
End-Functionalized Poly(N-isopropylacrylamide) with d-Glucosamine through Different Initiator from C-1 and C-2 Positions via Atom Transfer Radical Polymerization

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
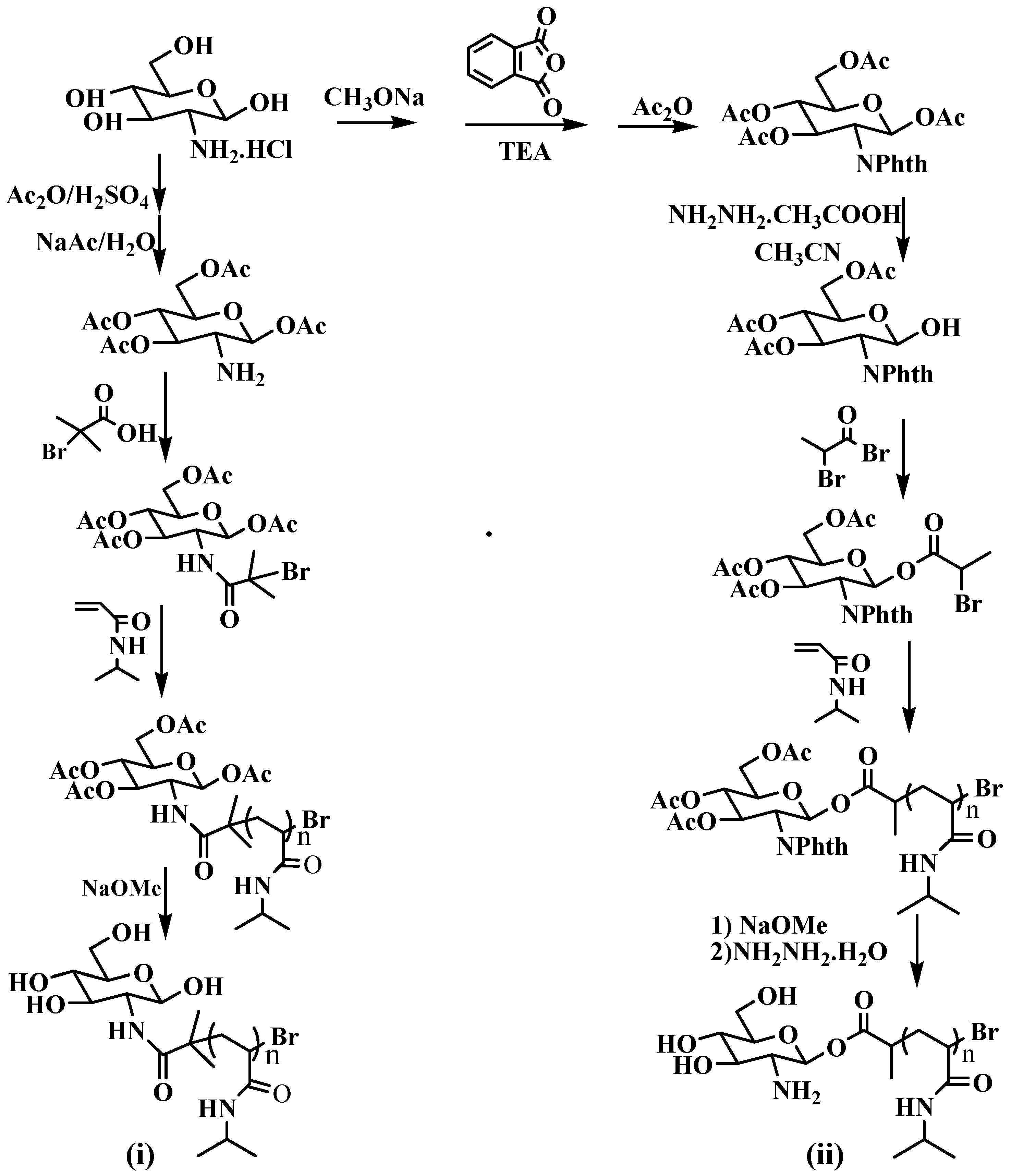
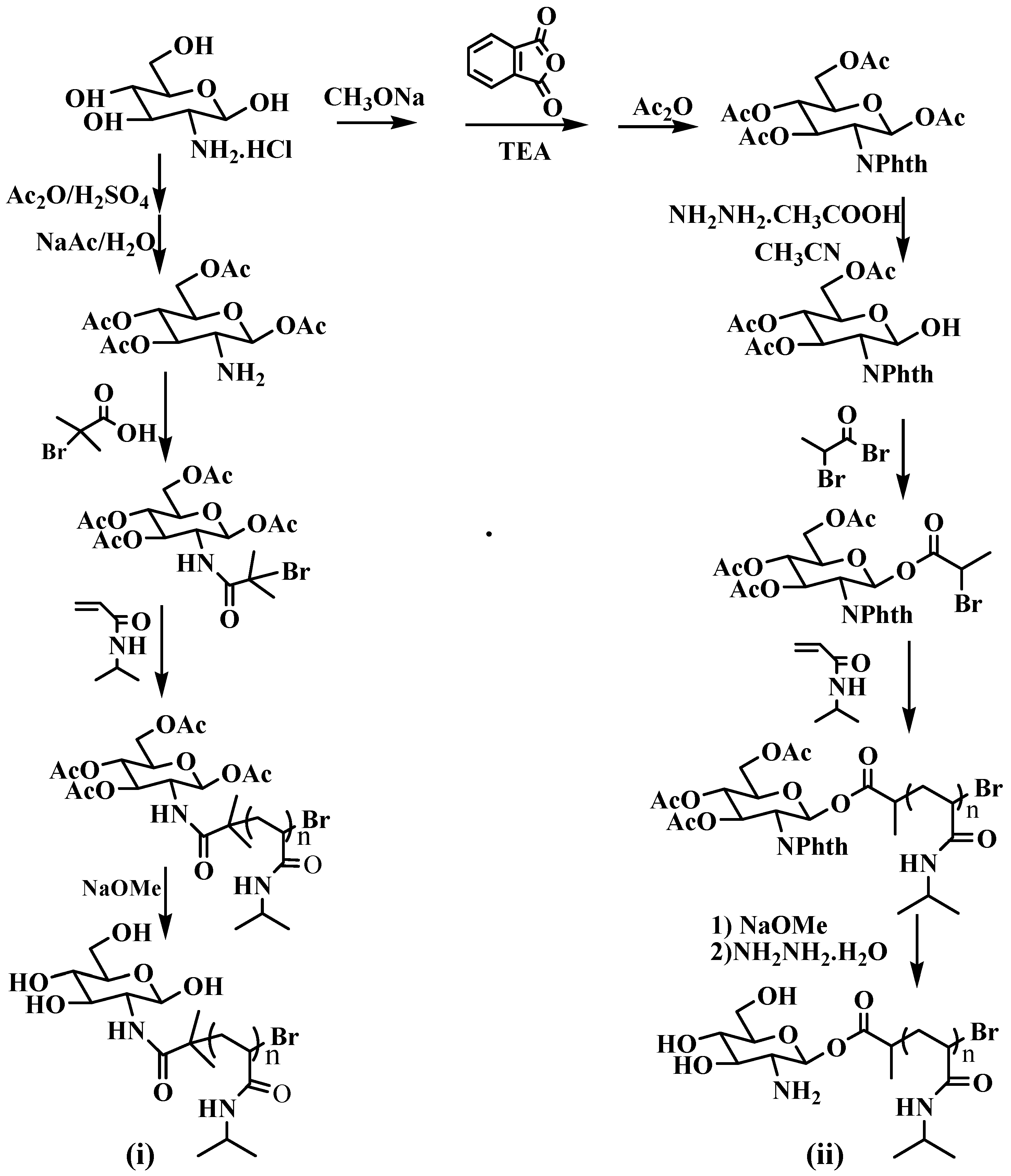
2.2. General Procedure for Synthesis of Polymers
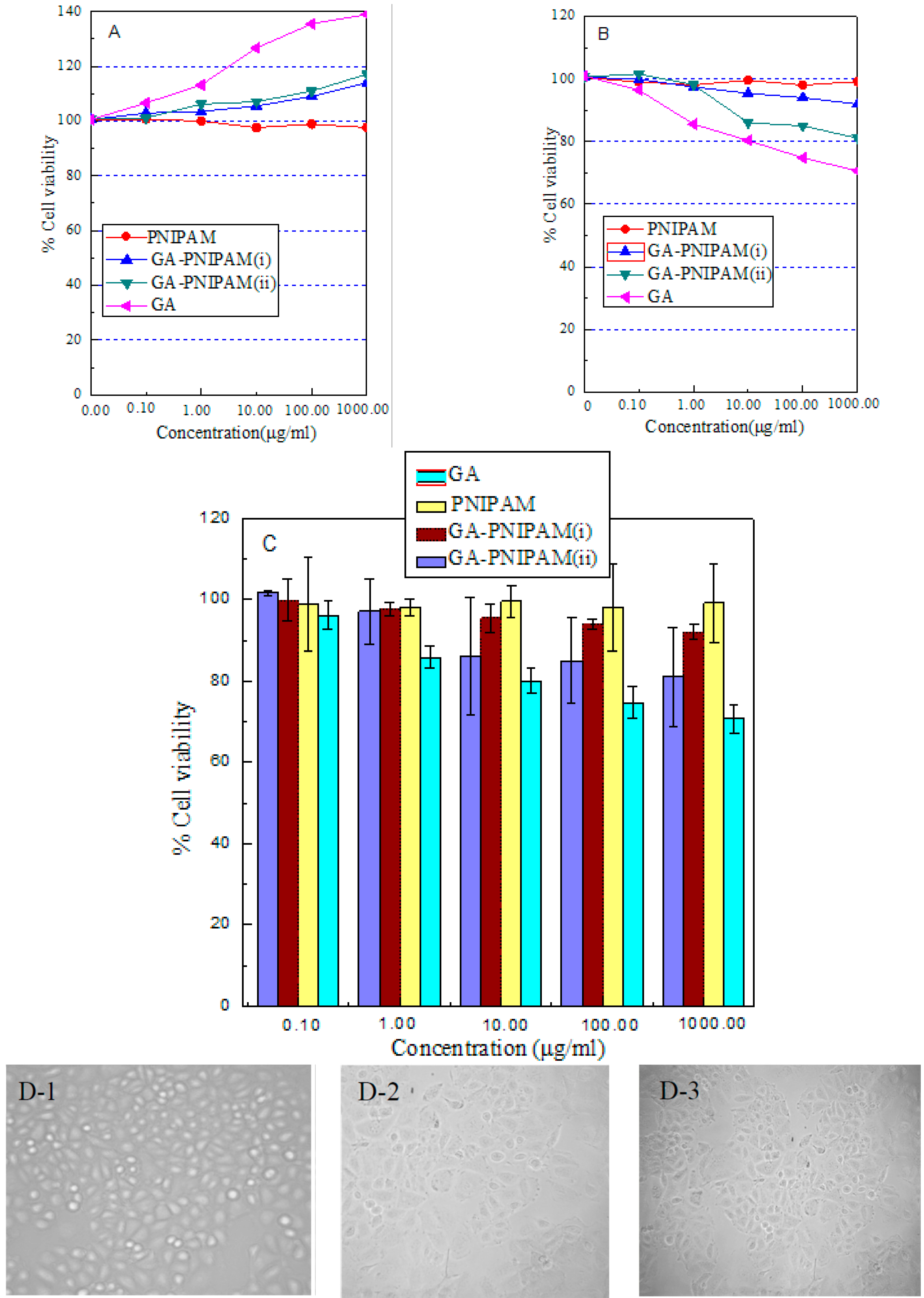
2.3. Biocompatibility Study
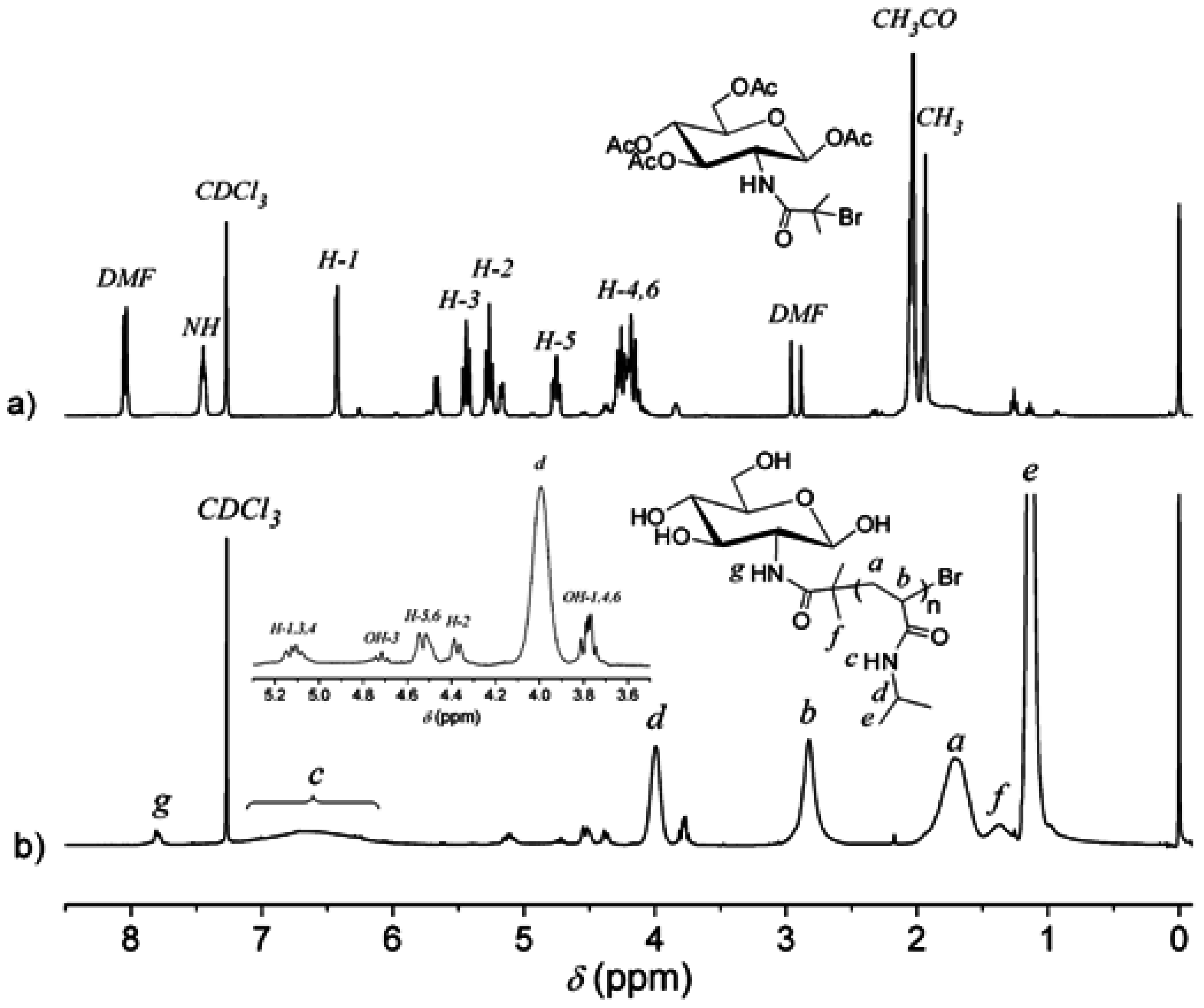
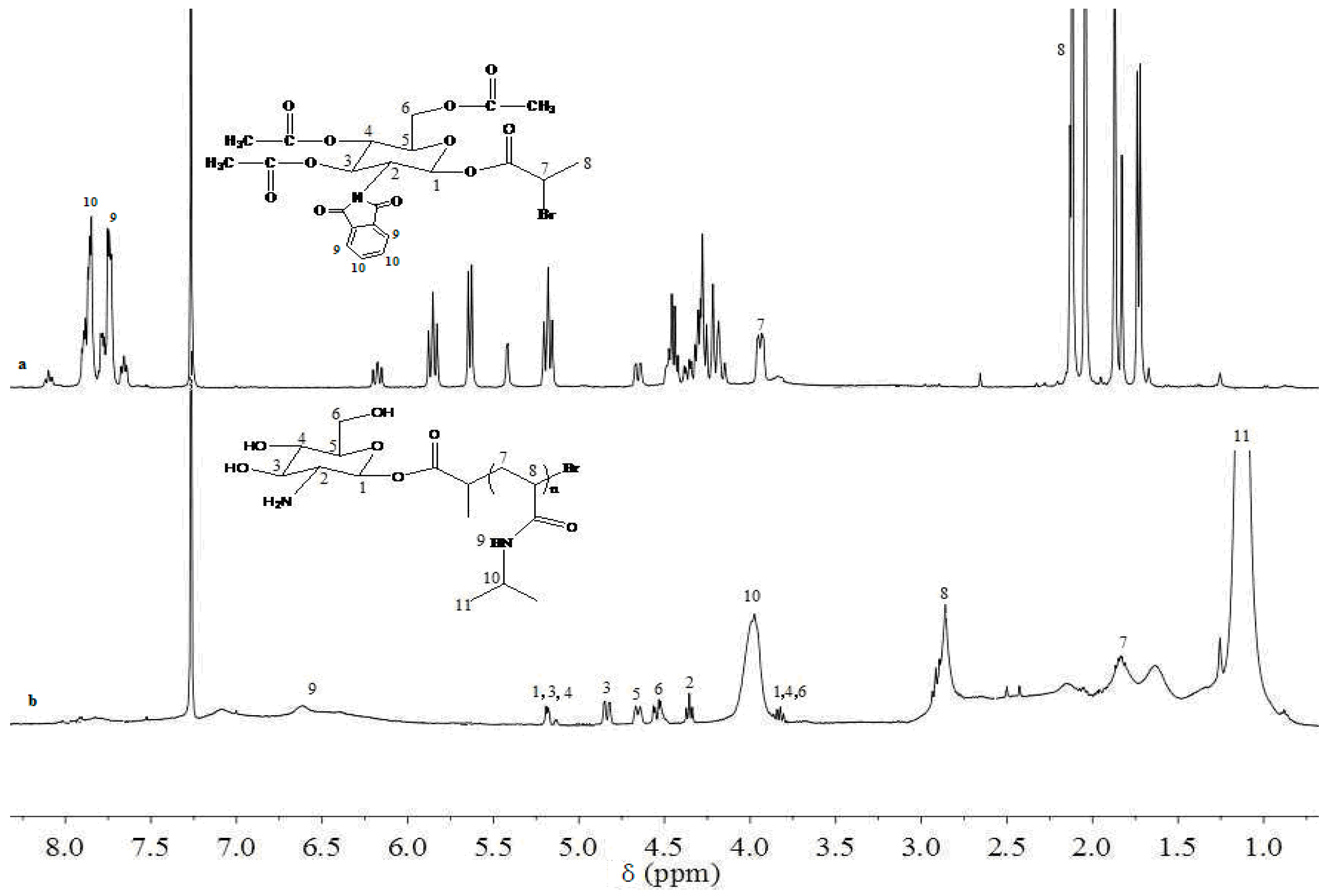
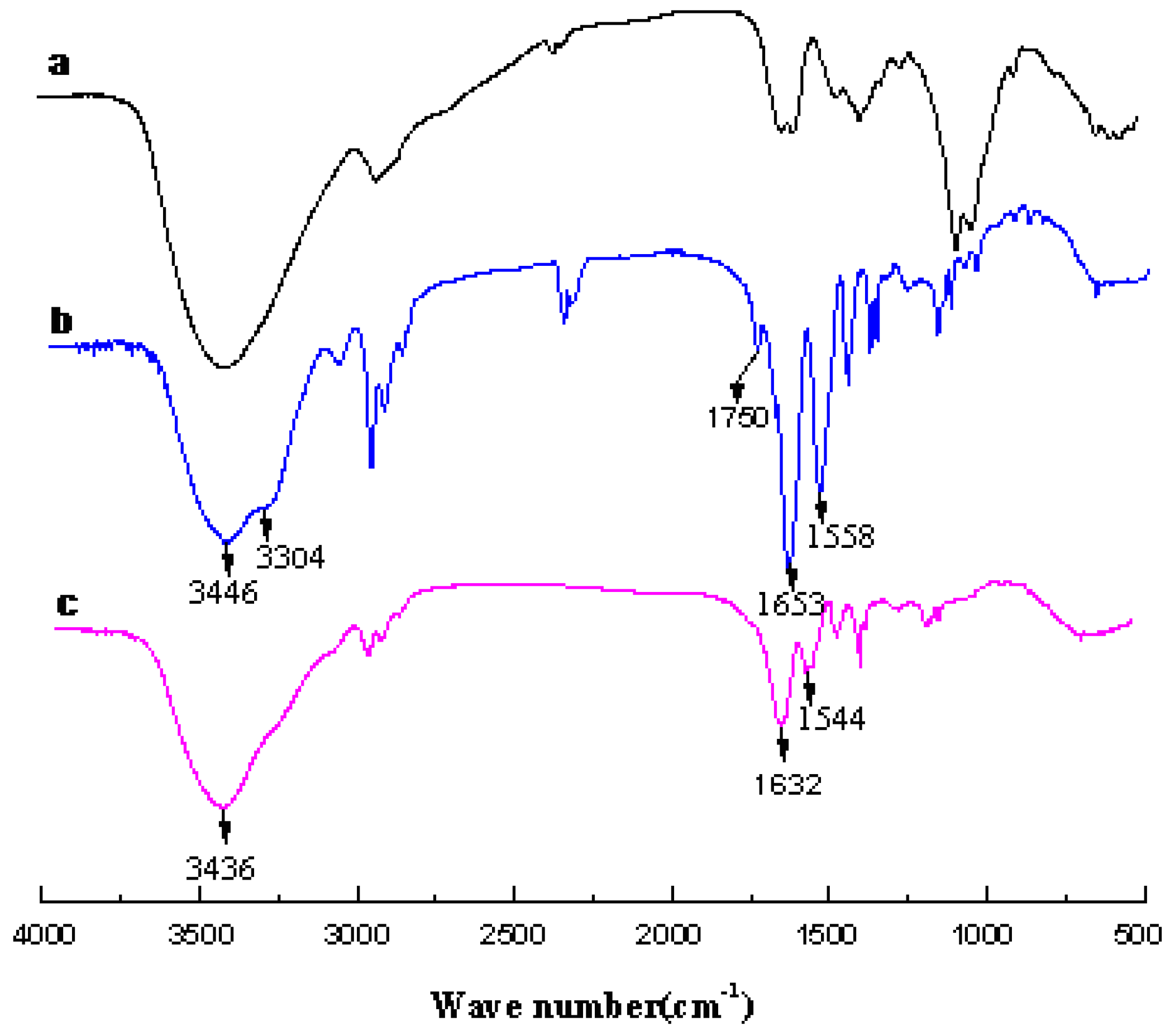
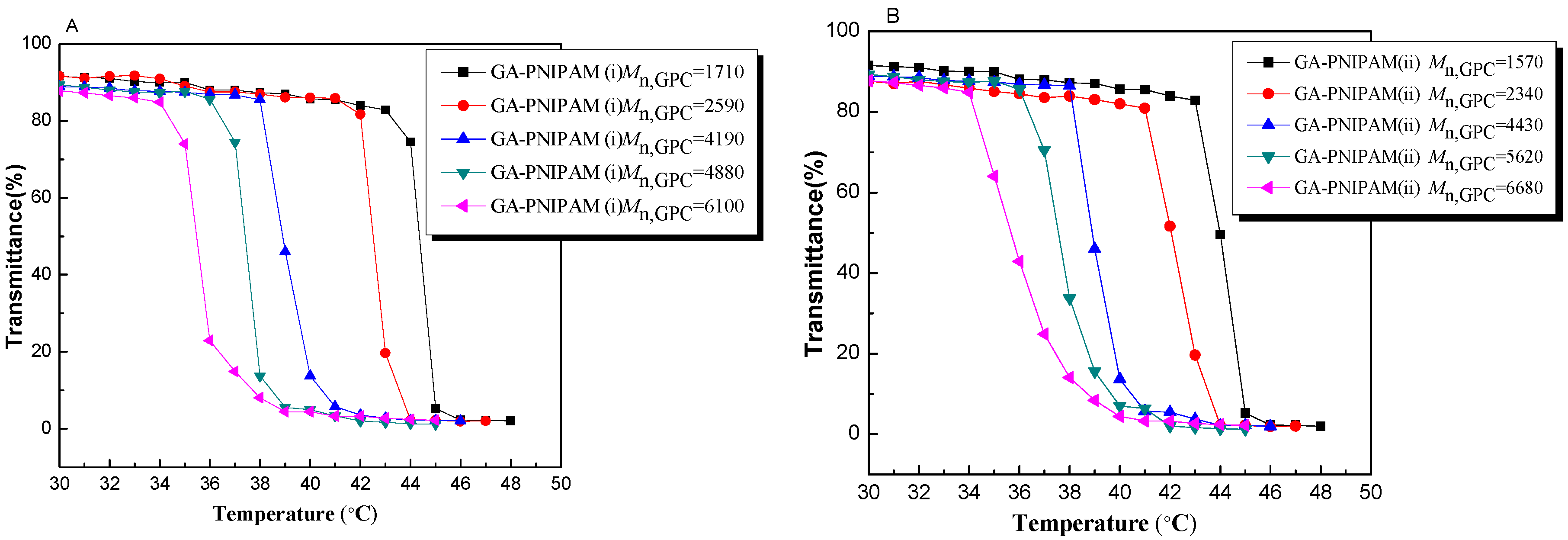
3. Results and Discussion
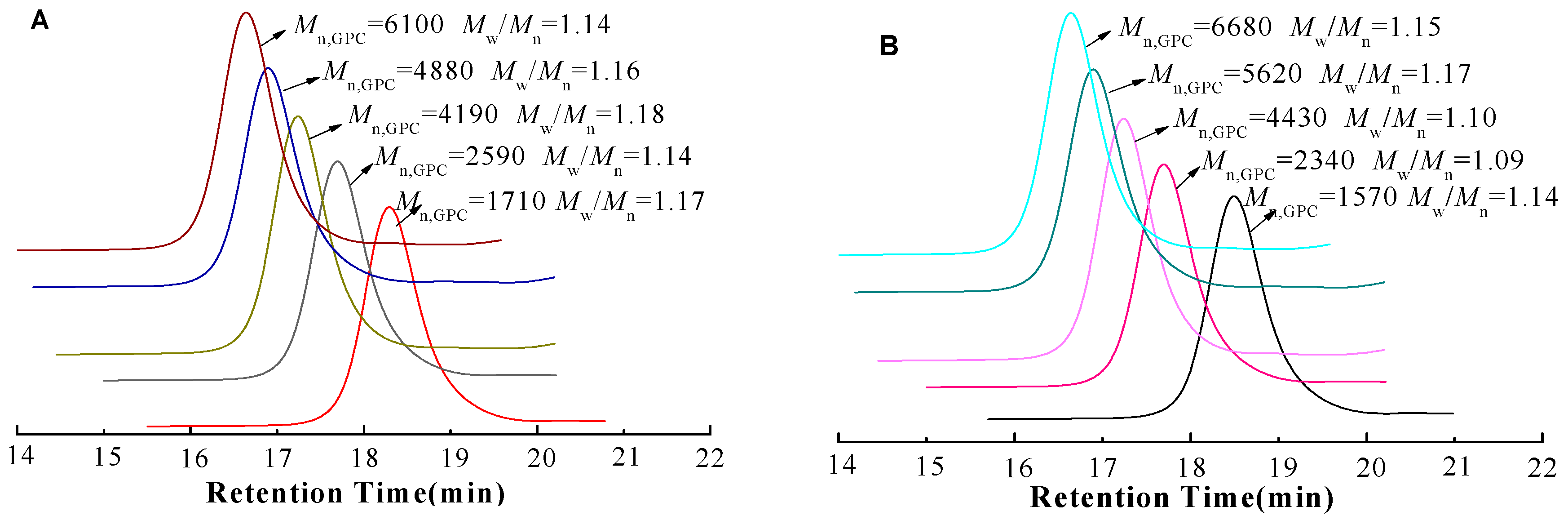
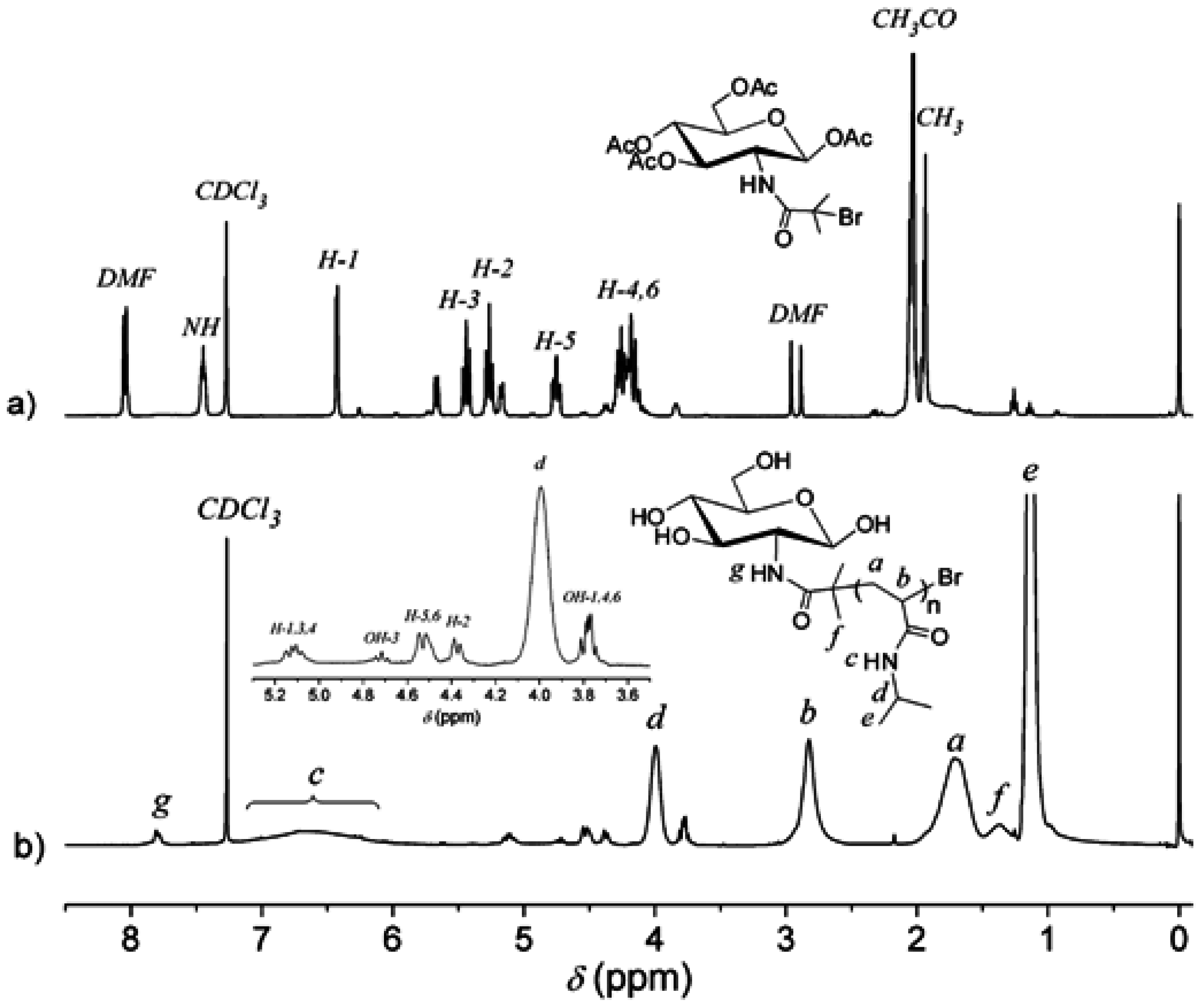
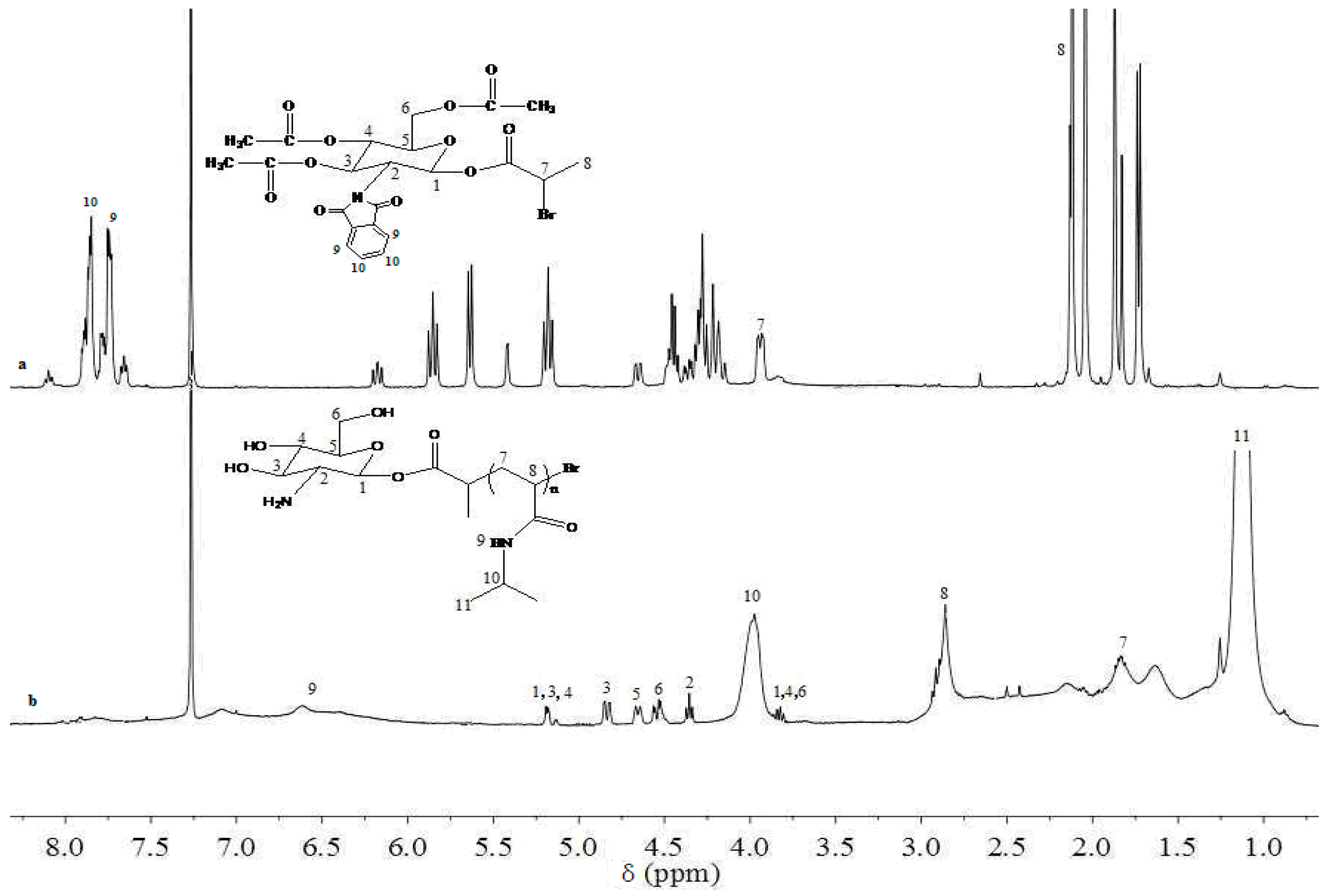
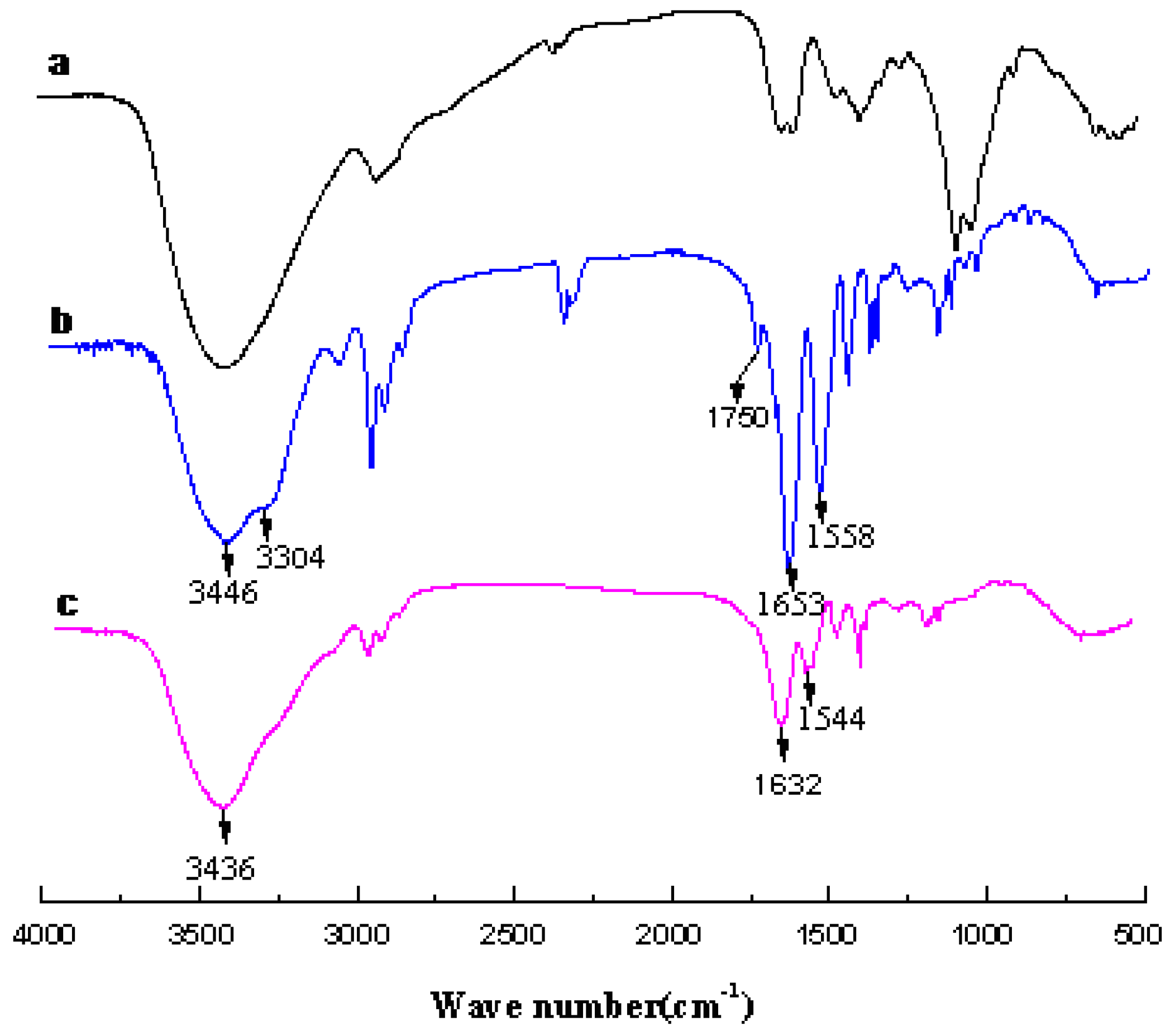
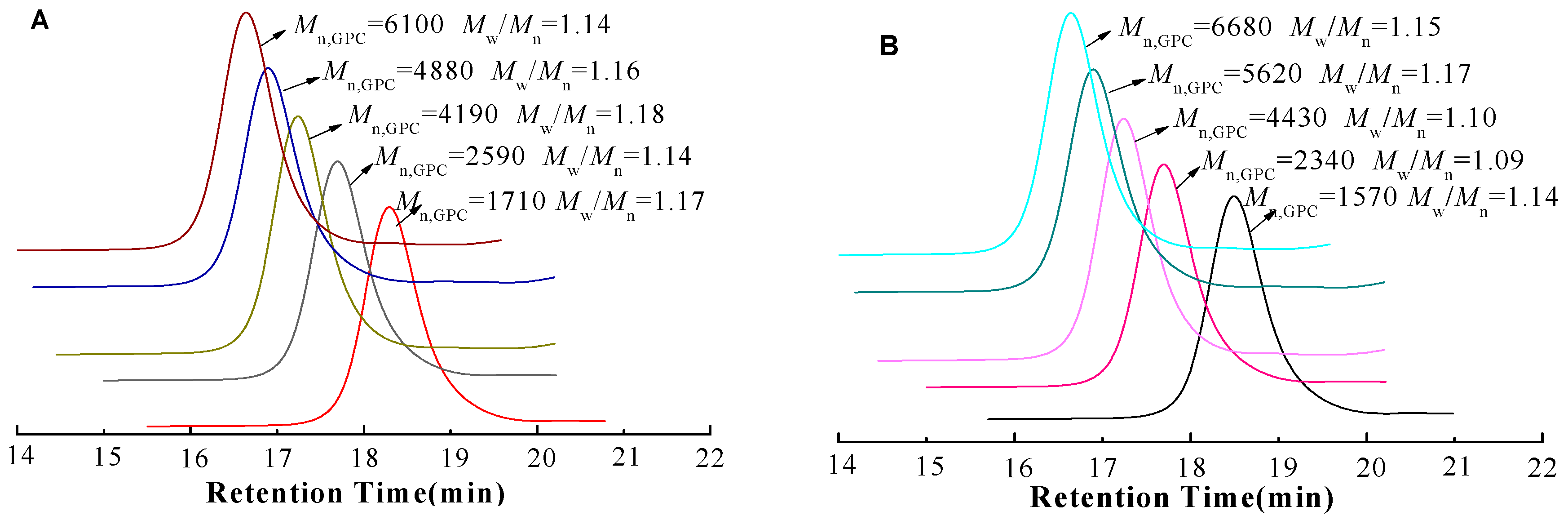
3.1. Analysis of Polymer Architecture
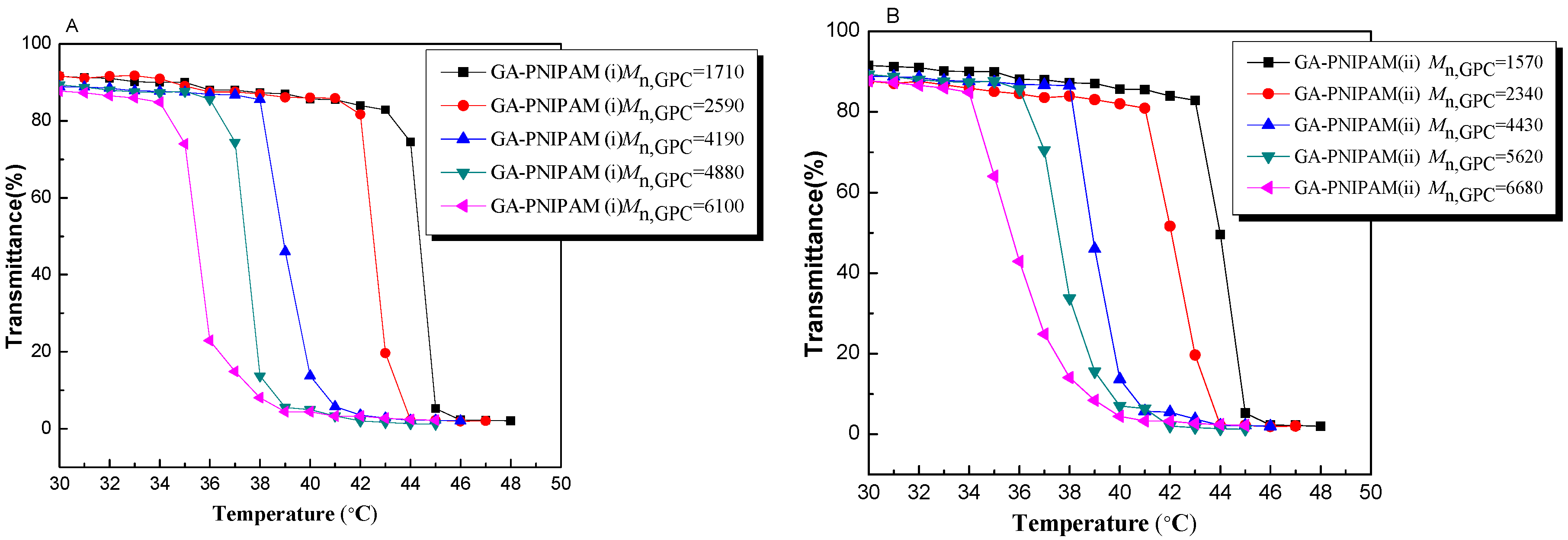
3.2. Thermo-Responsivity Property of GA-PNIPAM (i) and (ii)
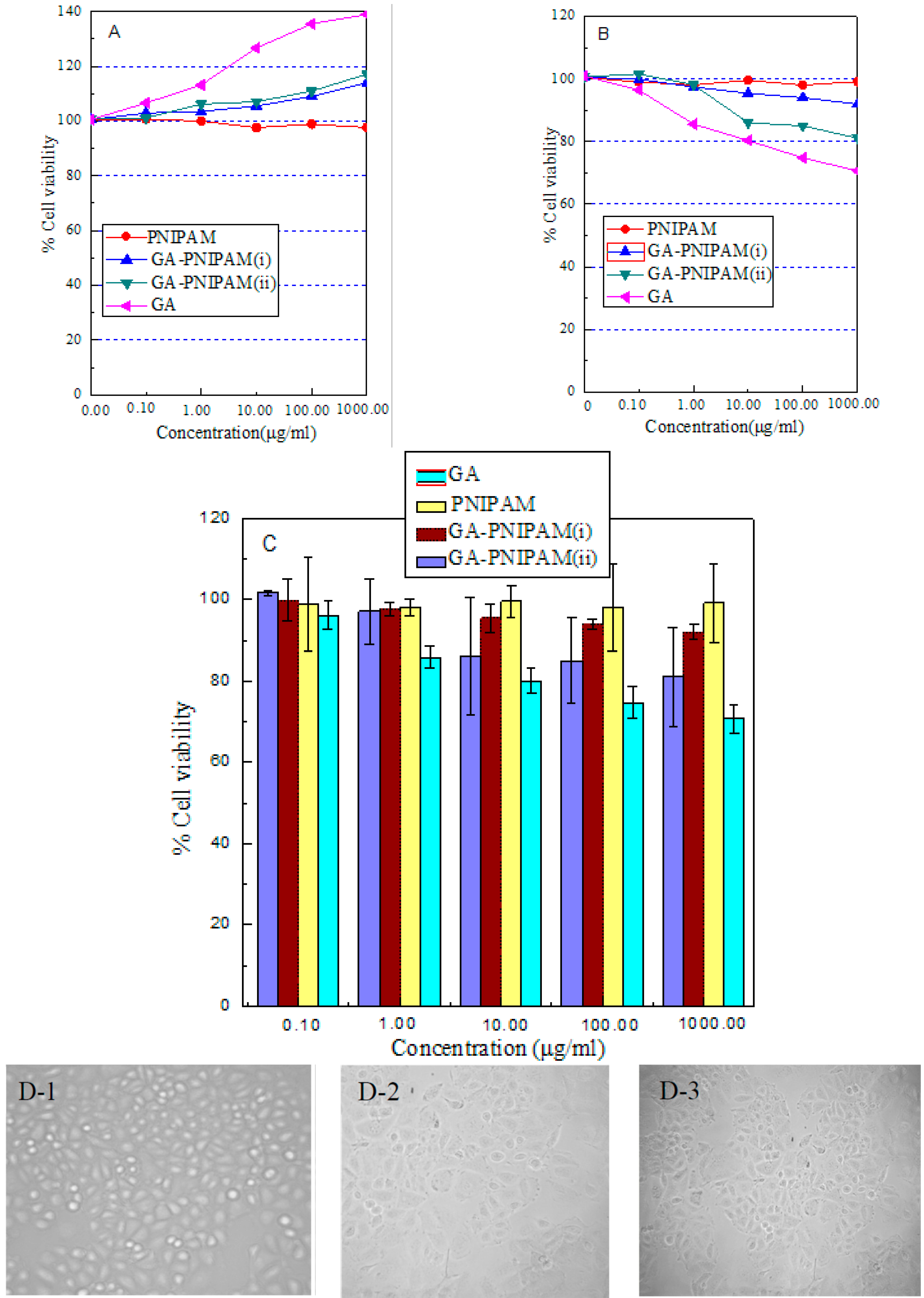
3.3. Assessment of Cell Viability
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kazuyuki, H.; Takashi, Y.; Toshiyuki, U. Ring-opening polymerization of new 1,6-anhydro-β-d-glucosamine derivatives. Carbohydr. Polym. 1998, 36, 129–135. [Google Scholar]
- Akiyama, K.; Kawazu, K.; Kobayashi, A. A novel method for chemo-enzymatic synthesis of elicitor-active chitosan oligomers and partially N-deacetylated chitin oligomers using N-acylated chitotrioses as substrates in a lysozyme-catalyzed trans-glycosylation reaction system. Carbohydr. Res. 1995, 279, 151–160. [Google Scholar] [CrossRef]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, L.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Sears, P.; Wong, C.H. Toward automated synthesis of oligosaccharides. Science 2001, 291, 2344–2350. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.J.; Skehan, P. Membrane-active drugs potentiate the killing of tumor cells by d-glucosamine. Proc. Natl. Acad. Sci. USA 1980, 77, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Molnar, Z.; Bekesi, J.G. Effects of d-glucosamine, d-mannosamine, and 2-deoxy-d-glucose on the ultrastructure of ascites tumor cells in vitro. Cancer Res. 1972, 32, 380–389. [Google Scholar] [PubMed]
- Molnar, Z.; Bekesi, J.G. Cytotoxic effects of d-glucosamine on the ultrastructures of normal and neoplastic tissues in vivo. Cancer Res. 1972, 32, 756–765. [Google Scholar] [PubMed]
- Reginster, J.Y.; Deroisy, R. Glucosamine for osteoarthritis: Dawn of a new era. Lancet 2001, 357, 251–256. [Google Scholar] [CrossRef]
- Posakony, J.J.; Adrian, R.F. Glucosamine and glucosamine-6-phosphate derivatives: Catalytic Cofactor Analogues for the glmS Ribozyme. J. Org. Chem. 2013, 78, 4730–4743. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Rethwisch, D.G. Scale-up of pseudo solid-phase enzymatic synthesis of alpha-methyl glucoside acrylate. Biotech. Bioeng. 2002, 79, 15–22. [Google Scholar] [CrossRef]
- Nisao, M.D.; Pedatella, S.; Bektas, S.; Nucci, A.; Caputo, R. d-Glucosamine in a chimeric prolinamide organocatalyst for direct asymmetric aldol addition. Carbohydr. Res. 2012, 356, 273–277. [Google Scholar]
- Wang, X.; Lin, Y.; Zeng, Y.; Sun, X.; Gong, T.; Zhang, Z. Nano-lipoidal carriers of tretinoin with enhanced percutaneous absorption, photostability, biocompatibility and anti-psoriatic activity. Int. J. Pharm. 2013, 456, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Hong, P.K.; Gottardi, D.; Ndagijimana, M.; Betti, M. Glycation and transglutaminase mediated glycosylation of fish gelatin peptides with glucosamine enhance bioactivity. Food Chem. 2014, 142, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Hrynets, Y.; Maurice, N.; Betti, M. Food hydrocolloids, Transglutaminase-catalyzed glycosylation of natural actomyosin (NAM) using glucosamine as amine donor: Functionality and gel microstructure. Food Hydrocoll. 2014, 36, 26–36. [Google Scholar] [CrossRef]
- Danac, R.; Ball, L.; Gurr, S.J.; Muller, T.; Fairbanks, A. Carbohydrate chain terminators: Rational design of novel carbohydrate-based antifungal agents. J. ChemBioChem 2007, 8, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.P.; Ramirez, C.M.; Miljkovic, N.; Li, H.; Rubin, J.P.; Marra, K.G. Thermosensitive injectable hyaluronic acid hydrogel for adipose tissue engineering. Biomaterials 2009, 30, 6844–6853. [Google Scholar] [CrossRef] [PubMed]
- Teng, D.Y.; Hou, J.L.; Zhang, X.G.; Wang, X.Z.; Li, C.X. Glucosamine-carrying temperature- and pH-sensitive microgels: Preparation, characterization, and in vitro drug release studies. J. Coll. Interface Sci. 2008, 322, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Schild, H.G. Poly(N-isopropylacrylamide)-experiment, theory and application. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Gil, E.S.; Hudson, S.M. Stimuli-reponsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Wang, D.; Liu, T.; Yin, J.; Liu, S.Y. Stimuli-responsive fluorescent Poly(N-isopropylacrylamide) microgels labeled with phenylboronic acid moieties as multifunctional ratiometric probes for glucose and temperatures. Macromolecules 2011, 44, 2282–2290. [Google Scholar] [CrossRef]
- Gody, G.; Boullanger, P.; Ladaviere, C.; Charreyre, M.T.; Delair, T. Biotin alpha-end-functionalized gradient glycopolymers synthesized by RAFT copolymerization. Macromol. Rapid Commun. 2008, 29, 511–519. [Google Scholar] [CrossRef]
- Cui, G.H.; Li, Y.H.; Shi, T.T.; Gao, Z.G.; Qiu, N.N.; Satoh, T.; Kakuchi, T.; Duan, Q. Synthesis and characterization on Eu(III) complexes of modified cellulose and poly(N-isopropylacrylamide). Carbohydr. Polym. 2013, 94, 77–81. [Google Scholar]
- Emmadi, M.; Kulkarni, S.S. Rapid transformation of d-Mannose into orthogonally protected d-Glucosamine and d-Galactosamine thioglycosides. J. Org. Chem. 2011, 76, 4703–4709. [Google Scholar] [CrossRef] [PubMed]
- Narain, R.; Armes, S.P. Synthesis of low polydispersity, controlled-structure sugar methacrylate polymers under mild conditions without protecting group chemistry. Chem. Commun. 2002, 23, 2776–2777. [Google Scholar] [CrossRef]
- Housni, A.; Cai, H.J.; Liu, S.Y.; Pun, S.H.; Narain, R. Facile preparation of glyconanoparticles and their bioconjugation to streptavidin. Langmuir 2007, 23, 5056–5061. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.S.; Nitz, M. syntheses of carbocyclic analogues of α-d-Glucosamine, α-d-Mannose, α-d-mannuronic acid, β-l-Idosamine, and β-l-Gulose. J. Org. Chem. 2012, 77, 7401–7410. [Google Scholar] [CrossRef] [PubMed]
- Nakamatu, K. Synthesis, luminescence quantum yields, and lifetimes of trischelated ruthenium (II) mixed-ligand complexes including 3,3′-Dimethyl-2,2′-bipyridyl. Bull. Chem. Soc. Jpn. 1982, 55, 2697–2702. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | GA-PNIPAM (i) | GA-PNIPAM (ii) | ||||
|---|---|---|---|---|---|---|
| Conversion (%) | GPC a | GPC a | Conversion (%) | GPC a | GPC a | |
| 1 | 12.7 | 1710 | 1.17 | 11.6 | 1570 | 1.14 |
| 3 | 22.5 | 2590 | 1.14 | 20.1 | 2340 | 1.09 |
| 6 | 48.3 | 4190 | 1.18 | 49.4 | 4430 | 1.10 |
| 9 | 58.8 | 4880 | 1.16 | 61.5 | 5620 | 1.17 |
| 12 | 69.6 | 6100 | 1.14 | 69.3 | 6680 | 1.15 |
| Sample | Time (h) | GA-PNIPAM (i) | GA-PNIPAM (ii) | ||||
|---|---|---|---|---|---|---|---|
| GPC a | GA (%) | LCST (°C) | GPC a | GA (%) | LCST (°C) | ||
| P1 | 1 | 1710 | 19.19 | 43.6 | 1570 | 20.01 | 43.7 |
| P2 | 3 | 2590 | 12.67 | 42.1 | 2340 | 13.42 | 41.8 |
| P3 | 6 | 4190 | 7.83 | 38.8 | 4430 | 7.09 | 38.1 |
| P4 | 9 | 4880 | 6.72 | 36.9 | 5620 | 5.59 | 36.6 |
| P5 | 12 | 6100 | 5.37 | 34.7 | 6680 | 4.70 | 34.5 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, G.; Gao, Z.; Qiu, N.; Satoh, T.; Kakuchi, T.; Duan, Q. End-Functionalized Poly(N-isopropylacrylamide) with d-Glucosamine through Different Initiator from C-1 and C-2 Positions via Atom Transfer Radical Polymerization. Materials 2016, 9, 913. https://doi.org/10.3390/ma9110913
Cui G, Gao Z, Qiu N, Satoh T, Kakuchi T, Duan Q. End-Functionalized Poly(N-isopropylacrylamide) with d-Glucosamine through Different Initiator from C-1 and C-2 Positions via Atom Transfer Radical Polymerization. Materials. 2016; 9(11):913. https://doi.org/10.3390/ma9110913
Chicago/Turabian StyleCui, Guihua, Zhengguo Gao, Nannan Qiu, Toshifumi Satoh, Toyoji Kakuchi, and Qian Duan. 2016. "End-Functionalized Poly(N-isopropylacrylamide) with d-Glucosamine through Different Initiator from C-1 and C-2 Positions via Atom Transfer Radical Polymerization" Materials 9, no. 11: 913. https://doi.org/10.3390/ma9110913
APA StyleCui, G., Gao, Z., Qiu, N., Satoh, T., Kakuchi, T., & Duan, Q. (2016). End-Functionalized Poly(N-isopropylacrylamide) with d-Glucosamine through Different Initiator from C-1 and C-2 Positions via Atom Transfer Radical Polymerization. Materials, 9(11), 913. https://doi.org/10.3390/ma9110913