profiles representing the input of the subsequent analysis. Each ORF can be associated to a specific Ribo–seq profile, a histogram that counts the number of reads that cover each nucleotide position. To realize the pairwise comparison of ORF–specific Ribo–seq profiles coming from independent datasets, we decided to proceed as described in the following.
3.1.2. Signal Digitalization Strategy
Firstly, we selected the ORFs in common between all the eight datasets highlighted in
Table 1. For each ORF of each dataset, we generated a Ribo–seq profile. The Ribo–seq profiles (
Figure 2, left side) are digitalized by comparing the profile heights at each nucleotide position (
coverage) with its median value, computed along the entire ORF. We assign
to the positions having a coverage value higher than the median,
otherwise. The result is a
digital Ribo–seq profile (
Figure 2, right side) for each Ribo–seq profile, i.e., a vector having the length of the associated ORF and containing a sequence of
and
.
3.1.3. Comparison of Digital Profiles
The digitalized profiles can be compared to detect matches, i.e., nucleotides characterized by an identical label (
Figure 2). Calculating the relative number of matches (the ratio between the number of matches and the length of the ORF) yields the
matching score (
). Intuitively, a matching score close to one could indicate a high degree of similarity between a pair of digitalized profiles, whereas a score around one-half could mean a very poor overlap because the observed matches are likely to have occurred by chance. Given each score has a certain probability of being obtained by chance, we decided to implement a statistical test able to assess the significance of the matching score [
6].
For any Ribo–seq profile involved in a pairwise comparison, a large number of randomized profiles is generated by re-distributing the reads in random positions on the reference ORF. The randomized profiles are, in turn, compared pairwise yielding a large number of random matching scores that build each null distribution. Reiterating this process, we generated pairs of random Ribo–seq profiles and an equal number of digitalized random profiles that, compared pairwise, yielded random matching scores. These scores are used to build an ORF–specific null distribution which allows us to estimate the probability of obtaining each similarity score just by chance.
Given a pair of Ribo–seq profiles, the similarity score resulting from their comparison is tested for significance by comparing it to the corresponding ORF–specific null distribution. For each and the corresponding null distribution, we computed a z-score , mapping each similarity score on a standard normal distribution.
Subsequently, we computed the
p-values
, as the integral:
where
is the standard normal distribution. Mapping the matching score on the null distribution will yield the
p-value. The results of this process can be summarised into a matrix (called
p-value matrix) containing all the computed
p-values and composed by one column for each pairwise comparison and one row for each considered ORF (for a total of 3588 rows and 28 columns). For the sake of simplicity,
Table 2 collects a small extract of such matrix. Each
quantifies the probability of obtaining a similarity score at least as extreme as the corresponding
, given that the null hypothesis is true. In our context, the lower the
p-value, the lower the probability that the similarity between the compared pairs of (digitalized) Ribo–seq profiles occurs by chance. If the
p-value will result below a given threshold, the compared Ribo–seq profiles will exhibit a significant degree of similarity.
3.1.4. Identification of Significantly Reproducible Ribo-seq Profiles
Our strategy consists in inspecting each row of the
p-value matrix. We define reproducible the Ribo–seq profiles referring to those rows featuring all the
p-values below a chosen significance threshold. To cast our strategy into a more rigorous statistical framework, we exploit the False Discovery Rate (FDR) concept and the Benjamini-Hockberg (BH) correction method for multiple tests. In this experiment, for any given row of the
p-value matrix, we set an FDR threshold of 0.01. This means that we accept that 1% of profiles are reproducible by chance. Then, we counted how many
p-values in each row resulted significant according to the BH method, and we defined reproducible those Ribo–seq profiles associated with the rows where 80% of the
p-values are significant. Following this strategy, we found that, out of 3588 genes that are common to the eight datasets, 40 genes, listed in
Table 3, have a significantly reproducible Ribo–seq profile.
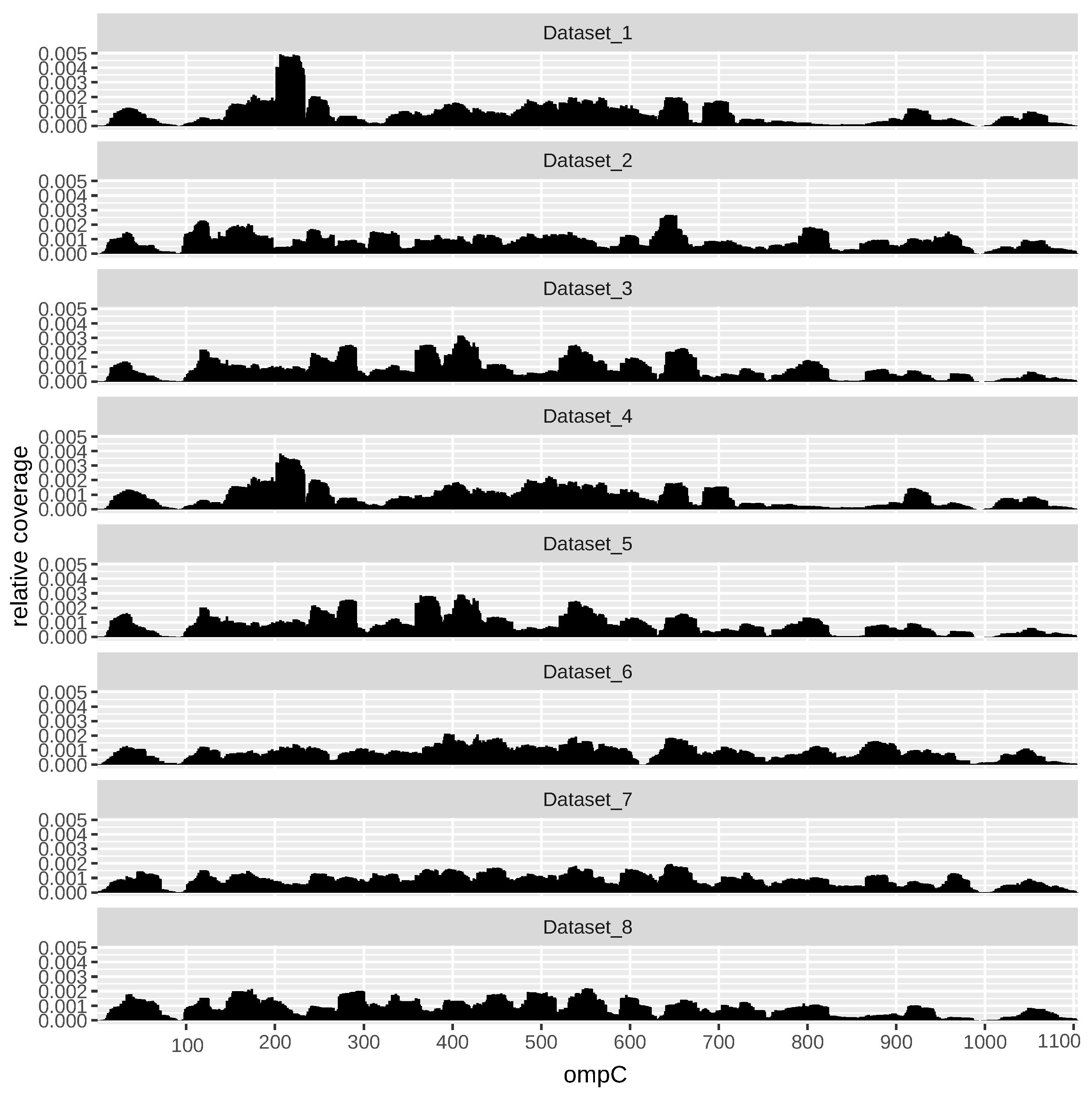
As an example,
Figure 3 shows the profile across all datasets of the gene ompC (EG10670). OmpC, also known as outer membrane (OM) protein C, is a porin of gram-negative bacteria tightly associated with the peptidoglycan layer. It has been recognized to have a crucial role in the non-specific diffusion of small solutes such as sugars, ions and amino acids across the outer membrane of the cell [
39].
To highlight which specific regions within the Ribo–seq profiles are similar to each other, we built a
consensus sequence. The consensus sequence is a character string representing the nucleotides of the reference ORF and, in
Figure 4, it is colored red in those positions where a peak is present in at least 80% of profiles (i.e., the digitalized profiles values are
and the ribosome proceeds slower), and green where a valley is located. The black color, instead, will be used in all other cases and the label assigned to these regions will be 0.
3.2. Dataset
The presented analysis has been applied to the prokaryotic organism E. coli on 3588 Ribo–seq profiles across eight independent datasets, revealing that only 40 profiles are significantly reproducible. The digitalization process described above produces a target belonging to for each nucleotide of the sequence. Based on the obtained profiles, a dataset (“SubsequencesDataset”) has been constructed, consisting in mRNA reproducible sub-sequences with a uniform target (since they are reproducible, the target is not 0). In particular, 459 sub-sequences of variable length have been obtained, of which 264 are characterized by a slow translation () and 195 by a fast translation (). In order to obtain a fixed-length vector of 36 elements, padding have been applied to the extracted regions, consisting of nucleotides of the original consensus sequence. Each nucleotide has been encoded with a 1-hot vector of 4 elements, representing one of the four possible nucleotides. The “SubsequencesDataset” is further splitted in a training set of 413 sub-sequences and a test set of 46 sub-sequences. Moreover, a validation set was built, randomly selecting of the training set sub-sequences, to give an unbiased evaluation of the model performance during training, and for keeping overfitting under control. The splitting was done ensuring that sub-sequences in the test set and in the training set are taken from different genes. Each sub-sequence is associated with a target which corresponds to its translation speed.
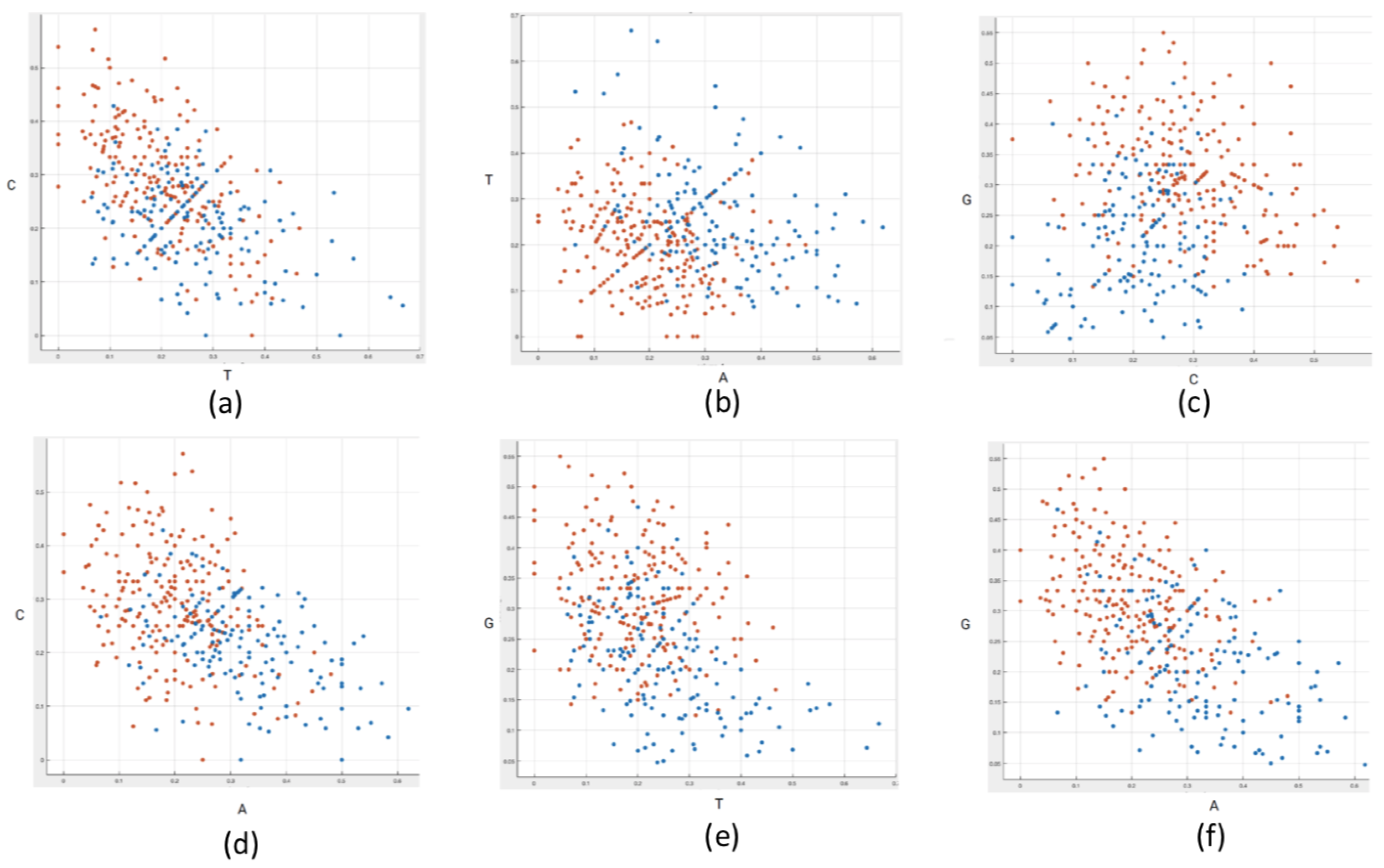
3.3. Statististical Analysis on the Nucleotide Composition of the Subsequences
Inferential statistics can provide insights on the specific patterns and characteristics of the data and highlight relationships between variables. The “SubsequencesDataset”, obtained with the procedure described in
Section 3.2, is analyzed to explore the nucleotide composition of the mRNA sub-sequences, relating to the assigned labels (
and
).
First, the relative frequency of each nucleotide (A, T, G, C) has been computed. To assess the significance of our results and to find out whether the obtained frequencies are typical of a certain speed or the correspondence is just obtained by chance, we built a statistical test. The aim of the test is to assign a probability value (p-value) to the following null hypothesis: the slow and fast sub-sequences are characterized by a random nucleotide composition. In order to test this hypothesis, new profiles are generated by randomizing the nucleotide sequence, keeping fixed the and labels and therefore the length of the original sub-sequences. In each of the obtained sequence, the relative frequencies of the four nucleotides within the fast and slow sub-sequences can be calculated. This procedure leads to eight null distributions of relative frequencies, one for each nucleotide both for slow and fast translation. Comparing the null distributions against the original relative frequencies allows to calculate the p-values. In particular, if the original frequency is lower than the mean of the null distribution, we define the p-value as the probability of a random sub-sequence to show a relative frequency smaller than that observed in the original sub-sequence. On the contrary, if the original frequency is higher than the mean of the null distribution, the p-value is calculated as the probability of finding by chance a higher relative frequency value. If the p-value lies under the significance threshold of , the null hypothesis of completely random nucleotide frequencies is rejected. This indicates a statistically significant difference in the nucleotide composition between slow/fast and randomly generated sub-sequences.
3.4. Data Validation with Neural Network Models
In this section, the informative content of the obtained data has been validated using a machine learning approach. This is achieved by employing common neural network architectures on the “SubsequencesDataset” (see
Section 3.2) to predict the translation speed class: “slow” or “fast”. In our specific problem, exploiting a network architecture can reveal whether there is enough information in the data to classify the sub-sequences into slow and fast with high accuracy. To perform this task, we used two different types of data: vectors and sequences. In fact, we initially considered four-dimensional vectors that collect the relative frequencies of occurrence of the four nucleotides and then we considered the entire sequence, to evaluate whether the order in which the nucleotides are arranged helps to capture the translation rate signal. Consistently, the experiments were carried out by applying two different neural network architectures:
Multilayer Perceptrons (MLPs) [
40] and
Convolutional Neural Networks (CNNs) [
41]. While MLPs are good at processing vectors, indeed, CNNs are powerful machine learning models that can be used to directly process complex data, sequences in our case. Hence, we have first predicted the translation speed based only on the nucleotide composition of the sub-sequence while, in a second set of experiments, we employed a CNN to process each sub-sequence.
3.4.1. MLP Analysis Based on the Nucleotide Frequencies
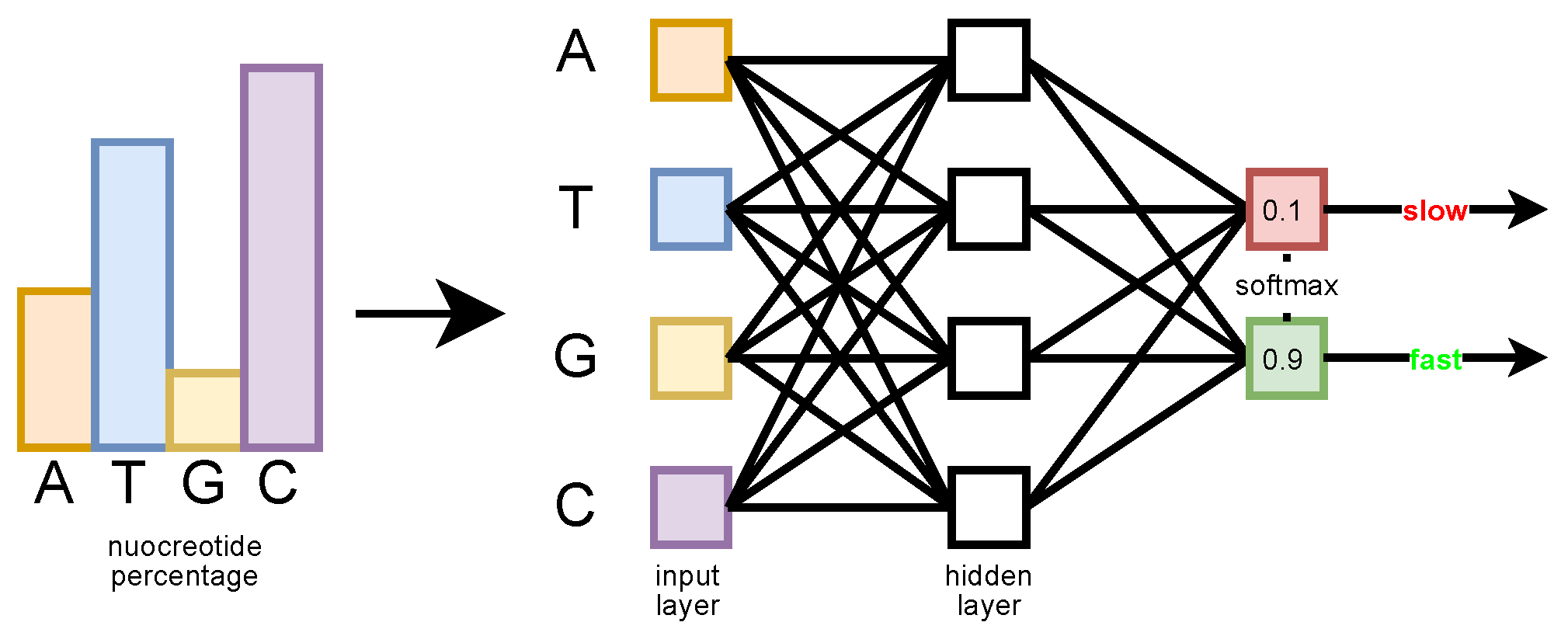
The MLP input is a four-dimensional vector whose elements correspond to the relative frequency value of each nucleotide in the sequence (in the order (A, T, G, C), see
Figure 5). The goal is to determine how much information is carried by this simple statistic, regardless of the order in which the nucleotides appear in the sequence. The MLP has a single hidden layer with four neurons and an output layer with two neurons. A
Softmax function is applied on the output layer, which produces the probability estimated by the network for each class (slow or fast) [
42]. The network has been trained with the Adam optimizer, with an initial learning rate of 0.05.
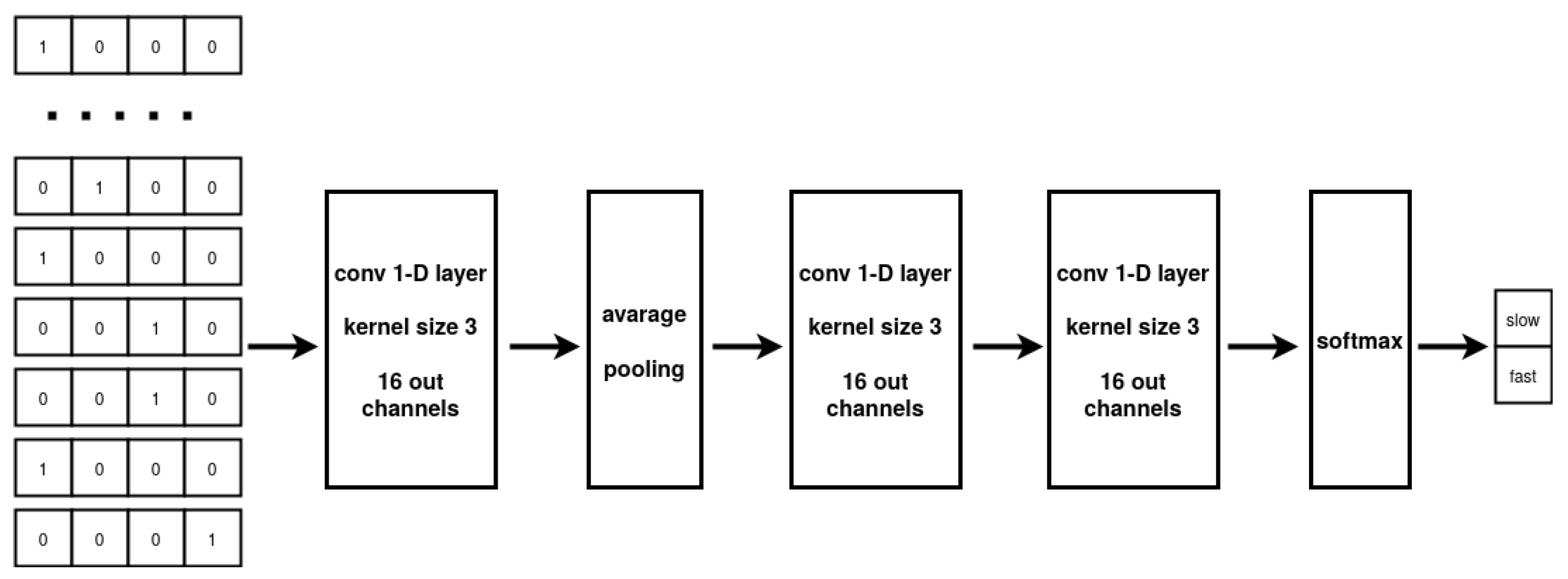
3.4.2. Convolutional Neural Network Analysis Based on Sub-Sequences
While the MLP architecture can only process vectorial data, CNNs can take full advantage of sequential information. The CNN multilayer design allows to extract a hierarchy of representations from the data while the implemented weight sharing guarantees to limit the number of parameters with respect to a fully connected architecture. In our experiments, the model input are the nucleotide sub-sequences collected in “SubsequencesDataset”. The CNN elaborates the input to produce a prediction about the translation speed associated with the sequence. Specifically, our CNN architecture comprises two successive 1-D convolutional layers, with average pooling employed between the layers, followed by two fully connected layers. The two-class probability distribution output of the network is produced by using a Softmax activation function on the output of the second fully-connected layer. The 1-D convolutional layers are based on 16 kernels with dimension three and a ReLU activation function, applied to each layer [
43]. The optimization of the network parameters is performed by the Adam optimizer using early stopping [
44]. The overall network architecture is described in
Figure 6.
3.4.3. Ensemble Convolutional Neural Networks
In the last experiment, an ensemble [
45] of seven CNNs has been employed to produce a more stable and accurate prediction. Each CNN receives as input the nucleotide sub-sequences contained in the “SubsequencesDataset”. The seven CNNs have been trained independently. In the test phase, their output has been averaged to produce the final result. More specifically, the Softmax CNN output for each class (slow and fast) is averaged among the seven networks that compose the ensemble.
5. Conclusions
In this paper, we have proposed an innovative method to characterize highly reproducibile Ribo–seq profiles. Ribo–seq data have been analysed through statistical analyses and state-of-the-art machine learning models, extremely effective in predicting the ribosome translation speed. In fact, using neural network architectures capable of processing both plain and sequential data, we have been able to obtain high accuracy, also proving that fundamental information is contained both in the nucleotide composition of the sequences and in the order in which nucleotides appear within each sequence. In this way, our work opens new exciting frontiers in the analysis of the ribosome translation dynamics in different organisms. Indeed, we have conducted a preliminary analysis of Ribo–seq profiles referring to liver tumours and their adjacent noncancerous normal liver tissues from ten patients with hepatocellular carcinoma (HCC) [
46], achieving promising results. For what concerns the machine learning analysis, once we have obtained the consensus sequences, we carried on a preliminary study exploiting the same neural architectures employed on the E. coli datasets. Nonetheless, given the increased complexity of the human data, we believe that a further analysis is necessary, in particular defining ad-hoc neural architectures and with a specialized hyperparameter search, to improve performance. Finally, it is worth mentioning that our method represents an effective approach for any kind of Ribo–seq data, to investigate an extremely relevant open questions in biology, i.e., which features can influence the speed of the ribosome during translation.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}