
The Role of Computer Simulation in Nanoporous Metals—A Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
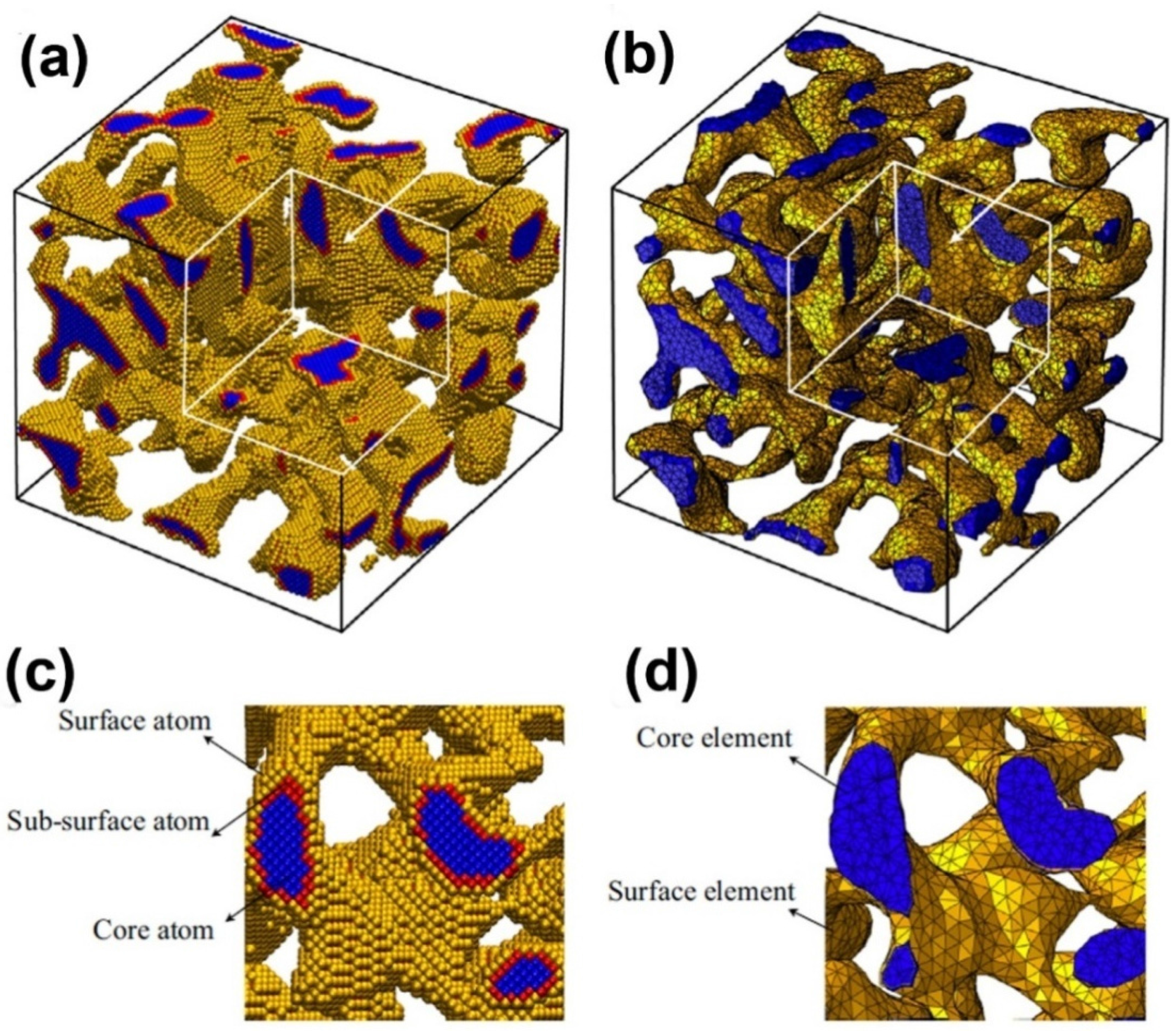
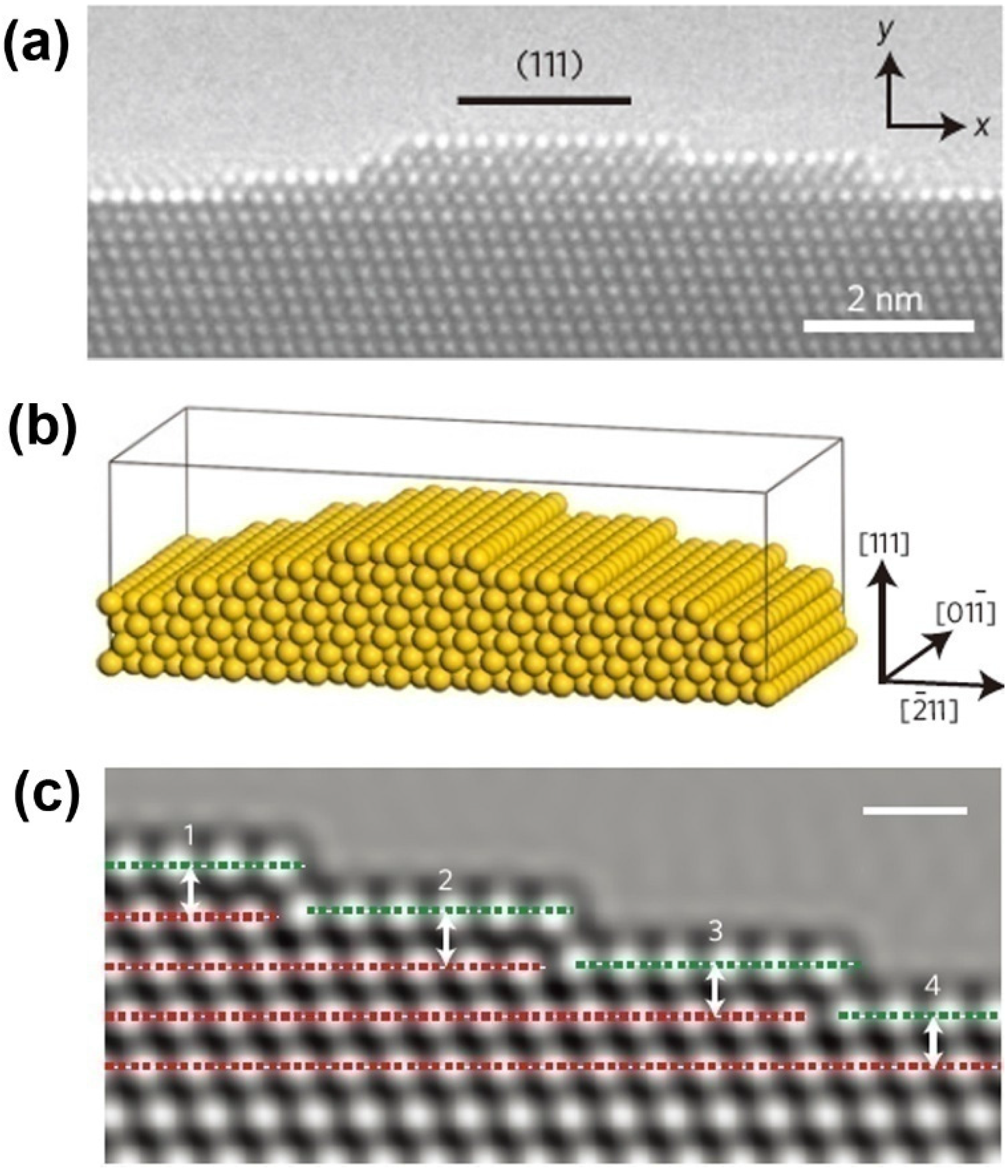
2. Simulation Methods
2.1. Ab Initio Calculation
2.2. Molecular Dynamics
2.3. Kinetic Monte Carlo Simulations
2.4. Finite Element Method
2.5. Phase Field Method
3. Simulation on Alloy Dealloying
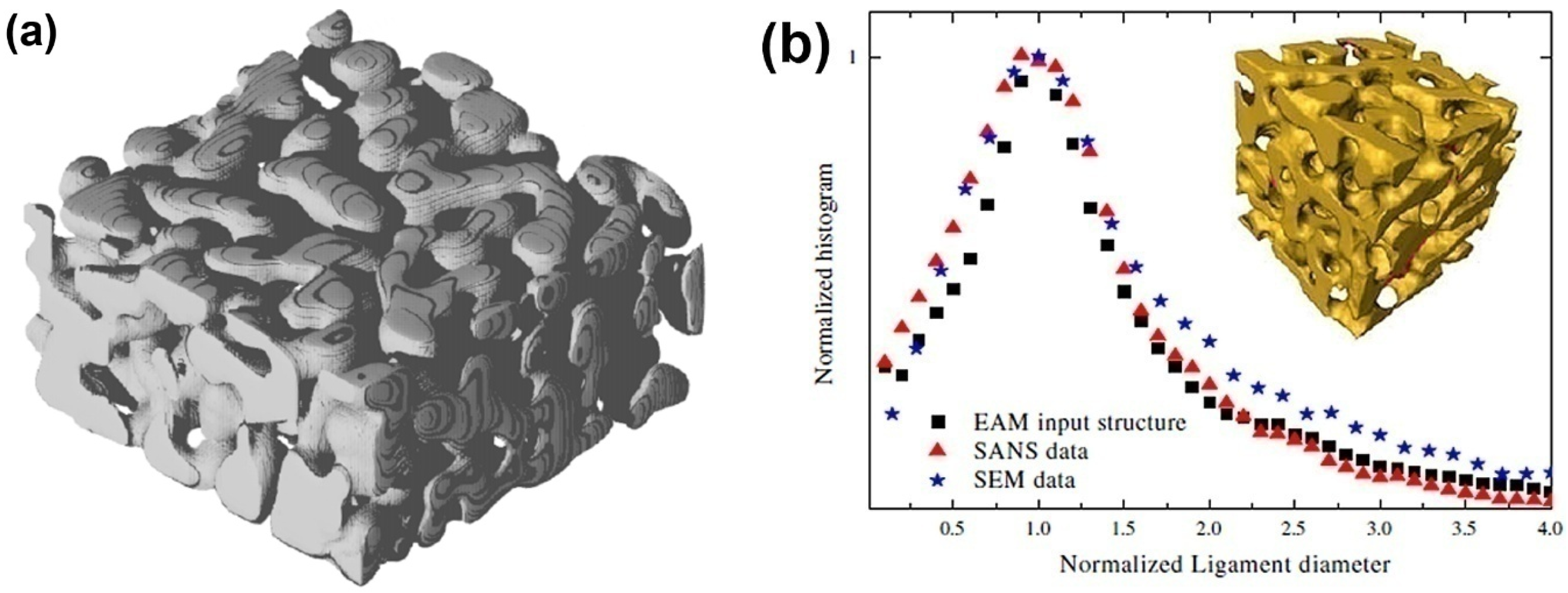
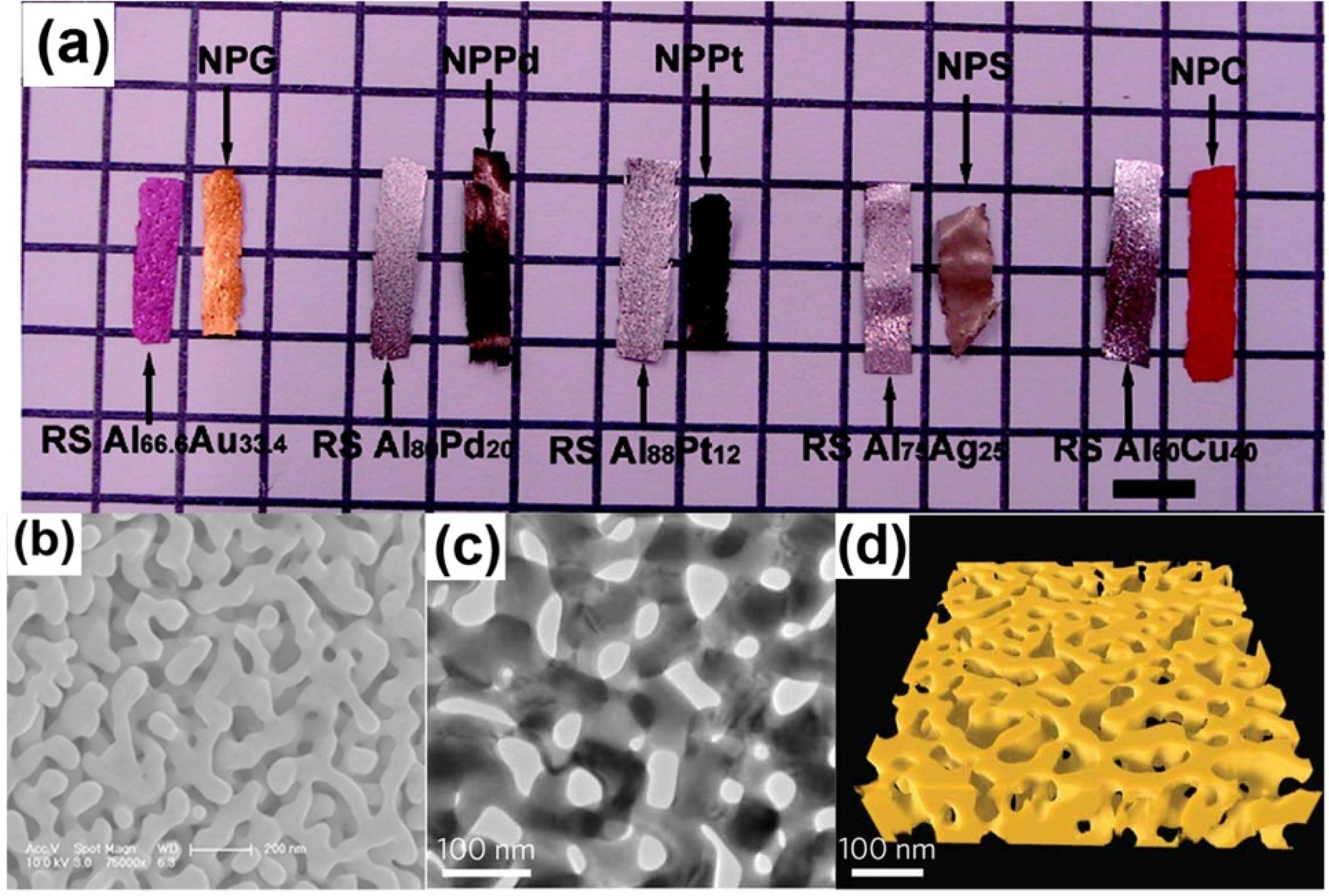
4. Mechanism of Coarsening
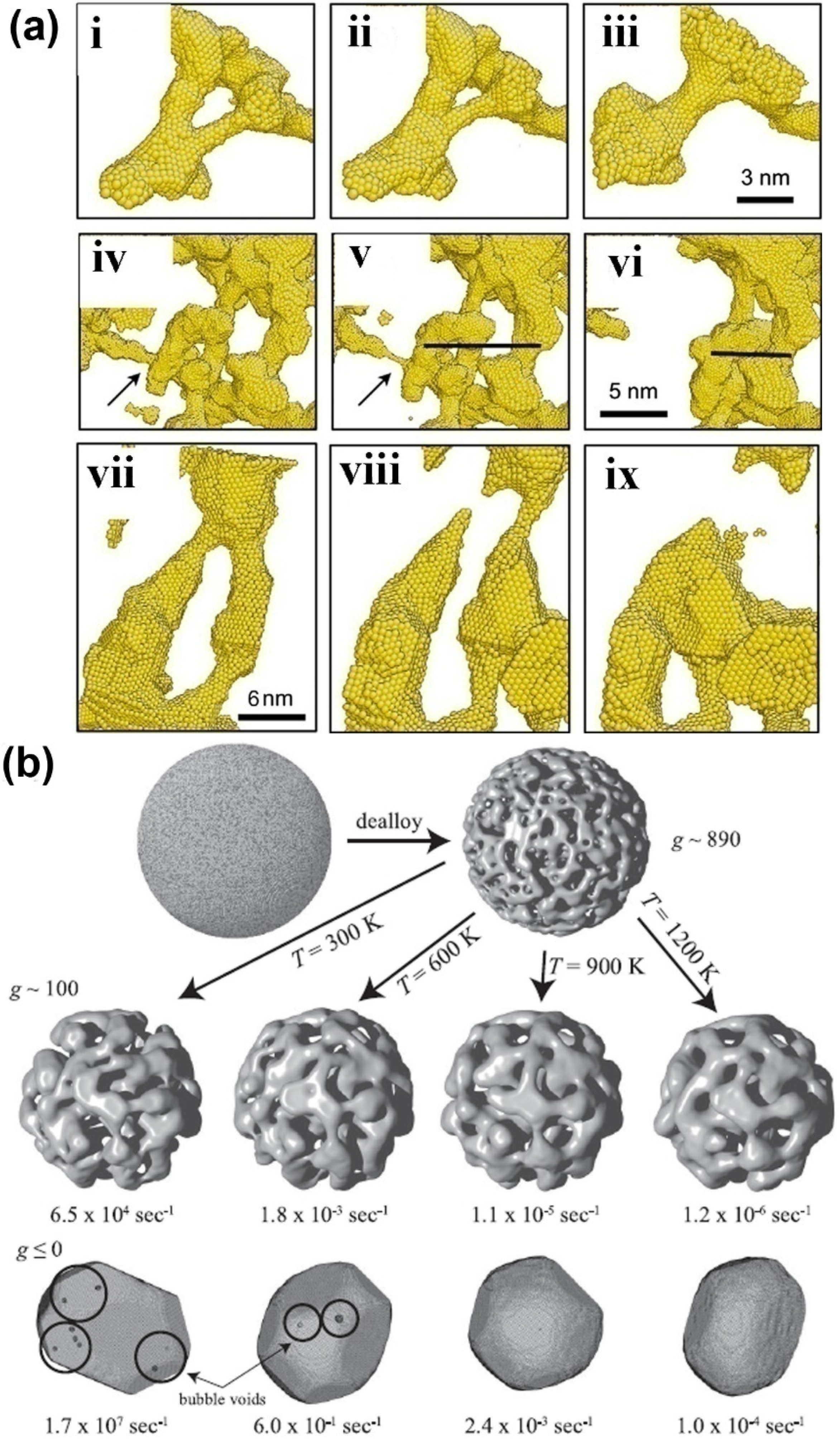
5. Mechanical Behavior of Nanoporous Metals
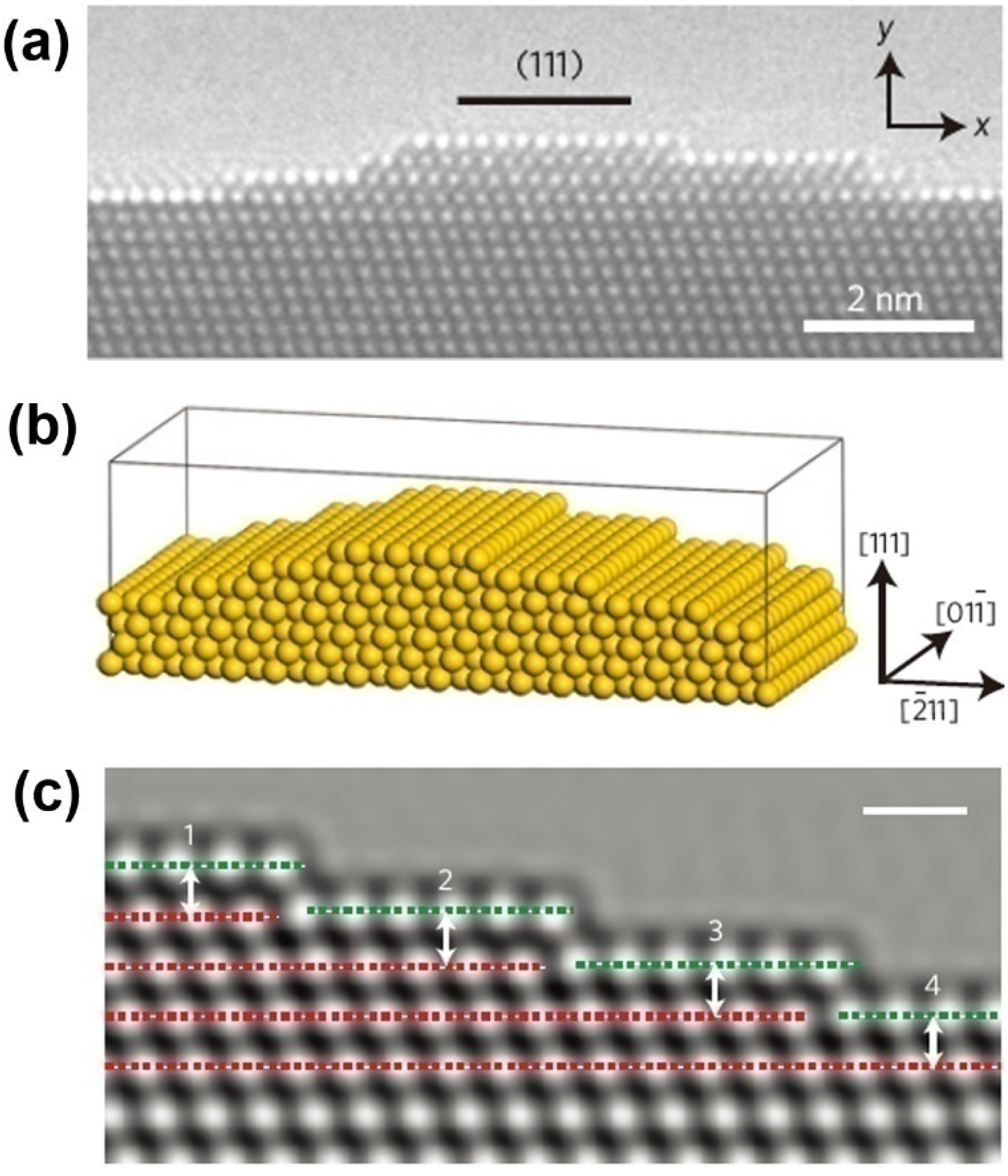
5.1. Surface Effect on Mechanical Properties
5.2. Deformation Mechanisms under Uniaxial Tension/Compression Loading
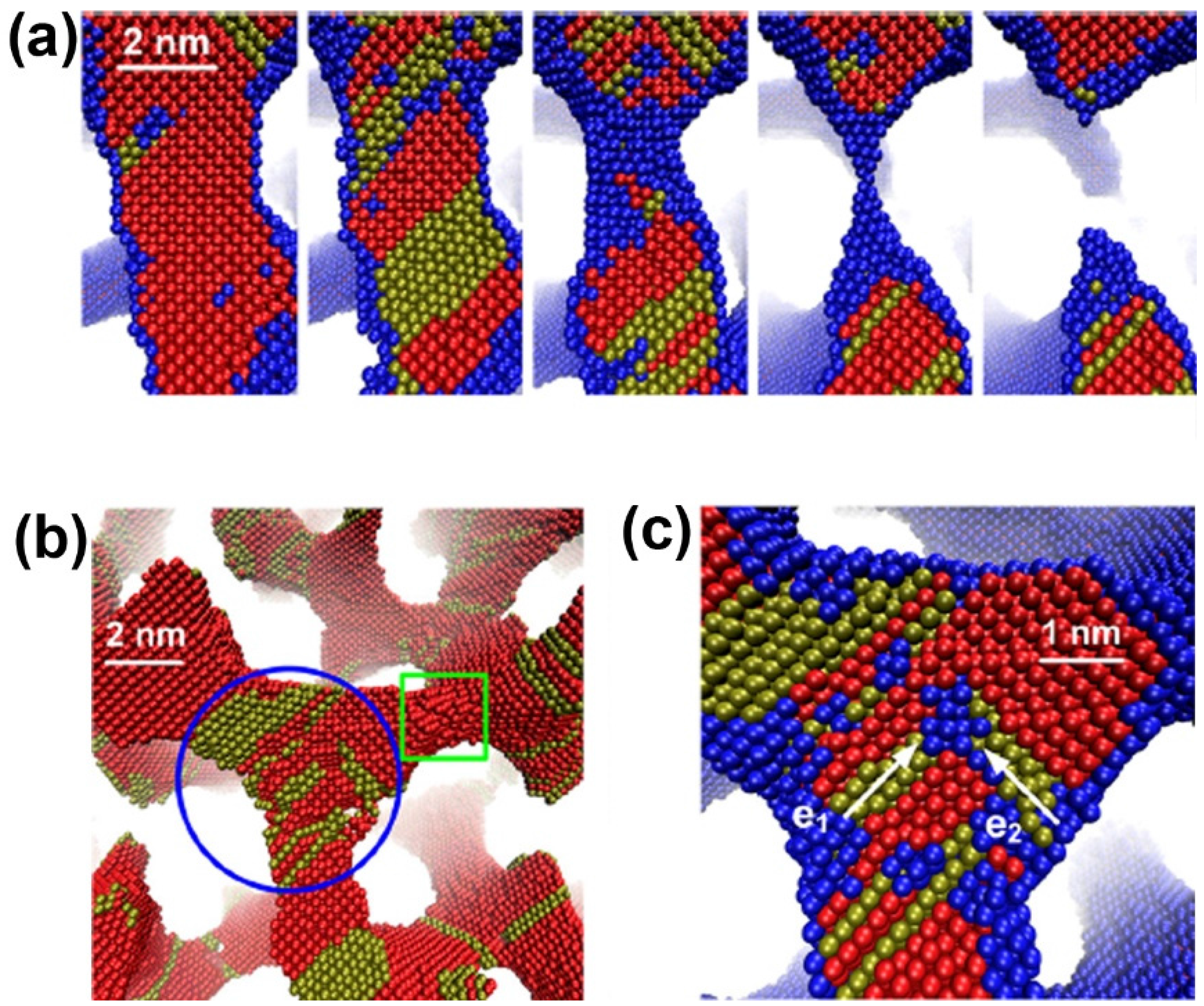
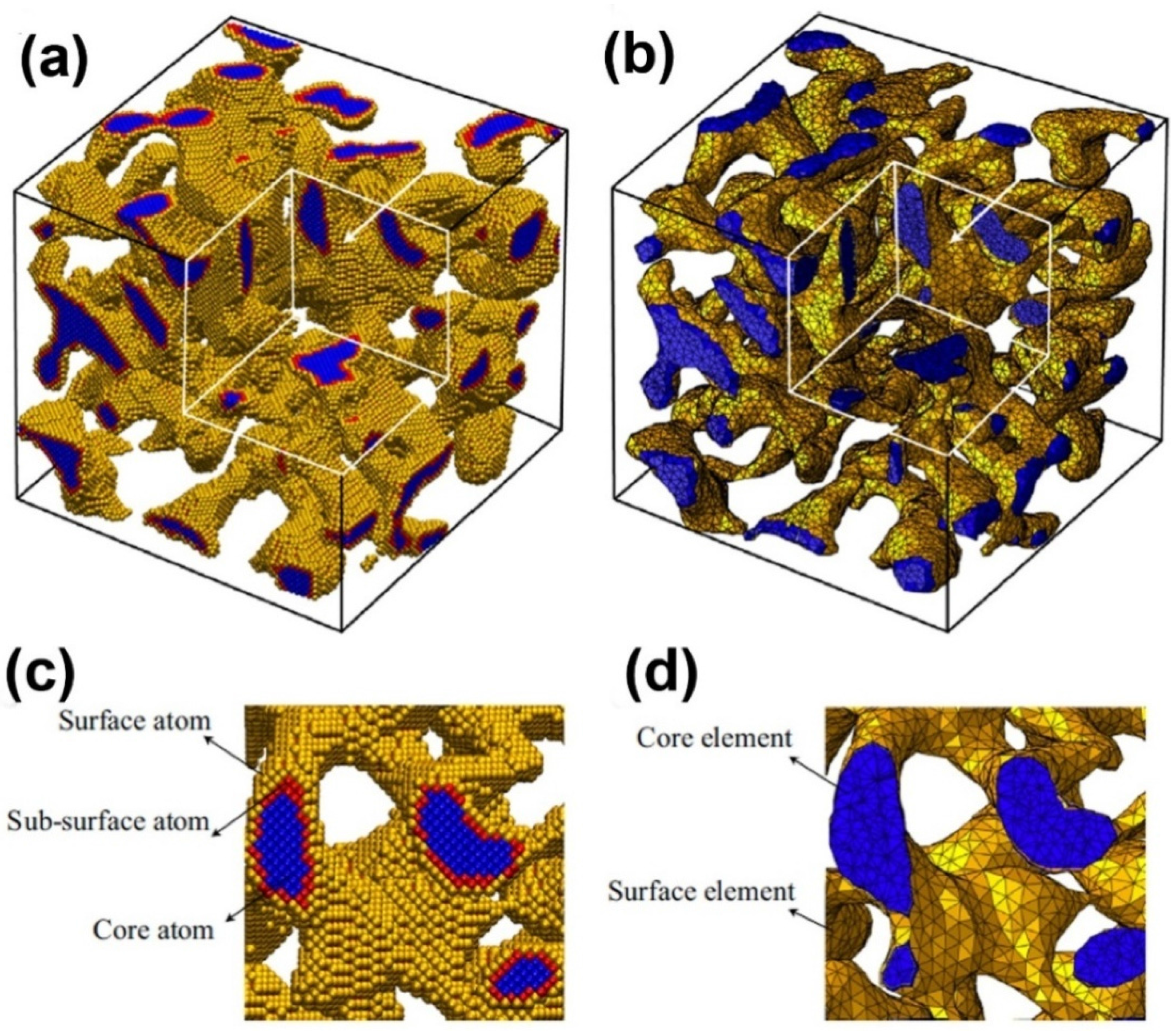
5.3. Scaling Laws
6. Other Properties
6.1. Thermal Properties
6.2. Optical and Magnetic Properties
6.3. High Catalytic Activity and Radiation Tolerance
7. Summary and Future Prospects
7.1. Summary
7.2. Future
- (1)
- The mechanical properties of NPMs can be significantly regulated through depositing only an atomic-layer-thick coating on its surface [112]. However, the underlying physical mechanisms are still open to further investigations. Thus, studying the surface modification effects of NPMs on mechanical properties by using multi-scale method is necessary.
- (2)
- The scaling laws for the mechanical properties of NPMs with surface adsorption/modification effects in terms of microstructure and coating parameters need to be developed. Efforts to explore the electric control of surface effects would be useful. In experiments, polycrystalline structural ligaments of NPMs are sometimes observed [113]. However, the ligaments in previous simulations are mostly considered to be monocrystal. It is necessary to find out the mechanical properties of NPMs, which are more sensitive to the ligament size or grain size.
- (3)
- Porous materials are widely used for energy absorption. Under compression, the energy is absorbed by the bending, buckling and fracture of the ligaments of the NPMs. The shock will induce dislocation nucleation in the NPMs. Therefore, the mechanical behavior of NPM under ultrahigh loading rates should be investigated.
- (4)
- The mechanical properties of NMPs are influenced considerably by their microstructures (geometry, topology, size, etc.). However, based on the current MD or KMC simulation, it is hard to predict the microstructure of practical NMPs from the beginning of the dealloying process. Large-scale (both spatial and temporal) computational frameworks which can capture the details of the atomic scale should be developed. Moreover, the macroscopic surface of NPMs is very rough, which makes for an ultrahigh frictional coefficient. Exploring the mechanism of the frictional behavior may provide guidelines to design optimal-performance NPMs.
- (5)
- Considering the complexity of the structures, simulations should also capture the plastic deformation localization at the weakest point of NMPs and the microscale heterogeneity of the structures. Besides, the chemical catalysis, thermal and electrical conductivity, and fluid transfer of NMPs are important for their potential applications, and these properties should be studied in future simulations.
- (6)
- In the future, multi-scale computational simulation methods should be further developed to capture the hierarchical structures of NMPs to predict the physical and mechanical properties from the bottom level. The chemical and electrical properties are also very important in applications for NPMs, such as catalysts, chemical adsorption, sensors, etc. Further computational simulations should also describe these properties of NPMs. Besides, the NMPs could be polycrystalline and work in multiple fields (e.g., electromagnetic field, chemical field, etc.) which should also be considered in computational simulations.
Acknowledgments
Conflicts of Interest
References
- Tappan, B.C.; Steiner, S.A., III; Luther, E.P. Nanoporous metal foams. Angew. Chem. Int. Ed. 2010, 49, 4544–4565. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.C.; Daniel, T.A.; Hieda, M.; Smilgies, D.M.; Chan, M.H.W.; Allara, D.L. Preparation, structure, and optical properties of nanoporous gold thin films. Langmuir 2007, 23, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Biener, J.; Erlebacher, J.; Bäumer, M. Nanoporous Gold: From an Ancient Technology to a High-Tech Material; Royal Society of Chemistry: London, UK, 2012. [Google Scholar]
- Ding, Y.; Chen, M.W. Nanoporous metals for catalytic and optical applications. MRS Bull. 2009, 34, 569–576. [Google Scholar] [CrossRef]
- Wittstock, A.; Biener, J.; Bäumer, M. Nanoporous gold: A new material for catalytic and sensor applications. Phys. Chem. Chem. Phys. 2010, 12, 12919–12930. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Ulstrup, J.; Li, H.; Wang, M.; Zhang, J.; Si, P. Nanoporous gold assembly of glucose oxidase for electrochemical biosensing. Electrochim. Acta 2014, 130, 559–567. [Google Scholar] [CrossRef]
- Ruffato, G.; Romanato, F.; Garoli, D.; Cattarin, S. Nanoporous gold plasmonic structures for sensing applications. Opt. Express 2011, 19, 13164–13170. [Google Scholar] [CrossRef] [PubMed]
- Hakamada, M.; Mabuchi, M. Fabrication, microstructure, and properties of nanoporousPd, Ni, and their alloys by dealloying. Crit. Rev. Solid State 2013, 38, 262–285. [Google Scholar] [CrossRef]
- Weissmüller, J.; Newman, R.C.; Jin, H.J.; Hodge, A.M.; Kysar, J.W. Nanoporous metals by alloy corrosion: Formation and mechanical properties. MRS Bull. 2009, 34, 577–586. [Google Scholar] [CrossRef]
- Kramer, D.; Viswanath, R.N.; Weissmüller, J. Surface-stress induced macroscopic bending of nanoporous gold cantilevers. Nano Lett. 2004, 4, 793–796. [Google Scholar] [CrossRef]
- Jin, H.J.; Weissmüller, J. A material with electrically tunable strength and flow stress. Science 2011, 332, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Okada, H.; Koyama, K.; Watanabe, K.; Maekawa, S.; Chen, M.W. Unusually small electrical resistance of three-dimensional nanoporous gold in external magnetic fields. Phys. Rev. Lett. 2008, 101, 166601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chang, H.; Hirata, A.; Wu, H.; Xue, Q.K.; Chen, M.W. Nanoporous gold based optical sensor for sub-ppt detection of mercury ions. ACS Nano 2013, 7, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhu, Y.; Wang, H.; Ding, Y. Nanoporous gold as an active low temperature catalyst toward CO oxidation in hydrogen-rich stream. Sci. Rep. UK 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Kashefi-Kheyrabadi, L.; Noori, A.; Mehrgardi, M.A. Biosensors Nanotechnology; Tiwari, A., Turner, A.P.F., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 347–369. [Google Scholar]
- Xu, Q. Nanoporous Materials: Synthesis and Applications; CRC Press: New York, NY, USA, 2013. [Google Scholar]
- Seker, E.; Reed, M.L.; Begley, M.R. Nanoporous gold: Fabrication, characterization, and applications. Materials 2009, 2, 2188–2215. [Google Scholar] [CrossRef]
- Biener, J.; Wittstock, A.; Baumann, T.F.; Weissmüller, J.; Bäumer, M.; Hamza, A.V. Surface chemistry in nanoscale materials. Materials 2009, 2, 2404–2428. [Google Scholar] [CrossRef]
- Kertis, F.; Snyder, J.; Govada, L.; Khurshid, S.; Chayen, N.; Erlebacher, J. Structure/processing relationships in the fabrication of nanoporous gold. JOM 2010, 62, 50–56. [Google Scholar] [CrossRef]
- Zheng, L.T.; Wei, Y.L.; Gong, H.Q.; Qian, L. Application progress of nanoporous gold in analytical chemistry. Chin. J. Anal. Chem. 2013, 41, 137–144. [Google Scholar] [CrossRef]
- Biener, J.; Wittstock, A.; Zepeda-Ruiz, L.A.; Biener, M.M.; Zielasek, V.; Kramer, D.; Viswanath, R.N.; Weissmüller, J.; Bäumer, M.; Hamza, A.V. Surface-chemistry-driven actuation in nanoporous gold. Nat. Mater. 2009, 8, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Wang, Y.; Qi, Z.; Zhuang, W.; Qin, J.; Frenzel, J. Generalized fabrication of nanoporous metals (Au, Pd, Pt, Ag, and Cu) through chemical dealloying. J. Phys. Chem. C 2009, 113, 12629–12636. [Google Scholar] [CrossRef]
- Fujita, T.; Guan, P.; McKenna, K.; Lang, X.; Hirata, A.; Zhang, L.; Tokunaga, T.; Arai, S.; Yamamoto, Y.; Nobuo Tanaka, N.; et al. Atomic origins of the high catalytic activity of nanoporous gold. Nat. Mater. 2012, 11, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, J.; Aziz, M.J.; Karma, A.; Dimitrov, N.; Sieradzki, K. Evolution of nanoporosity in dealloying. Nature 2001, 410, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.; Koo, E. Scale-and shape-dependent transport property of nanoporous materials. J. Appl. Phys. 2013, 113. [Google Scholar] [CrossRef]
- Crowson, D.A.; Farkas, D.; Corcoran, S.G. Mechanical stability of nanoporous metals with small ligament sizes. Scripta Mater. 2009, 61, 497–499. [Google Scholar] [CrossRef]
- Curtarolo, S.; Setyawan, W.; Wang, S.; Xue, J.; Yang, K.; Taylor, R.H.; Nelson, L.J.; Hart, G.L.W.; Sanvito, S.; Buongiorno Nardelli, M.; et al. AFLOWLIB.ORG: A distributed materials properties repository from high-throughput ab initio calculations. Comp. Mater. Sci. 2012, 58, 227–235. [Google Scholar] [CrossRef]
- Epelbaum, E.; Krebs, H.; Lee, D.; Meißner, U.-G. Ab initio calculation of the Hoyle State. Phys. Rev. Lett. 2011, 106. [Google Scholar] [CrossRef]
- Nazarov, R.; Hickel, T.; Neugebauer, J. Ab initio study of H-vacancy interactions in fcc metals: Implications for the formation of superabundant vacancies. Phys. Rev. B 2014, 89. [Google Scholar] [CrossRef]
- Jansen, G.R.; Engel, J.; Hagen, G.; Navratil, P.; Signoracci, A. Ab initio coupled-cluster effective interactions for the shell model: Application to neutron-rich oxygen and carbon isotopes. Phys. Rev. Lett. 2014, 113. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Schaeffer, H.F. Quantum Chemistry: The Development of Ab Initio Methods in Molecular Electronic Structure Theory; Clarendon Press: Oxford, UK, 1984. [Google Scholar]
- Arita, R.; Kuneš, J.; Kozhevnikov, A.V.; Equiluz, A.G.; Imada, M. Ab initio studies on the interplay between spin-orbit interaction and Coulomb correlation in Sr2Ir4 and Ba2IrO4. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef]
- Ohno, K.; Esfarjani, K.; Kawazoe, Y. Computational Materials Science: From Ab Initio to Monte Carlo Methods; Springer-Verlag: Heidelberg, Germany, 1999. [Google Scholar]
- Marx, D.; Hutter, J. Ab initio molecular dynamics: Theory and Implementation. In Modern Methods and Algorithms of Quantum Chemistry; NIC Series: Forschungszentrum, Germany, 2000; Volume 1, pp. 301–449. [Google Scholar]
- Saane, S.S.R.; Mangipudi, K.R.; Loos, K.U.; de Hosson, J.T.M.; Onck, P.R. Multiscale modeling of charge-induced deformation of nanoporous gold structures. J. Mech. Phys. Solids 2014, 66, 1–15. [Google Scholar] [CrossRef]
- Liu, Y.L.; Xu, Z.P. Multimodal and self-healable interfaces enable strong and tough grapheme-derived materials. J. Mech. Phys. Solids 2014, 70, 30–41. [Google Scholar] [CrossRef]
- Sun, X.Y.; Li, Q.; Gu, Y.; Feng, X.Q. Mechanical properties of bioinspired bicontinuous nanocomposites. Comp. Mater. Sci. 2013, 80, 71–78. [Google Scholar] [CrossRef]
- Liu, Y.L.; Chen, X. Mechanical properties of nanoporousgraphene membrane. J. Appl. Phys. 2014, 115. [Google Scholar] [CrossRef]
- Allen, M.P. Introduction to Molecular Dynamics Simulation. In Computational Soft Matter: From Synthetic Polymers to Proteins; NIC Series: Jülich, Germany, 2004; Volume 23, pp. 1–28. [Google Scholar]
- Wang, C.C.; Mao, Y.W.; Shan, Z.W.; Dao, M.; Li, J.; Sun, J.; Ma, E.; Suresh, S. Real-time, high-resolution study of nanocrystallization and fatigue cracking in a cyclically strained metallic glass. Proc. Natl. Acad. Sci. USA 2013, 110, 19725–19730. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, C.R.; Cai, W. Plasticity of metal wires in torsion: Molecular dynamics and dislocation dynamics simulations. J. Mech. Phys. Solids 2010, 58, 1011–1025. [Google Scholar] [CrossRef]
- Rao, S.I.; Dimiduk, D.M.; El-Awady, J.A.; Parthasarathy, T.A.; Uchic, M.D.; Woodward, C. Spontaneous athermal cross-slip nucleation at screw dislocation intersections in FCC metals and L1 2 intermetallics investigated via atomistic simulations. Philos. Mag. 2013, 93, 3012–3028. [Google Scholar] [CrossRef]
- Higginbotham, A.; Suggit, M.J.; Bringa, E.M.; Erhart, P.; Hawreliak, J.A.; Mogni, G.; Park, N.; Remington, B.A.; Wark, J.S. Molecular dynamics simulations of shock-induced deformation twinning of a body-centered-cubic metal. Phys. Rev. B 2013, 88. [Google Scholar] [CrossRef]
- Ravelo, R.; Germann, T.C.; Guerrero, O.; An, Q.; Holian, B.L. Shock-induced plasticity in tantalum single crystals: Interatomic potentials and large-scale molecular-dynamics simulations. Phys. Rev. B 2013, 88. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Lu, L.; Lu, K.; Gao, H. Dislocation nucleation governed softening and maximum strength in nano-twinned metals. Nature 2010, 464, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Gao, H. Plastic deformation mechanism in nanotwinned metals: An insight from molecular dynamics and mechanistic modeling. Scr. Mater. 2012, 66, 843–848. [Google Scholar] [CrossRef]
- Rodriguez-Nieva, J.F.; Bringa, E.M. Molecular dynamics and Monte Carlo simulations of the sputtering of a nanoporous solid. Nuclear Instrum. Methods B 2013, 304, 23–26. [Google Scholar] [CrossRef]
- Chen, S.; Liang, J.; Mo, Y.; Luo, D.; Jiang, S. Onset of shadowing-dominated growth of Ag films in glancing angle deposition: Kinetic Monte Carlo simulation. Appl. Surf. Sci. 2013, 264, 552–556. [Google Scholar] [CrossRef]
- Schulze, T.P.; Smereka, P. Kinetic Monte Carlo simulation of heteroepitaxial growth: Wetting layers, quantum dots, capping, and nanorings. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Castin, N.; Pascuet, M.I.; Malerba, L. Mobility and stability of large vacancy and vacancy–copper clusters in iron: An atomistic kinetic Monte Carlo study. J. Nuclear Mater. 2012, 429, 315–324. [Google Scholar] [CrossRef]
- Dholabhai, P.P.; Anwar, S.; Adams, J.B.; Crozier, P.; Sharma, R. Kinetic lattice Monte Carlo model for oxygen vacancy diffusion in praseodymium doped ceria: Applications to materials design. J. Solid State Chem. 2011, 184, 811–817. [Google Scholar] [CrossRef]
- Molnar, D.; Mukherjee, R.; Choudhury, A.; Mora, A.; Binkele, P.; Selzer, M.; Nestler, B.; Schmauder, S. Multiscale simulations on the coarsening of Cu-rich precipitates in α-Fe using kinetic Monte Carlo, molecular dynamics and phase-field simulations. Acta Mater. 2012, 60, 6961–6971. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, W.; Bellon, P.; Averback, R.S.; Zuo, J.M. A kinetic Monte Carlo study of coarsening resistance of novel core/shell precipitates. Acta Mater. 2014, 79, 37–46. [Google Scholar] [CrossRef]
- Erlebacher, J. An atomistic description of dealloying porosity evolution, the critical potential, and rate-limiting behavior. J. Electrochem. Soc. 2004, 151, C614–C626. [Google Scholar] [CrossRef]
- Zinchenko, O.; De Raedt, H.A.; Detsi, E.; Onck, P.R.; de Hosson, J.T.M. Nanoporous gold formation by dealloying: A Metropolis Monte Carlo study. Comput. Phys. Commun. 2013, 184, 1562–1569. [Google Scholar] [CrossRef]
- Erlebacher, J.; McCue, I. Geometric characterization of nanoporous metals. Acta Mater. 2012, 60, 6164–6174. [Google Scholar] [CrossRef]
- Wang, J.; Lam, D.C. Model and analysis of size-stiffening in nanoporouscellular solids. J. Mater. Sci. 2009, 44, 985–991. [Google Scholar] [CrossRef]
- Huang, Y. A User-Material Subroutine Incorporating Single Crystal Plasticity in the ABAQUS Finite Element Program, Mech Report 178; Harvard University: Cambridge, MA, USA, 1991; pp. 1–46. [Google Scholar]
- Lee, D.; Wei, X.; Zhao, M.; Chen, X.; Jun, S.C.; Hone, J.; Kysar, J.W. Plastic deformation in nanoscale gold single crystals and open-celled nanoporous gold. Model. Simul. Mater. Sci. 2007, 15, S181–S192. [Google Scholar] [CrossRef]
- Zhu, H.X.; Zhang, H.C.; You, J.F.; Kennedy, D.; Wang, Z.B.; Fan, T.X.; Zhang, D. The elastic and geometrical properties of micro-and nano-structured hierarchical random irregular honeycombs. J. Mater. Sci. 2014, 49, 5690–5702. [Google Scholar] [CrossRef]
- Fujita, T.; Qian, L.H.; Inoke, K.; Erlebacher, J.; Chen, M.W. Three-dimensional morphology of nanoporous gold. Appl. Phys. Lett. 2008, 92. [Google Scholar] [CrossRef]
- Newman, R.C.; Corcoran, S.G.; Erlebacher, J.; Aziz, M.J.; Sieradzki, K. Alloy corrosion. MRS Bull. 1999, 24, 24–28. [Google Scholar]
- Pugh, D.V. Nanoporous Platinum. Ph.D. Thesis, Virginia Polytechnic Institute and State University, Montgomery County, VA, USA, 2003. [Google Scholar]
- Chan, J.W.; Hilliard, J.E. Free energy of a non-uniform system I. interfacial free energy. J. Chem. Phys. 1958, 28, 258–267. [Google Scholar]
- Rashed, J. Coarsening Dynamics for the Cahn-Hilliard Equation. Master’s Thesis, Technion-Israel Institute of Technology, Haifa, Israel, 2009. [Google Scholar]
- Rugolo, J.; Erlebacher, J.; Sieradzki, K. Length scales in alloy dissolution and measurement of absolute interfacial free energy. Nat. Mater. 2006, 5, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Artymowicz, D.M.; Erlebacher, J.; Newman, R.C. Relationship between the parting limit for de-alloying and a particular geometric high-density site percolation threshold. Philos. Mag. 2009, 89, 1663–1693. [Google Scholar] [CrossRef]
- Santiago-Cortés, C.U.; Mejía-Mendoza, L.M.; Valladares, R.M.; Valladares, A.; Valladares, A.A. Computational alternatives to generate amorphous nanoporous structures using ab initio molecular dynamics. J. Non-Cryst. Solids 2012, 358, 596–603. [Google Scholar] [CrossRef]
- Shruti, M.B. Statistical Analysis of Factors Affecting Nanoporous Gold and its Sensitivity in Comparison with Bulk Gold. Master’s Thesis, The University of Toledo, Toledo, OH, USA, 2010. [Google Scholar]
- Chen-Wiegart, Y.K.; Wang, S.; McNulty, I.; Dunand, D.C. Effect of Ag-Au composition and acid concentration on dealloying front velocity and cracking during nanoporous gold formation. Acta Mater. 2013, 61, 5561–5570. [Google Scholar] [CrossRef]
- Adisoejoso, J.; Tahara, K.; Lei, S.; Szabelski, P.; Rzysko, W.; Inukai, K.; Blunt, M.O.; Tobe, Y.; De Feyter, S. One building block, two different nanoporousself-assembled monolayers: A combined STM and Monte Carlo study. ACS Nano 2012, 6, 897–903. [Google Scholar] [CrossRef] [PubMed]
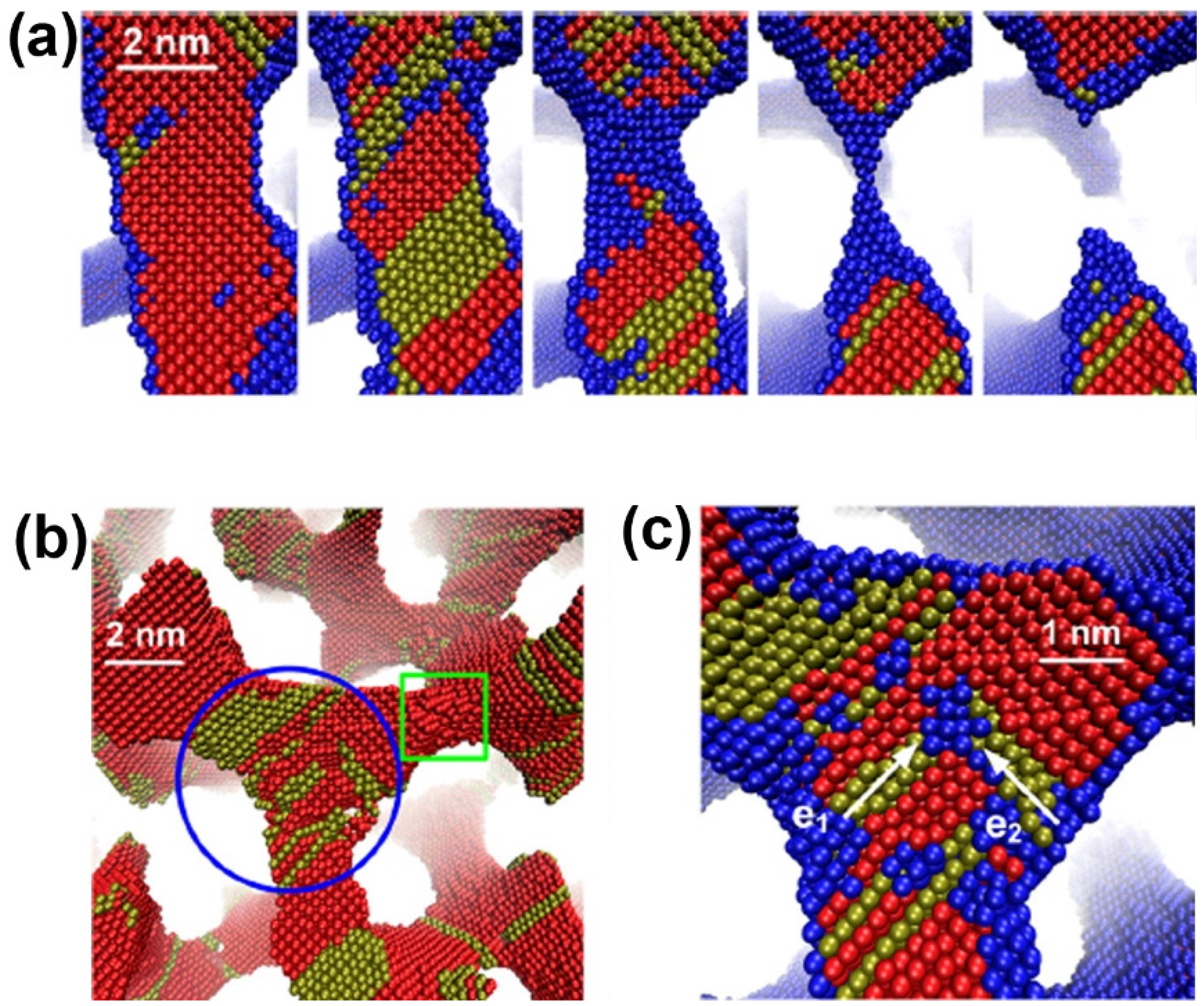
- Kolluri, K.; Demkowicz, M.J. Coarsening by network restructuring in model nanoporous gold. Acta Mater. 2011, 59, 7645–7653. [Google Scholar] [CrossRef]
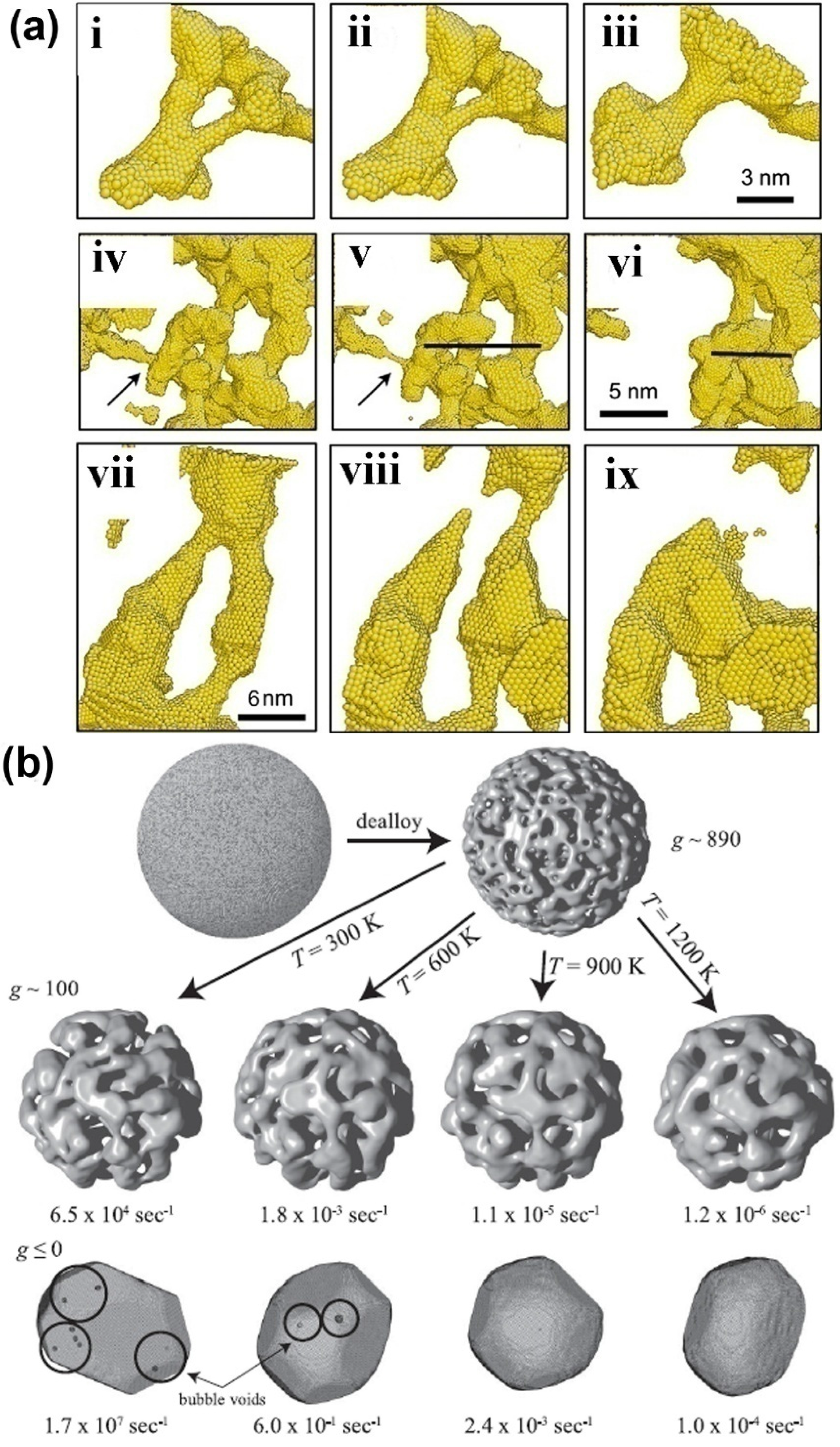
- Erlebacher, J. Mechanism of coarsening and bubble formation in high-genus nanoporous metals. Phys. Rev. Lett. 2011, 106. [Google Scholar] [CrossRef]
- Detsi, E.; de Jong, E.; Zinchenko, A.; Vuković, Z.; Vuković, I.; Punzhin, S.; Loos, K.; Ten Brinke, G.; de Raedt, H.A.; Onck, P.R.; et al. On the specific surface area of nanoporous materials. Acta Mater. 2011, 59, 7488–7497. [Google Scholar] [CrossRef]
- Xia, R.; Feng, X.Q.; Wang, G.F. Effective elastic properties of nanoporous materials with hierarchical structure. Acta Mater. 2011, 59, 6801–6808. [Google Scholar] [CrossRef]
- Crowson, D.A.; Farkas, D.; Corcoran, S.G. Geometric relaxation of nanoporous metals: The role of surface relaxation. Scripta Mater. 2007, 56, 919–922. [Google Scholar] [CrossRef]
- Weissmüller, J.; Duan, H.L.; Farkas, D. Deformation of solids with nanoscale pores by the action of capillary forces. Acta Mater. 2010, 58, 1–13. [Google Scholar] [CrossRef]
- Abdolrahim, N.; Bahr, D.F.; Revard, B.; Reilly, C.; Ye, J.; Balk, T.J.; Zbib, H.M. The mechanical response of core-shell structures for nanoporous metallic materials. Philos. Mag. 2013, 93, 736–748. [Google Scholar] [CrossRef]
- Cantrell, C. Molecular Dynamics Simulation of Mechanical Behavior of Nanoporous Copper Foams. Bachelor’s Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 2006. [Google Scholar]
- Ruestes, C.J.; Bringa, E.M.; Stukowski, A.; Rodríguez Nieva, J.F.; Tang, Y.; Meyers, M.A. Plastic deformation of a porous bcc metal containing nanometer sized voids. Comp. Mater. Sci. 2014, 88, 92–102. [Google Scholar] [CrossRef]
- Ruestes, C.J.; Bringa, E.M.; Stukowski, A.; Rodríguez Nieva, J.F.; Bertolino, G.; Tang, Y.; Meyers, M.A. Atomistic simulation of the mechanical response of a nanoporous body-centered cubic metal. Scr. Mater. 2013, 68, 817–820. [Google Scholar] [CrossRef]
- Erhart, P.; Bringa, E.M.; Kumar, M.; Albe, K. Atomistic mechanism of shock-induced void collapse in nanoporous metals. Phys. Rev. B 2005, 72. [Google Scholar] [CrossRef]
- To, A.C.; Tao, J.; Kirca, M.; Schalk, L. Ligament and joint sizes govern softening in nanoporous aluminum. Appl. Phys. Lett. 2011, 98. [Google Scholar] [CrossRef]
- Giri, A. Molecular Dynamics Simulations of the Mechanical Deformation of Nanoporous Gold. Bachelor’s Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2012. [Google Scholar]
- Giri, A.; Tao, J.; Wang, L.; Kirca, M.; To, A.C. Compressive behavior and deformation mechanism of nanoporous open-cell foam with ultrathin ligaments. J. Nanomech. Micromech. 2013, 4. [Google Scholar] [CrossRef]
- Sun, X.Y.; Xu, G.K.; Li, X.; Feng, X.Q.; Gao, H. Mechanical properties and scaling laws of nanoporous gold. J. Appl. Phys. 2013, 113. [Google Scholar] [CrossRef]
- Farkas, D.; Caro, A.; Bringa, E.; Crowson, D. Mechanical response of nanoporous gold. Acta Mater. 2013, 61, 3249–3256. [Google Scholar] [CrossRef]
- Biener, J.; Hodge, A.M.; Hayes, J.R.; Volkert, C.A.; Zepeda-Ruiz, L.A.; Hamza, A.V.; Abraham, F.F. Size effects on the mechancial behavior of nanoporous Au. Nano Lett. 2006, 6, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Wang, J.L.; Wang, R.; Zhang, X.; Feng, X.Q.; Ding, Y. Correlation of the thermal and electrical conductivities of nanoporous gold. Nanotechnology 2010, 21. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, A.I.; Vasić, R.; Dukesz, B.; Li, D.; Chen, M.W.; Zhang, L.; Fujita, T. Thermal properties of nanoporous gold. Phys. Rev. B 2012, 85. [Google Scholar] [CrossRef]
- Cheng, I.C.; Hodge, A.M. Morphology, oxidation, and mechanical behavior of nanoporous Cu foams. Adv. Eng. Mater. 2012, 14, 219–226. [Google Scholar] [CrossRef]
- Wang, J.L.; Xia, R.; Zhu, J.; Ding, Y.; Zhang, X.; Chen, Y. Effect of thermal coarsening on the themal conductivity of nanoporous gold. J. Mater. Sci. 2012, 47, 5013–5018. [Google Scholar] [CrossRef]
- Seker, E.; Gaskins, J.T.; Bart-Smith, H.; Zhu, J.; Reed, M.L.; Zangari, G.; Kelly, R.; Begley, M.R. The effects of annealing prior to dealloying on the mechanical properties of nanoporous gold microbeams. Acta Mater. 2008, 56, 324–332. [Google Scholar] [CrossRef]
- Luke, J.R.; Tien, C.L. Molecular dynamics simulation of thermal conduction in nanoporous thin films. Microscale Therm. Eng. 2004, 8, 341–359. [Google Scholar] [CrossRef]
- Bera, C.; Mingo, N.; Volz, S. Marked effects of alloying on the thermal conductivity of nanoporous materials. Phys. Rev. Lett. 2010, 104. [Google Scholar] [CrossRef]
- Sardana, N.; Birr, T.; Schlenker, S.; Reinhardt, C.; Schilling, J. Surface plasmons on ordered and bi-continuous spongy nanoporous gold. New J. Phys. 2014, 16. [Google Scholar] [CrossRef]
- Li, R.; Liu, X.J.; Wang, H.; Wu, X.; Chu, X.M.; Lu, Z.P. Nanoporous silver with tunable pore characteristics and superior surface enhanced Raman scattering. Corros. Sci. 2014, 84, 159–164. [Google Scholar] [CrossRef]
- Qi, J.; Motwani, P.; Gheewala, M.; Brennan, C.; Wolfe, J.C.; Shih, W.-C. Surface-enhanced Raman spectroscopy with monolithic nanoporous gold disk substrates. Nanoscale 2013, 5, 4105–4109. [Google Scholar] [CrossRef] [PubMed]
- Detsi, E.; Salverda, M.; Onck, P.R.; De Hosson, J.T.M. On the localized sufaceplasmon resonance modes in nanoporous gold films. J. Appl. Phys. 2014, 115. [Google Scholar] [CrossRef]
- Bosman, M.; Anstis, G.R.; Keast, V.J.; Clarke, J.D.; Cortie, M.B. Light splitting in nanoporous gold and silver. ACS Nano 2011, 6, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, J.; Nowaczyk, K.; Lakowicz, J.R. Enhanced single molecule fluorescence and reduced observation volumes on nanoporous gold (NPG) films. Chem. Commun. 2013, 49, 10874–10876. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Gu, Y.; Wu, Z.; Wang, Y.; Xu, S.; Xu, W. Surface-enhanced Raman scattering (SERS) biosensing based on nanoporous dielectric waveguide resonance. Sens. Actuators B Chem. 2014, 201, 173–176. [Google Scholar] [CrossRef]
- Zhao, F.; Zeng, J.; Parvez, A.M.; Sun, P.; Qi, J.; Motwani, P.; Gheewala, M.; Li, C.; Paterson, A.; Strych, U.; et al. Monolithic NPG nanoparticles with large surface area, tunable plasmonics, and high-density internal hot-spots. Nanoscale 2014, 6, 8199–8207. [Google Scholar] [CrossRef] [PubMed]
- Hakamada, M.; Hirashima, F.; Takahashi, M.; Nakazawa, T.; Mabuchi, M. Large-strain-induced magnetic properties of Co electrodeposited on nanoporous Au. J. Appl. Phys. 2011, 109. [Google Scholar] [CrossRef] [Green Version]
- Wittstock, A.; Wichmann, A.; Bäumer, M. Nanoporous gold as a platform for a building block catalyst. ACS Catal. 2012, 2, 2199–2215. [Google Scholar] [CrossRef]
- Xu, C.; Su, J.; Xu, X.; Liu, P.; Zhao, H.; Tian, F.; Ding, Y. Low temperature CO oxidation over unsupported nanoporous gold. J. Am. Chem. Soc. 2007, 129, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, I.J.; Caro, A.; Demkowicz, M.J.; Mara, N.A.; Misra, A.; Uberuaga, B.P. Radiation damage tolerant nanomaterials. Mater. Today 2013, 16, 443–449. [Google Scholar] [CrossRef]
- Bringa, E.M.; Monk, J.D.; Caro, A.; Misra, A.; Zepeda-Ruiz, L.; Duchaineau, M.; Nastasi, M.; Picraux, S.T.; Wang, Y.Q.; Farkas, D. Are nanoporous materials radiation resistant? Nano Lett. 2011, 12, 3351–3355. [Google Scholar] [CrossRef] [PubMed]
- Fu, E.G.; Caro, M.; Zepeda-Ruiz, L.A.; Wang, Y.Q.; Baldwin, K.; Nastasi, M.; Bringa, E.; Nastasi, M.; Caro, A. Surface effects on the radiation response of nanoporous Au foams. Appl. Phys. Lett. 2012, 101. [Google Scholar] [CrossRef]
- Sun, C.; Bufford, D.; Chen, Y.; Kirk, M.A.; Wang, Y.Q.; Li, M.; Wang, H.; Maloy, S.A.; Zhang, X. In situ study of defect migration kinetics in nanoporous Ag with enhanced radiation tolerance. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Biener, M.M.; Biener, J.; Wichmann, A.; Wittstock, A.; Baumann, T.F.; Bäumer, M.; Hamza, A.V. ALD functionalized nanoporous gold: Thermal stability, mechanical properties, and catalytic activity. Nano Lett. 2011, 11, 3085–3090. [Google Scholar] [CrossRef] [PubMed]
- Hakamada, M.; Mabuchi, M. Nanoporous gold prism microassembly through a self-organizing route. Nano Lett. 2006, 6, 882–885. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, R.; Wu, R.N.; Liu, Y.L.; Sun, X.Y. The Role of Computer Simulation in Nanoporous Metals—A Review. Materials 2015, 8, 5060-5083. https://doi.org/10.3390/ma8085060
Xia R, Wu RN, Liu YL, Sun XY. The Role of Computer Simulation in Nanoporous Metals—A Review. Materials. 2015; 8(8):5060-5083. https://doi.org/10.3390/ma8085060
Chicago/Turabian StyleXia, Re, Run Ni Wu, Yi Lun Liu, and Xiao Yu Sun. 2015. "The Role of Computer Simulation in Nanoporous Metals—A Review" Materials 8, no. 8: 5060-5083. https://doi.org/10.3390/ma8085060
APA StyleXia, R., Wu, R. N., Liu, Y. L., & Sun, X. Y. (2015). The Role of Computer Simulation in Nanoporous Metals—A Review. Materials, 8(8), 5060-5083. https://doi.org/10.3390/ma8085060