A Brain-Inspired Hyperdimensional Computing Approach for Classifying Massive DNA Methylation Data of Cancer



Abstract
1. Introduction
2. Materials and Methods
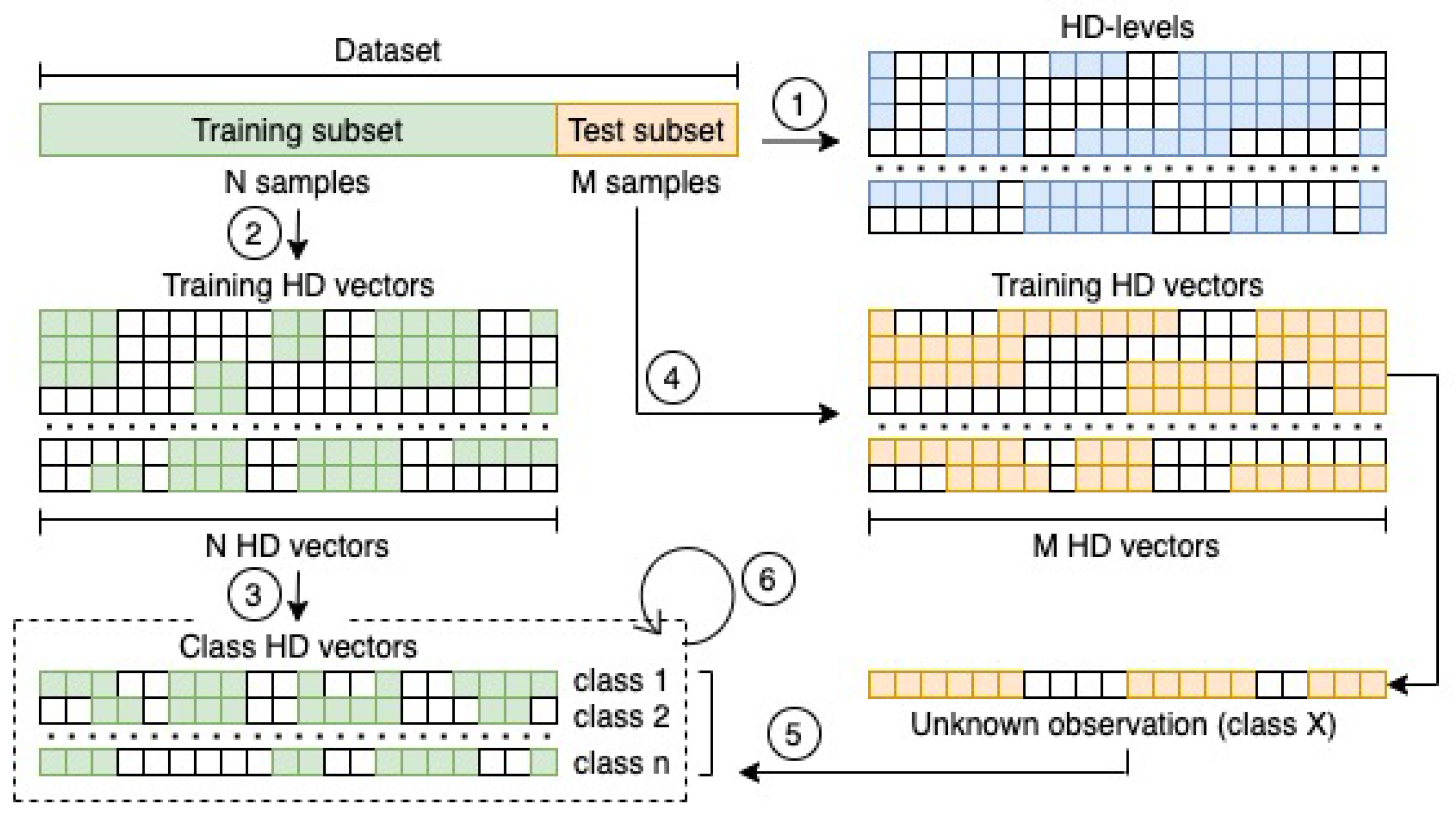
2.1. Identifying HD-Levels
2.2. Encoding Data and Building the Classification Model
| Algorithm 1 Encoding observations to class hypervectors |
|
2.3. Class Prediction
3. Results
4. Discussion, Conclusions, and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BRCA | Breast Invasive Carcinoma |
| DM | DNA methylation |
| DT | Decision Tree |
| GDC | Genomic Data Commons |
| HD | Hyperdimensional |
| KIRP | Kidney renal papillary cell carcinoma |
| ML | Machine Learning |
| SVM | Support Vector Machine |
| TCGA | The Cancer Genome Atlas |
| THCA | Thyroid carcinoma |
References
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2007, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Rodriguez-Antolin, C.; Vallespin, E.; De Castro Carpeno, J.; De Caceres, I.I. The impact of next-generation sequencing on the DNA methylation–based translational cancer research. Transl. Res. 2016, 169, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Aravanis, A.M.; Lee, M.; Klausner, R.D. Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell 2017, 168, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1985, 321, 209–213. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Li, Z.; Lei, H.; Luo, M.; Wang, Y.; Dong, L.; Ma, Y.; Liu, C.; Song, W.; Wang, F.; Zhang, J.; et al. DNA methylation downregulated mir-10b acts as a tumor suppressor in gastric cancer. Gastric Cancer 2015, 18, 43–54. [Google Scholar] [CrossRef]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.; Polyak, K.; et al. RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef]
- Deng, S.P.; Cao, S.; Huang, D.S.; Wang, Y.P. Identifying Stages of Kidney Renal Cell Carcinoma by Combining Gene Expression and DNA Methylation Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 14, 1147–1153. [Google Scholar] [CrossRef]
- Cappelli, E.; Felici, G.; Weitschek, E. Combining DNA methylation and RNA sequencing data of cancer for supervised knowledge extraction. BioData Min. 2018, 11, 22. [Google Scholar] [CrossRef]
- Wadapurkar, R.M.; Vyas, R. Computational analysis of next generation sequencing data and its applications in clinical oncology. Inform. Med. Unlocked 2018, 11, 75–82. [Google Scholar] [CrossRef]
- Weitschek, E.; Cumbo, F.; Cappelli, E.; Felici, G. Genomic data integration: A case study on next generation sequencing of cancer. In Proceedings of the 2016 27th International Workshop on Database and Expert Systems Applications (DEXA), Porto, Portugal, 5–8 September 2016; pp. 49–53. [Google Scholar]
- Jabbari, K.; Bernardi, G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 2004, 333, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.A.; Ferretti, V.; Grossman, R.L.; Staudt, L.M. The NCI Genomic Data Commons as an engine for precision medicine. Blood 2017, 130, 453–459. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar] [CrossRef]
- Cruz, J.A.; Wishart, D.S. Applications of machine learning in cancer prediction and prognosis. Cancer Inform. 2006, 2, 117693510600200030. [Google Scholar] [CrossRef]
- Weitschek, E.; Cumbo, F.; Cappelli, E.; Felici, G.; Bertolazzi, P. Classifying big DNA methylation data: A gene-oriented approach. In Proceedings of the International Conference on Database and Expert Systems Applications, Regensburg, Germany, 3–6 September 2018; pp. 138–149. [Google Scholar]
- Polychronopoulos, D.; Weitschek, E.; Dimitrieva, S.; Bucher, P.; Felici, G.; Almirantis, Y. Classification of selectively constrained dna elements using feature vectors and rule-based classifiers. Genomics 2014, 104, 79–86. [Google Scholar] [CrossRef]
- Tan, P.; Steinbach, M.; Kumar, V. Introduction to Data Mining; Addison Wesley: Boston, MA, USA, 2005. [Google Scholar]
- Celli, F.; Cumbo, F.; Weitschek, E. Classification of large DNA methylation datasets for identifying cancer drivers. Big Data Res. 2018, 13, 21–28. [Google Scholar] [CrossRef]
- Cestarelli, V.; Fiscon, G.; Felici, G.; Bertolazzi, P.; Weitschek, E. CAMUR: Knowledge extraction from RNA-seq cancer data through equivalent classification rules. Bioinformatics 2016, 32, 697–704. [Google Scholar] [CrossRef]
- Mašić, N.; Gagro, A.; Rabatić, S.; Sabioncello, A.; Dašić, G.; Jakšić, B.; Vitale, B. Decision-tree approach to the immunophenotype-based prognosis of the B-cell chronic lymphocytic leukemia. Am. J. Hematol. 1998, 59, 143–148. [Google Scholar] [CrossRef]
- Li, Y.; Tang, X.Q.; Bai, Z.; Dai, X. Exploring the intrinsic differences among breast tumor subtypes defined using immunohistochemistry markers based on the decision tree. Sci. Rep. 2016, 6, 35773. [Google Scholar] [CrossRef]
- Rahimi, A.; Kanerva, P.; Rabaey, J.M. A robust and energy-efficient classifier using brain-inspired hyperdimensional computing. In Proceedings of the 2016 International Symposium on Low Power Electronics and Design, San Francisco, CA, USA, 8–10 August 2016; pp. 64–69. [Google Scholar]
- Kanerva, P. Hyperdimensional computing: An introduction to computing in distributed representation with high-dimensional random vectors. Cogn. Comput. 2009, 1, 139–159. [Google Scholar] [CrossRef]
- Ge, L.; Parhi, K.K. Classification Using Hyperdimensional Computing: A Review. IEEE Circuits Syst. Mag. 2020, 20, 30–47. [Google Scholar] [CrossRef]
- Imani, M.; Kong, D.; Rahimi, A.; Rosing, T. Voicehd: Hyperdimensional computing for efficient speech recognition. In Proceedings of the 2017 IEEE International Conference on Rebooting Computing (ICRC), Washington, DC, USA, 8–9 November 2017; pp. 1–8. [Google Scholar]
- Imani, M.; Huang, C.; Kong, D.; Rosing, T. Hierarchical hyperdimensional computing for energy efficient classification. In Proceedings of the 2018 55th ACM/ESDA/IEEE Design Automation Conference (DAC), San Francisco, CA, USA, 24–28 June 2018; pp. 1–6. [Google Scholar]
- Gupta, S.; Imani, M.; Rosing, T. Felix: Fast and energy-efficient logic in memory. In Proceedings of the 2018 IEEE/ACM International Conference on Computer-Aided Design (ICCAD), San Diego, CA, USA, 5–8 November 2018; pp. 1–7. [Google Scholar]
- Imani, M.; Kim, Y.; Riazi, S.; Messerly, J.; Liu, P.; Koushanfar, F.; Rosing, T. A framework for collaborative learning in secure high-dimensional space. In Proceedings of the 2019 IEEE 12th International Conference on Cloud Computing (CLOUD), Milan, Italy, 8–13 July 2019; pp. 435–446. [Google Scholar]
- Kim, Y.; Imani, M.; Rosing, T.S. Efficient human activity recognition using hyperdimensional computing. In Proceedings of the 8th International Conference on the Internet of Things, Santa Barbara, CA, USA, 15–18 October 2018; pp. 1–6. [Google Scholar]
- Datta, S.; Antonio, R.A.; Ison, A.R.; Rabaey, J.M. A programmable hyper-dimensional processor architecture for human-centric IoT. IEEE J. Emerg. Sel. Top. Circuits Syst. 2019, 9, 439–452. [Google Scholar] [CrossRef]
- Burrello, A.; Schindler, K.; Benini, L.; Rahimi, A. One-shot learning for iEEG seizure detection using end-to-end binary operations: Local binary patterns with hyperdimensional computing. In Proceedings of the 2018 IEEE Biomedical Circuits and Systems Conference (BioCAS), Cleveland, OH, USA, 17–19 October 2018; pp. 1–4. [Google Scholar]
- Imani, M.; Nassar, T.; Rahimi, A.; Rosing, T. Hdna: Energy-efficient dna sequencing using hyperdimensional computing. In Proceedings of the 2018 IEEE EMBS International Conference on Biomedical & Health Informatics (BHI), Las Vegas, NV, USA, 4–7 March 2018; pp. 271–274. [Google Scholar]
- Kim, Y.; Imani, M.; Moshiri, N.; Rosing, T. GenieHD: Efficient DNA pattern matching accelerator using hyperdimensional computing. In Proceedings of the 2020 Design, Automation & Test in Europe Conference & Exhibition (DATE), Grenoble, France, 9–13 March 2020; pp. 115–120. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Salamat, S.; Imani, M.; Khaleghi, B.; Rosing, T. F5-hd: Fast flexible fpga-based framework for refreshing hyperdimensional computing. In Proceedings of the 2019 ACM/SIGDA International Symposium on Field-Programmable Gate Arrays, Seaside, CA, USA, 24–26 February 2019; pp. 53–62. [Google Scholar]
- Imani, M.; Messerly, J.; Wu, F.; Pi, W.; Rosing, T. A binary learning framework for hyperdimensional computing. In Proceedings of the 2019 Design, Automation & Test in Europe Conference & Exhibition (DATE), Florence, Italy, 25–29 March 2019; pp. 126–131. [Google Scholar]


{kind=link}
| Dataset | Tumor Samples | Normal Samples | Features |
|---|---|---|---|
| BRCA | 799 | 98 | 485,512 |
| KIRP | 276 | 45 | 485,512 |
| THCA | 515 | 56 | 485,512 |
| Sample | CpG Site | CpG Site | ⋯ | CpG Site | Class Label |
|---|---|---|---|---|---|
| bv | bv | ⋯ | bv | normal | |
| bv | bv | ⋯ | bv | tumor | |
| ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
| bv | bv | ⋯ | bv | normal |
| Dataset | Retraining Iterations | Avg. Training Time ± Std.Dev (h) | Avg. Classification Time ± Std.Dev (s) | Avg. Accuracy ± Std.Dev (%) |
|---|---|---|---|---|
| BRCA | 4 | 5.44 ± 0.37 | 3 ± 0.68 | 97.7 ± 0.54 |
| KIRP | 1 | 2.97 ± 0.12 | 1 ± 0.32 | 98.4 ± 1.36 |
| THCA | 7 | 3.68 ± 0.47 | 2 ± 0.53 | 100 ± 0.002 |
| Dataset | HD Vectors Dimensionality | HD-Levels | Retraining Iterations | Training Time (h) | Classification Time (s) | Accuracy (%) |
|---|---|---|---|---|---|---|
| BRCA | 9000 | 900 | 100 | 10.53 | 6 | 56.11 |
| 9000 | 600 | 25 | 3.08 | 1 | 54.44 | |
| 9000 | 500 | 100 | 3.07 | 6 | 57.78 | |
| 7000 | 900 | 40 | 13.85 | 2 | 56.67 | |
| 7000 | 700 | 52 | 5.08 | 3 | 57.78 | |
| 7000 | 500 | 82 | 2.94 | 4 | 59.44 | |
| 5000 | 900 | 5 | 3.51 | <1 | 51.11 | |
| 5000 | 500 | 7 | 8.30 | <1 | 48.89 | |
| 3000 | 900 | 84 | 2.79 | 3 | 58.33 | |
| 3000 | 500 | 85 | 3.34 | 3 | 61.11 | |
| 1000 | 900 | 3 | 2.33 | <1 | 88.89 | |
| 1000 | 500 | 65 | 11.41 | 2 | 60.00 | |
| KIRP | 9000 | 900 | 94 | 3.67 | 2 | 64.06 |
| 9000 | 600 | 12 | 1.07 | <1 | 57.81 | |
| 9000 | 500 | 29 | 1.05 | <1 | 59.38 | |
| 7000 | 900 | 100 | 1.00 | 1 | 62.50 | |
| 7000 | 700 | 69 | 1.00 | 1 | 62.50 | |
| 7000 | 500 | 2 | 0.98 | <1 | 60.94 | |
| 5000 | 900 | 25 | 0.94 | <1 | 64.06 | |
| 5000 | 500 | 38 | 0.93 | <1 | 62.50 | |
| 3000 | 900 | 94 | 0.82 | 1 | 62.50 | |
| 3000 | 500 | 8 | 0.82 | <1 | 60.94 | |
| 1000 | 900 | 3 | 0.78 | <1 | 85.94 | |
| 1000 | 500 | 24 | 0.77 | <1 | 59.38 | |
| THCA | 9000 | 900 | 32 | 2.00 | 1 | 54.39 |
| 9000 | 600 | 18 | 2.10 | <1 | 57.89 | |
| 9000 | 500 | 38 | 2.07 | 1 | 59.65 | |
| 7000 | 900 | 100 | 1.91 | 3 | 64.04 | |
| 7000 | 700 | 50 | 1.95 | 1 | 61.40 | |
| 7000 | 500 | 43 | 2.39 | 1 | 61.40 | |
| 5000 | 900 | 22 | 1.71 | <1 | 56.14 | |
| 5000 | 500 | 26 | 1.69 | <1 | 56.14 | |
| 3000 | 900 | 5 | 1.87 | <1 | 100.0 | |
| 3000 | 500 | 61 | 1.62 | 1 | 62.28 | |
| 1000 | 900 | 3 | 1.47 | <1 | 90.35 | |
| 1000 | 500 | 6 | 4.95 | <1 | 99.12 |
| Dataset | HD-Levels | Retraining Iterations | Training Time (h) | Classification Time (s) | Accuracy (%) |
|---|---|---|---|---|---|
| BRCA | 1000 | 2 | 2.30 | <1 | 88.89 |
| 900 | 3 | 2.33 | <1 | 88.89 | |
| 800 | 3 | 6.77 | <1 | 88.89 | |
| 700 | 3 | 2.73 | <1 | 88.89 | |
| 600 | 3 | 4.76 | <1 | 88.89 | |
| 500 | 65 | 11.41 | 2 | 60.00 | |
| KIRP | 1000 | 3 | 0.78 | <1 | 85.94 |
| 900 | 3 | 0.78 | <1 | 85.94 | |
| 800 | 3 | 0.77 | <1 | 85.94 | |
| 700 | 3 | 0.77 | <1 | 85.94 | |
| 600 | 3 | 0.77 | <1 | 85.94 | |
| 500 | 24 | 0.77 | <1 | 59.38 | |
| THCA | 1000 | 3 | 1.38 | <1 | 90.35 |
| 900 | 3 | 1.47 | <1 | 90.35 | |
| 800 | 3 | 1.37 | <1 | 90.35 | |
| 700 | 2 | 1.38 | <1 | 90.35 | |
| 600 | 2 | 1.35 | <1 | 90.35 | |
| 500 | 6 | 4.95 | <1 | 99.12 |
| Dataset | HD-Classifier Avg. Accuracy ± Std.Dev (%) | BIGBIOCL Avg. Accuracy ± Std.Dev (%) |
|---|---|---|
| BRCA | 97.7 ± 0.54 | 98.0 ± 0.94 |
| KIRP | 98.4 ± 1.36 | 97.0 ± 2.04 |
| THCA | 100 ± 0.002 | 97.0 ± 0.03 |
| Memory | Threads | Regularization Method | Regularization Parameter | Iterations | Avg. Execution Time (h) | Avg. Accuracy ± Std.Dev (%) |
|---|---|---|---|---|---|---|
| 12 GB | 7 | L2 | 1.0 | 100 | 2.05 ± 0.11 | 98.95 ± 1.54 |
| 12 GB | 7 | L2 | 1.0 | 200 | 3.53 ± 0.02 | 98.97 ± 2.04 |
| 12 GB | 7 | L1 | 0.1 | 100 | 1.67 ± 0.07 | 95.46 ± 0.63 |
| 12 GB | 7 | L1 | 0.1 | 200 | 1.32 ± 0.04 | 99.16 ± 1.35 |
| Memory | Threads | Max Depth | Max Bins | Impurity | Avg. Execution Time ± Std.Dev (min) | Avg. Accuracy ± Std.Dev |
|---|---|---|---|---|---|---|
| 5 GB | 4 | 5 | 16 | Gini | 55.2 ± 3.02 | 98.51 ± 1.34 |
| 5 GB | 4 | 5 | 32 | Gini | Out of Memory | - |
| 12 GB | 7 | 5 | 32 | Gini | 68.19 ± 2.74 | 98.76 ± 0.83 |
| 12 GB | 7 | 10 | 32 | Gini | 69.88 ± 3.68 | 99.20 ± 0.38 |
| 12 GB | 7 | 5 | 8 | Gini | 11.52 ± 0.86 | 98.03 ± 2.17 |
| 18 GB | 7 | 5 | 128 | Gini | Out of Memory | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cumbo, F.; Cappelli, E.; Weitschek, E. A Brain-Inspired Hyperdimensional Computing Approach for Classifying Massive DNA Methylation Data of Cancer. Algorithms 2020, 13, 233. https://doi.org/10.3390/a13090233
Cumbo F, Cappelli E, Weitschek E. A Brain-Inspired Hyperdimensional Computing Approach for Classifying Massive DNA Methylation Data of Cancer. Algorithms. 2020; 13(9):233. https://doi.org/10.3390/a13090233
Chicago/Turabian StyleCumbo, Fabio, Eleonora Cappelli, and Emanuel Weitschek. 2020. "A Brain-Inspired Hyperdimensional Computing Approach for Classifying Massive DNA Methylation Data of Cancer" Algorithms 13, no. 9: 233. https://doi.org/10.3390/a13090233
APA StyleCumbo, F., Cappelli, E., & Weitschek, E. (2020). A Brain-Inspired Hyperdimensional Computing Approach for Classifying Massive DNA Methylation Data of Cancer. Algorithms, 13(9), 233. https://doi.org/10.3390/a13090233