
Porous Materials for Hydrolytic Dehydrogenation of Ammonia Borane

Abstract
:1. Introduction
2. Porous Support Materials
2.1. Microporous and Mesoporous Inorganic Support Materials

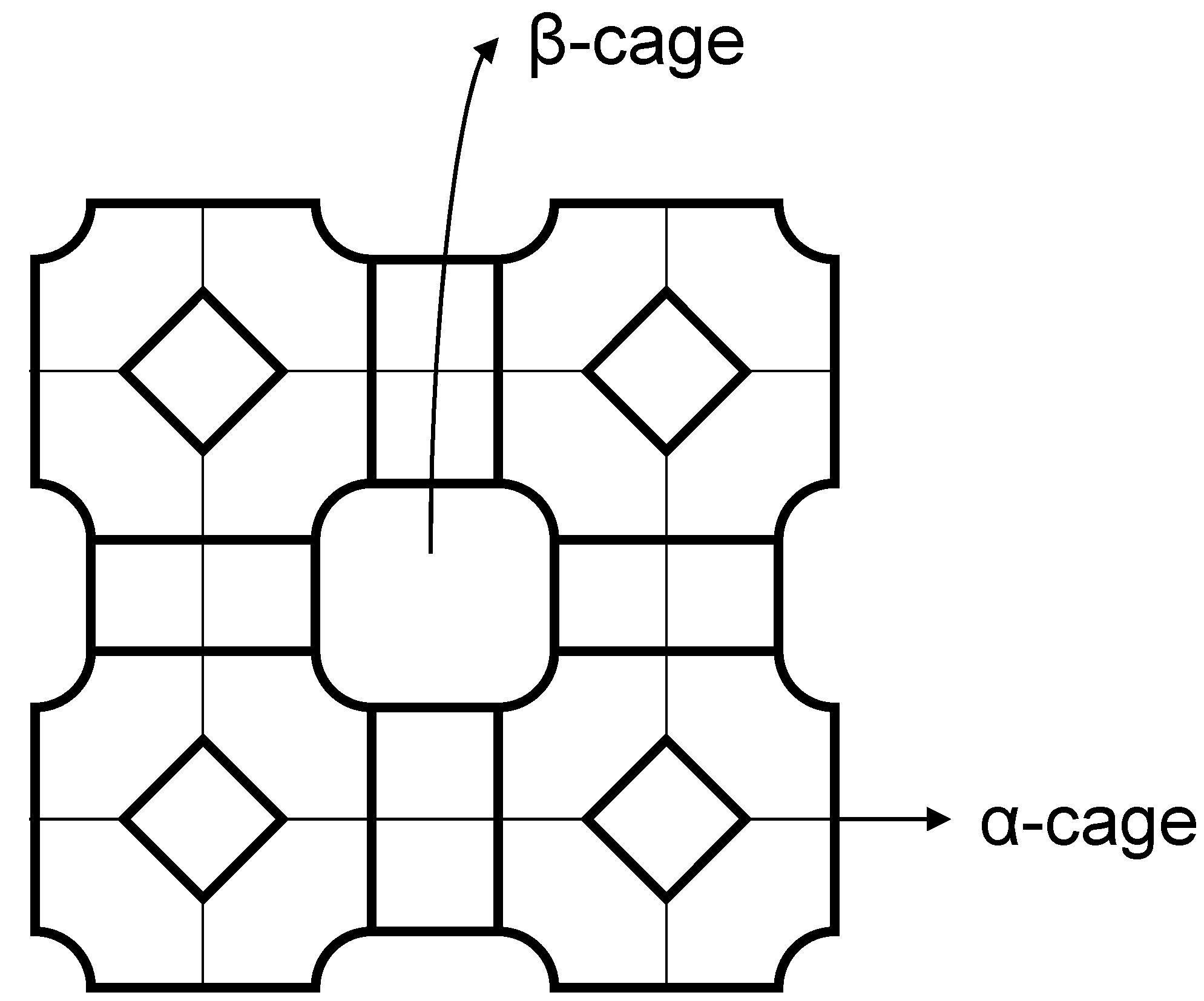
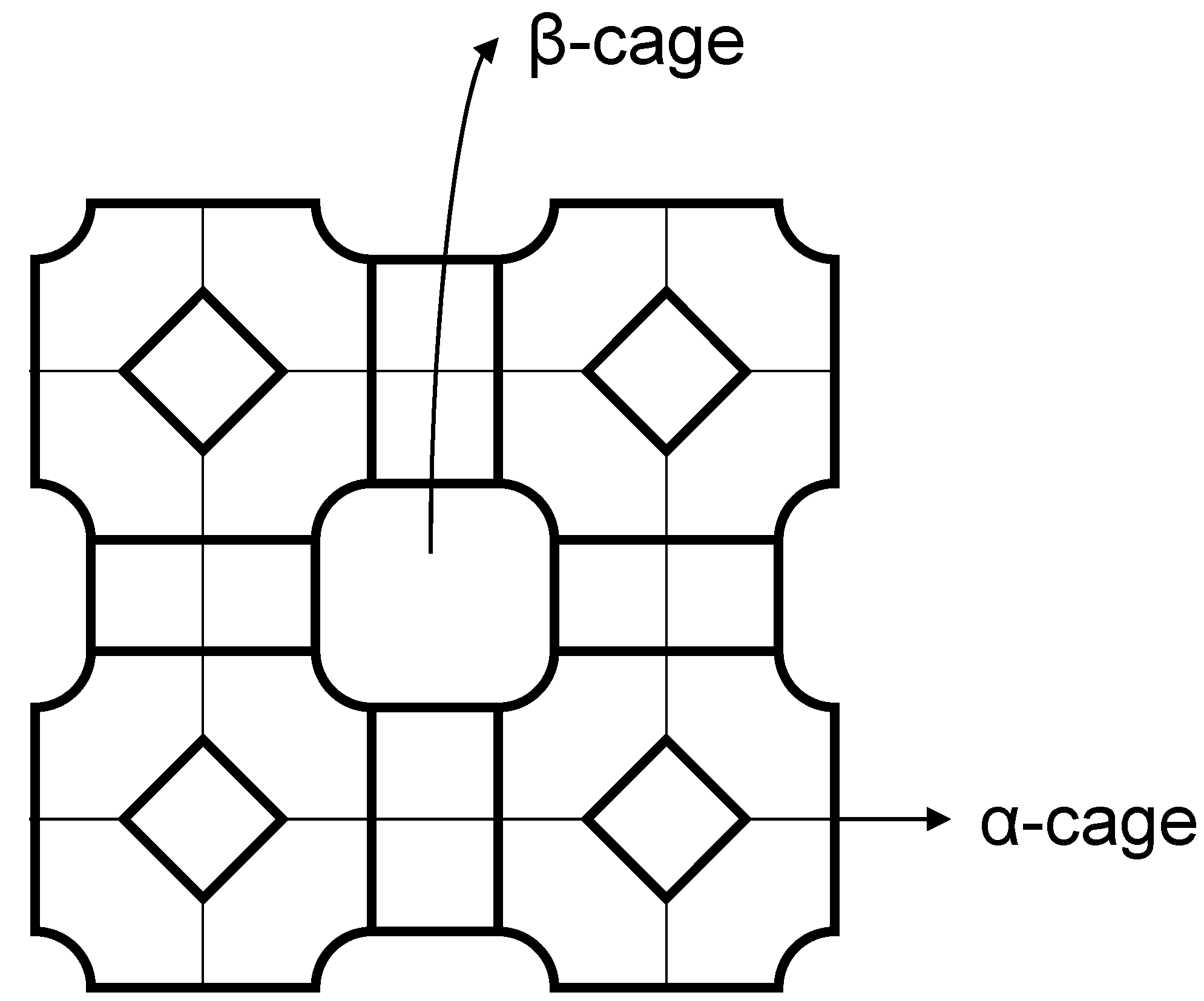
2.2. Polymer Gels for Support or Immobilization of Active Species
2.3. Metal-Organic Frameworks for Immobilization of Active Species


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Active Species | Particle Size (nm) | TOF (mol-H2 min−1 mol-Active Species−1) | Ref. |
|---|---|---|---|---|
| Co–B/SBA-15 | Co–B | 6–12 | 3.4 | [55] |
| Co–B/MCM-41 | - | 3–30 | 2 | - |
| Co–B/FSM-16 | - | 3–30 | 2.1 | - |
| Co–B/non-porous | - | 30 | 0.9 | - |
| unsupported Co–B | - | 30–40 | 0.6 | - |
| Co–B/C-film | - | 50–300 | 0.6 | [62] |
| unsupported Co–B film | - | ~250 | 0.1 | - |
| Co–W–P–B/Ni foam | - | 200–400 | - | [64] |
| Ru@ZK-4 | Ru | 2.9 | 90 | [66] |
| Ni@3D-(N)GFs | Ni | 2–4 | 41.7 | [67] |
| p(HEMA)-Co | Co | - | 3.8 | [81] |
| p(HEMA)-Ni | Ni | - | 0.8 | - |
| p(HEMA)-Cu | Cu | - | 1.1 | - |
| p(SPM)-Co | Co | - | 5.8 | [82] |
| p(SPM)-Ni | Ni | - | 3.8 | - |
| p(SPM)-Cu | Cu | - | 1.8 | - |
| p(VPA)-Co | Co | - | 7.7 | [85] |
| p(VPA)-Ni | Ni | - | 3.6 | - |
| p(VPA)-Cu | Cu | - | 1.1 | - |
| Co-MOF | Co | <10 | 20.8 | [98] |
| Ni-ZIF-8 | Ni | 2.7 | 14.2 | [99] |
| Pt@MIL-101(Cr) | Pt | 1.2–3 | 446.4 | [46] |
| AuNi@MIL-101(Cr) | AuNi | 2.9–3.4 | 66.2 | [103] |
| AuCo@MIL101(Cr) | AuCo | 1.8 | 23.5 | [102] |
3. Nanostructured Materials
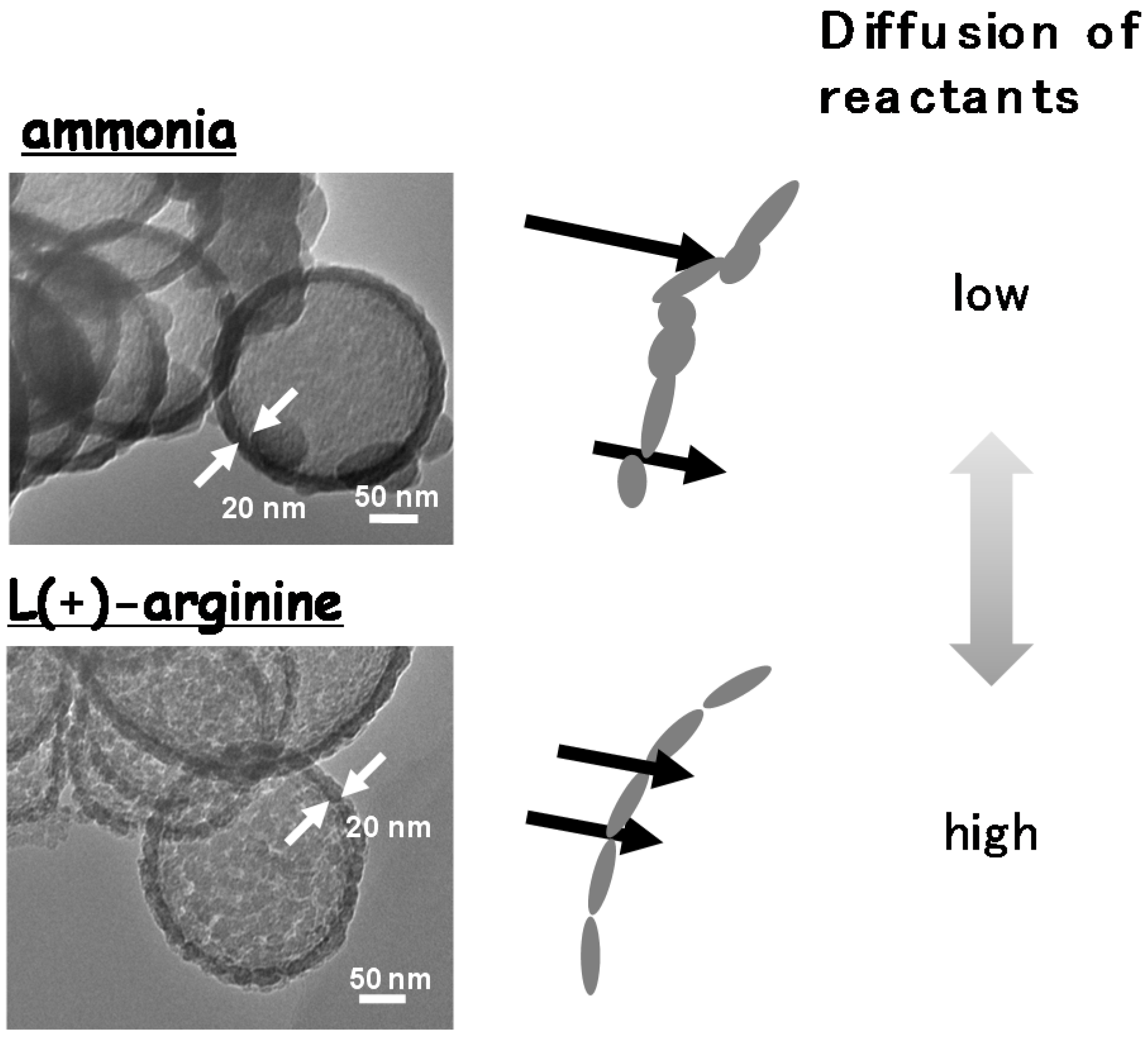
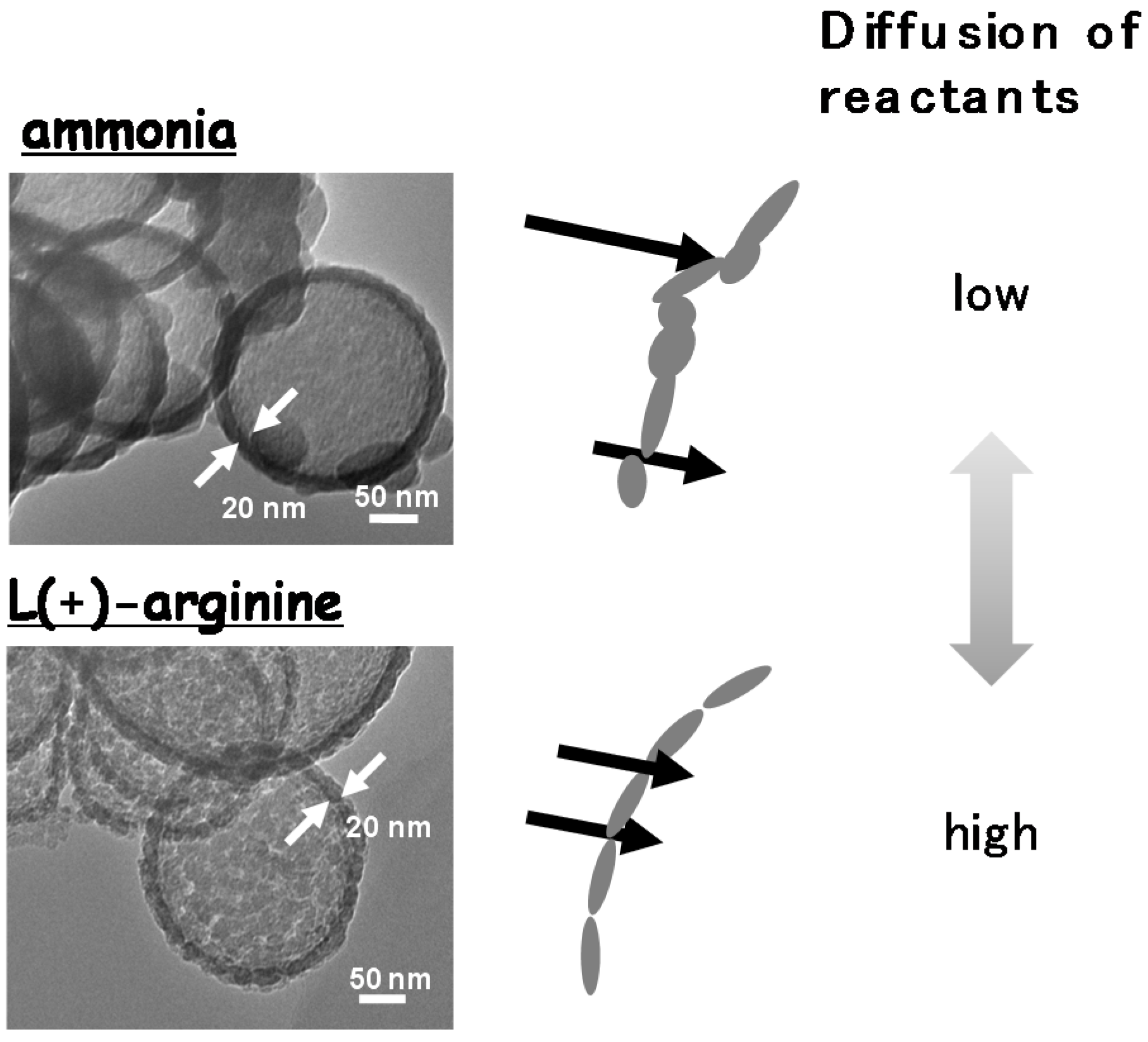
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schlapbach, L.; Zuttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Siblerud, R. Our Future is Hydrogen: Energy, Environment, and Economy; New Science Publications: Wellington, FL, USA, 2001. [Google Scholar]
- Barreto, L.; Makihira, A.; Riahi, K. The hydrogen economy in the 21st century: A sustainable development scenario. Int. J. Hydrogen Energy 2003, 28, 267–284. [Google Scholar] [CrossRef]
- Van den Berg, A.W.C.; Arean, C.O. Materials for hydrogen storage: Current research trends and perspectives. Chem. Commun. 2008, 48, 668–681. [Google Scholar] [CrossRef]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Moriarty, P.; Honnery, D. Hydrogen’s role in an uncertain energy future. Int. J. Hydrogen Energy 2009, 34, 31–39. [Google Scholar] [CrossRef]
- Kelly, N.A.; Gibson, T.L.; Cai, M.; Spearot, J.A.; Ouwerkerk, D.B. Development of a renewable hydrogen economy: Optimization of existing technologies. Int. J. Hydrogen Energy 2010, 35, 892–899. [Google Scholar] [CrossRef]
- Tiwari, A.; Pandey, A. Cyanobacterial hydrogen production—A step towards clean environment. Int. J. Hydrogen Energy 2012, 37, 139–150. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, H.M.; Yi, B.L. Recent advances in hydrogen generation with chemical methods. Prog. Chem. 2005, 17, 423–429. [Google Scholar]
- Yamada, O. Generation of hydrogen gas by reforming biomass with superheated steam. Thin Solid Films 2006, 509, 207–211. [Google Scholar] [CrossRef]
- Satyapala, S.; Petrovic, J.; Read, C.; Thomas, G.; Ordaz, G. The U.S. Department of energy’s national hydrogen storage project: Progress towards meeting hydrogen-powered vehicle requirements. Catal. Today 2007, 120, 246–256. [Google Scholar] [CrossRef]
- Stephens, F.H.; Pons, V.; Baker, R.T. Ammonia-borane: The hydrogen source par excellence? Dalton Trans. 2007, 25, 2613–2626. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chandra, M. A portable hydrogen generation system: Catalytic hydrolysis of ammonia-borane. J. Alloys Compd. 2007, 446–447, 729–732. [Google Scholar] [CrossRef]
- Biniwale, R.B.; Rayalu, S.; Devotta, S.; Ichikawa, M. Chemical hydrides: A solution to high capacity hydrogen storage and supply. Int. J. Hydrogen Energy 2008, 33, 360–365. [Google Scholar] [CrossRef]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B–N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Kang, X.D. Hydrogen-rich boron-containing materials for hydrogen storage. Dalton Trans. 2008, 40, 5400–5413. [Google Scholar] [CrossRef] [PubMed]
- Umegaki, T.; Yan, J.M.; Zhang, X.B.; Shioyama, H.; Kuriyama, N.; Xu, Q. Boron- and nitrogen-based chemical hydrogen storage materials. Int. J. Hydrogen Energy 2009, 34, 2303–2311. [Google Scholar] [CrossRef]
- Sanyal, U.; Demirci, U.B.; Jagirdar, B.R.; Miele, P. Hydrolysis of ammonia borane as a hydrogen source: Fundamental issues and potential solutions towards implementation. ChemSusChem 2011, 4, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Xu, Q. Liquid-phase chemical hydrogen storage materials. Energy Environ. Sci. 2012, 5, 9698–9725. [Google Scholar] [CrossRef]
- Lu, Z.-H.; Xu, Q. Recent progress in boron- and nitrogen-based chemical storage. Funct. Mater. Lett. 2012, 5, 1230001:1–1230001:9. [Google Scholar] [CrossRef]
- Xu, Q.; Chandra, M. Catalytic activities of non-noble metals for hydrogen generation from aqueous ammonia-borane at room temperature. J. Power Sources 2006, 163, 364–370. [Google Scholar] [CrossRef]
- Patel, N.; Guella, G.; Kale, A.; Miotello, A.; Patton, B.; Zanchetta, C.; Mirenghi, L.; Rotolo, R. Thin films of Co–B prepared by pulsed laser deposition as efficient catalysts in hydrogen producing reactions. Appl. Catal. A 2007, 323, 18–24. [Google Scholar] [CrossRef]
- Chandra, M.; Xu, Q. Room temperature hydrogen generation from aqueous ammonia-borane using noble metal nanoclusters as highly active catalysts. J. Power Sources 2007, 168, 135–142. [Google Scholar] [CrossRef]
- Yan, J.M.; Zhang, X.B.; Han, S.; Shioyama, H.; Xu, Q. Iron nanoparticle-catalyzed hydrolytic dehydrogenation of ammonia borane for chemical hydrogen storage. Angew. Chem. Int. Ed. 2008, 47, 2287–2289. [Google Scholar] [CrossRef] [PubMed]
- Kalidindi, S.B.; Indirani, M.; Jagirdar, B.R. First row transition metal ion-assisted ammonia-borane hydrolysis for hydrogen generation. Inorg. Chem. 2008, 47, 7424–7429. [Google Scholar] [CrossRef] [PubMed]
- Diwan, M.; Hanna, D.; Varma, A. Method to release hydrogen from ammonia borane for portable fuel cell applications. Int. J. Hydrogen Energy 2010, 35, 577–584. [Google Scholar] [CrossRef]
- Yan, J.M.; Zhang, X.B.; Akita, T.; Haruta, M.; Xu, Q. One-step seeding growth of magnetically recyclable Au@Co core-shell nanoparticles: Highly efficient catalyst for hydrolytic dehydrogenation of ammonia borane. J. Am. Chem. Soc. 2010, 132, 5326–5327. [Google Scholar] [CrossRef] [PubMed]
- Figen, A.K.; Pişkin, M.B.; Coşkuner, B.; İmamoğlu, V. Synthesis, structural characterization, and hydrolysis of ammonia borane (NH3BH3) as a hydrogen storage carrier. Int. J. Hydrogen Energy 2013, 38, 16215–16228. [Google Scholar] [CrossRef]
- Cao, N.; Hua, K.; Luo, W.; Cheng, G. RuCu nanoparticles supported on graphene: A highly efficient catalyst for hydrolysis of ammonia borane. J. Alloys Compd. 2014, 590, 241–246. [Google Scholar] [CrossRef]
- Veeraraghavan, P.; Gagare, P.D. Preparation of ammonia borane in high yield and purity, methanolysis and regeneration. Inorg. Chem. 2007, 46, 7810–7817. [Google Scholar]
- Rakap, M.; Özkar, S. Zeolite confined palladium(0) nanoclusters as effective and reusable catalyst for hydrogen generation from the hydrolysis of ammonia borane. Int. J. Hydrogen Energy 2010, 35, 1305–1312. [Google Scholar] [CrossRef]
- Chandra, M.; Xu, Q. Dissociation and hydrolysis of ammonia-borane with solid acids and carbon dioxide: An efficient hydrogen generation system. J. Power Sources 2006, 159, 855–860. [Google Scholar] [CrossRef]
- Chandra, M.; Xu, Q. A high-performance hydrogen generation system: Transition metal-catalyzed dissociation and hydrolysis of ammonia-borane. J. Power Sources 2006, 156, 190–194. [Google Scholar] [CrossRef]
- Cheng, F.Y.; Ma, H.; Li, Y.M.; Chen, J. Ni1−xPtx (x = 0–0.12) hollow spheres as catalysts for hydrogen generation from ammonia borane. Inorg. Chem. 2007, 46, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Kalidindi, S.B.; Sanyal, U.; Jagirdar, B.R. Nanostructured Cu and Cu@Cu2O core shell catalysts for hydrogen generation from ammonia-borane. Phys. Chem. Chem. Phys. 2008, 10, 5870–5874. [Google Scholar] [CrossRef] [PubMed]
- Zahmaklran, M.; Özkar, S. Zeolite framework stabilized rhodium(0) nanoclusters catalyst for the hydrolysis of ammonia-borane in air: Outstanding catalytic activity, reusability and lifetime. Appl. Catal. B 2009, 89, 104–110. [Google Scholar] [CrossRef]
- Umegaki, T.; Yan, J.M.; Zhang, X.B.; Shioyama, H.; Kuriyama, N.; Xu, Q. Preparation and catalysis of poly(N-vinyl-2-pyrrolidone) (PVP) stabilized nickel catalyst for hydrolytic dehydrogenation of ammonia borane. Int. J. Hydrogen Energy 2009, 34, 3816–3822. [Google Scholar] [CrossRef]
- Durap, F.; Zahmakiran, M.; Özkar, S. Water soluble laurate stabilized ruthenium(0) nanoclusters catalyst for hydrogen generation from the hydrolysis of ammonia-borane: High activity and long lifetime. Int. J. Hydrogen Energy 2009, 34, 7223–7230. [Google Scholar] [CrossRef]
- Basu, S.; Brockman, A.; Gagare, P.; Zheng, Y.; Ramachandran, P.V.; Delgass, W.N.; Gore, J.P. Chemical kinetics of Ru-catalyzed ammonia borane hydrolysis. J. Power Sources 2009, 188, 238–243. [Google Scholar] [CrossRef]
- Umegaki, T.; Yan, J.M.; Zhang, X.B.; Shioyama, H.; Kuriyama, N.; Xu, Q. Hollow Ni–SiO2 nanosphere-catalyzed hydrolytic dehydrogenation of ammonia borane for chemical hydrogen storage. J. Power Sources 2009, 191, 209–216. [Google Scholar] [CrossRef]
- Yan, J.M.; Zhang, X.B.; Han, S.; Shioyama, H.; Xu, Q. Magnetically recyclable Fe–Ni alloy catalyzed dehydrogenation of ammonia borane in aqueous solution under ambient atmosphere. J. Power Sources 2009, 194, 478–481. [Google Scholar] [CrossRef]
- Brockman, A.; Zheng, Y.; Gore, J. A study of catalytic hydrolysis of concentrated ammonia borane solutions. Int. J. Hydrogen Energy 2010, 35, 7350–7356. [Google Scholar] [CrossRef]
- Metin, Ö.; Mazumder, V.; Özkar, S.; Sun, S.S. Monodisperse nickel nanoparticles; and their catalysis in hydrolytic dehydrogenation of ammonia borane. J. Am. Chem. Soc. 2010, 132, 1468–1469. [Google Scholar]
- Sun, D.H.; Mazumder, V.; Metin, O.; Sun, S.H. Catalytic hydrolysis of ammonia borane via cobalt palladium nanoparticles. ACS Nano 2011, 8, 6458–6464. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Z.; Aijaz, A.; Xu, Q. Highly dispersed surfactant-free nickel nanoparticles and their remarkable catalytic activity in the hydrolysis of ammonia borane for hydrogen generation. Angew. Chem. Int. Ed. 2012, 51, 6753–6756. [Google Scholar] [CrossRef] [PubMed]
- Aijaz, A.; Karkamkar, A.; Choi, Y.J.; Tsumori, N.; Rönnebro, E.; Autrey, T.; Shioyama, H.; Xu, Q. Immobilizing highly catalytically active Pt Nanoparticles inside the pores of metal-organic framework: A double solvents approach. J. Am. Chem. Soc. 2012, 134, 13926–13929. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.C.; Liu, Y.H.; Hung, Y.; Liu, X.Y.; Mou, C.Y. Mesoporous silica supported cobalt catalysts for hydrogen generation in hydrolysis of ammonia borane. Int. J. Hydrogen Energy 2013, 38, 7280–7290. [Google Scholar] [CrossRef]
- Seven, F.; Sahiner, N. Metal ion-imprinted hydrogel with magnetic properties and enhanced catalytic performances in hydrolysis of NaBH4 and NH3BH3. Int. J. Hydrogen Energy 2013, 38, 15275–15284. [Google Scholar] [CrossRef]
- Hoa, M.L.K.; Lu, M.H.; Zhang, Y. Preparation of porous materials with ordered hole structure. Adv. Colloid Interface Sci. 2006, 121, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Gokmen, M.T.; Prez, F.E.D. Porous polymer particles—A comprehensive guide to synthesis, characterization, functionalization and application. Prog. Polym. Sci. 2012, 37, 365–405. [Google Scholar] [CrossRef]
- Rakap, M.; Özkar, S. Hydroxyapatite-supported cobalt(0) nanoclusters as efficient and cost-effective catalyst for hydrogen generation from the hydrolysis of both sodium borohydride and ammonia-borane. Catal. Today 2012, 183, 17–25. [Google Scholar] [CrossRef]
- Doner, A.; Karci, I.; Kardas, G. Effect of C-felt supported Ni, Co and NiCo catalysts to produce hydrogen. Int. J. Hydrogen Energy 2012, 37, 9470–9476. [Google Scholar] [CrossRef]
- Seidel, A.; Loos, J.; Boddenberg, B. Copper nanoparticles in zeolite Y. J. Mater. Chem. 1999, 9, 2495–2498. [Google Scholar] [CrossRef]
- Tang, Q.; Zhang, Q.; Wang, P.; Wang, Y.; Wan, H. Characterizations of cobalt oxide nanoparticles within faujasite zeolites and the formation of metallic cobalt. Chem. Mater. 2004, 16, 1967–1976. [Google Scholar] [CrossRef]
- Patel, N.; Fernandes, R.; Gupta, S.; Edla, R.; Kothari, D.C.; Miotello, A. Co–B catalyst supported over mesoporous silica for hydrogen production by catalytic hydrolysis of Ammonia Borane: A study on influence of pore structure. Appl. Catal. B 2013, 140–141, 125–132. [Google Scholar] [CrossRef]
- Rakap, M.; Özkar, S. Hydrogen generation from the hydrolysis of ammonia-borane using intrazeolite cobalt(0) nanoclusters catalyst. Int. J. Hydrogen Energy 2010, 35, 3341–3346. [Google Scholar] [CrossRef]
- Eom, K.S.; Cho, K.W.; Kwon, H.S. Hydrogen generation from hydrolysis of NH3BH3 by an electroplated Co–P catalyst. Int. J. Hydrogen Energy 2010, 35, 181–186. [Google Scholar] [CrossRef]
- Dai, H.B.; Gao, L.L.; Liang, Y.; Kang, X.D.; Wang, P. Promoted hydrogen generation from ammonia borane aqueous solution using cobalt-molybdenum-boron/nickel foam catalyst. J. Power Sources 2010, 195, 307–312. [Google Scholar] [CrossRef]
- Patel, N.; Femandes, R.; Edla, R.; Lihitkar, P.B.; Kothari, D.C.; Miotello, A. Superior hydrogen production rate by catalytic hydrolysis of ammonia borane using Co–B nanoparticles supported over mesoporous silica particles. Catal. Commun. 2012, 23, 39–42. [Google Scholar] [CrossRef]
- Patel, N.; Fernandes, R.; Guella, G.; Miotello, A. Nanoparticle assembled Co–B thin film for the hydrolysis of ammonia borane: A highly active catalyst for hydrogen production. Appl. Catal. B 2010, 95, 137–143. [Google Scholar] [CrossRef]
- Ossi, P.M.; Bottani, C.E.; Miotello, A. Pulsed-laser deposition of carbon: From DLC to cluster-assembled films. Thin Solid Films 2005, 482, 2–8. [Google Scholar] [CrossRef]
- Patel, N.; Fernandes, R.; Santini, A.; Miotello, A. Co–B nanoparticles supported on carbon film synthesized by pulsed laser deposition for hydrolysis of ammonia borane. Int. J. Hydrogen Energy 2012, 37, 2007–2013. [Google Scholar] [CrossRef]
- Fernandes, R.; Patel, N.; Miotello, A.; Jaiswal, R.; Kothari, D.C. Dehydrogenation of ammonia borane with transition metal-doped Co–B alloy catalysts. Int. J. Hydrogen Energy 2012, 37, 2397–2406. [Google Scholar] [CrossRef]
- Yang, J.; Cheng, F.; Liang, J.; Chen, J. Hydrogen generation by hydrolysis of ammonia borane with a nanoporous cobalt-tungsten-boron-phosphorous catalyst supported on Ni foam. Int. J. Hydrogen Energy 2011, 36, 1411–1417. [Google Scholar] [CrossRef]
- Zahmakıran, M.; Durap, F.; Ozkar, S. Zeolite confined copper(0) nanoclusters as cost-effective and reusable catalyst in hydrogen generation from the hydrolysis of ammonia-borane. Int. J. Hydrogen Energy 2010, 35, 187–197. [Google Scholar] [CrossRef]
- Zahmakiran, M. Preparation and characterization of LTA-type zeolite framework dispersed ruthenium nanoparticles and their catalytic application in the hydrolytic dehydrogenation of ammonia-borane for efficient hydrogen generation. Mater. Sci. Eng. B 2012, 177, 606–613. [Google Scholar] [CrossRef]
- Mahyari, M.; Shaabani, A. Nickel nanoparticles immobilized on three-dimensional nitrogen-doped graphene as a superb catalyst for the generation of hydrogen from the hydrolysis of ammonia borane. J. Mater. Chem. A 2014, 2, 16652–16659. [Google Scholar] [CrossRef]
- Sahiner, N. Soft and flexible hydrogel templates of different sizes and various functionalities for metal nanoparticle preparation and their use in catalysis. Prog. Polym. Sci. 2013, 38, 1329–1356. [Google Scholar] [CrossRef]
- Guizard, C.; Bac, A.; Barboiu, M.; Hovnanian, N. Hybrid organic-inorganic membranes with specific transport properties: Applications in separation and sensors technologies. Sep. Purif. Technol. 2001, 25, 167–180. [Google Scholar] [CrossRef]
- Lu, Z.H.; Liu, G.J.; Duncan, S. Poly(2-hydroxyethyl acrylate-co-methyl acrylate)/SiO2/TiO2 hybrid membranes. J. Membr. Sci. 2003, 221, 113–122. [Google Scholar] [CrossRef]
- Chiang, P.C.; Whang, W.T.; Tsai, M.H. Physical and mechanical properties of polyimide/titania hybrid films. Thin Solid Films 2004, 447–448, 359–364. [Google Scholar] [CrossRef]
- Yu, L.Y.; Xu, Z.L.; Shen, H.M.; Yang, H. Preparation and characterization of PVDF-SiO2 composite hollow fiber UF membrane by sol-gel method. J. Membr. Sci. 2009, 337, 257–265. [Google Scholar] [CrossRef]
- Oha, J.K.; Leea, D.I.; Park, J.M. Biopolymer-based microgels/nanogels for drug delivery application. Prog. Polym. Sci. 2009, 34, 1261–1282. [Google Scholar] [CrossRef]
- Chang, C.; Zhang, L. Cellulose-based hydrogels: Present status and application prospects. Carbohydr. Polym. 2011, 84, 40–53. [Google Scholar] [CrossRef]
- Jesionowski, T.; Krysztafkiewicz, A. Preparation of the hydrophilic/hydrophobic silica particles. Colloid Surf. A 2002, 207, 49–58. [Google Scholar] [CrossRef]
- Schexnailder, P.; Schmidt, G. Nanocomposite polymer hydrogels. Colloid Polym. Sci. 2009, 287, 1–11. [Google Scholar] [CrossRef]
- Tuzmen, N.; Kalburcu, T.; Denizli, A. Immobilization of catalase via adsorption onto metal-chelated affinity cryogels. Process Biochem. 2012, 47, 26–33. [Google Scholar] [CrossRef]
- Dragan, E.S.; Loghin, D.F.A. Enhanced sorption of methylene blue from aqueous solutions by semi-IPN composite cryogels with anionically modified potato starch entrapped in PAAm matrix. Chem. Eng. J. 2013, 234, 211–222. [Google Scholar] [CrossRef]
- Tuncaboylu, D.C.; Okay, O. Hierarchically macroporous cryogels of polyisobutylene and silica nanoparticles. Langmuir 2010, 26, 7574–7581. [Google Scholar] [CrossRef] [PubMed]
- Bilici, C.; Karayel, S.; Demir, T.T.; Okay, O. Self-oscillating pH responsive cryogels as possible candidates of soft materials for generating mechanical energy. J. Appl. Polym. Sci. 2010, 118, 2981–2988. [Google Scholar] [CrossRef]
- Seven, F.; Sahiner, N. Superporous P(2-hydroxyethyl methacrylate) cryogel-M (M: Co, Ni, Cu) composites as highly effective catalysts in H2 generation from hydrolysis of NaBH4 and NH3BH3. Int. J. Hydrogen Energy 2014, 39, 15455–15463. [Google Scholar] [CrossRef]
- Yildiz, S.; Aktas, N.; Sahiner, N. Metal nanoparticle-embedded super porous poly(3-sulfopropyl methacrylate) cryogel for H2 production from chemical hydride hydrolysis. Int. J. Hydrogen Energy 2014, 39, 14690–14700. [Google Scholar] [CrossRef]
- Sahiner, N.; Turhan, T. Lyon LAILC. (ionic liquid colloids) based on p(4-VP) (poly(4-vinyl pyridine)) microgels: Synthesis, characterization and use in hydrogen production. Energy 2014, 66, 256–263. [Google Scholar] [CrossRef]
- Sahiner, N.; Sagbas, S. The preparation of poly(vinyl phosphonic acid) hydrogels as new functional materials for in situ metal nanoparticles. Colloid Surf. A 2013, 418, 76–83. [Google Scholar] [CrossRef]
- Sahiner, N.; Sagbas, S. The use of poly(vinyl phosphonic acid) microgels for the preparation of inherently magnetic Co metal catalyst particles in hydrogen production. J. Power Sources 2014, 246, 55–62. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, H.C. A metal-organic framework with entatic metal centers exhibiting high gas adsorption affinity. J. Am. Chem. Soc. 2006, 128, 11734–11735. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Bureekaew, S.; Kitagawa, S. A dynamic, isocyanurate-functionalized porous coordination polymer. Angew. Chem. Int. Ed. 2008, 47, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Xie, L.; Li, Y.; Zheng, J.; Li, X.G. Metal-organic-framework-based catalyst for highly efficient H2 generation from aqueous NH3BH3 solution. Chem. Eur. J. 2009, 15, 8951–8954. [Google Scholar] [CrossRef] [PubMed]
- Férey, G.; Serre, C. Large breathing effects in three-demensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev. 2009, 38, 1380–1399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.L.; Tatsu, Y.; Lu, Z.H.; Xu, Q. Non-, micro, and mesoporous metal-organic framework isomers: Reversible transformation, fluorescence sensing, and large molecule separation. J. Am. Chem. Soc. 2010, 132, 5586–5587. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, S.; Makiura, R.; Sakata, O.; Kitagawa, H. Highly crystalline nanofilm by layering of porphyrin metal-organic framework sheets. J. Am. Chem. Soc. 2011, 133, 5640–5643. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.L.; Xu, Q. Porous metal-organic frameworks as platforms for functional applications. Chem. Commun. 2011, 47, 3351–3370. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Li, S.; Guo, Z.; Farha, O.K.; Hauser, B.G.; Qi, X.; Wang, Y.; Wang, X.; Han, S.; Liu, X.; et al. Imparting functionality to a metal-organic framework material by controlled nanoparticle encapsulation. Nat. Chem. 2012, 4, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Li, S.L.; Xu, Q. Metal-organic frameworks as platforms for clean energy. Energy Environ. Sci. 2013, 6, 1656–1683. [Google Scholar] [CrossRef]
- Gu, X.; Lu, Z.-H.; Jiang, H.L.; Akita, T.; Xu, Q. Synergistic catalysis of metal-organic framework-immobilized Au–Pd nanoparticles in dehydrogenation of formic acid for chemical hydrogen storage. J. Am. Chem. Soc. 2011, 133, 11822–11825. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.L.; Xu, Q. Metal-organic framework composites. Chem. Soc. Rev. 2014, 43, 5468–5512. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, Y.; Li, W.; He, B.; Yang, J.; Li, X. A highly efficient Co(0) catalyst derived from metal-organic framework for the hydrolysis of ammonia borane. Int. J. Hydrogen Energy 2011, 36, 10468–10473. [Google Scholar] [CrossRef]
- Li, P.Z.; Aranishi, K.; Xu, Q. ZIF-8 immobilized nickel nanoparticles: Highly effective catalysts for hydrogen generation from hydrolysis of ammonia borane. Chem. Commun. 2012, 48, 3173–3175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.L.; Xu, Q. Recent progress in synergistic catalysis over heterometallic nanoparticles. J. Mater. Chem. 2011, 21, 13705–13725. [Google Scholar] [CrossRef]
- Singh, A.K.; Xu, Q. Synergistic catalysis over bimetallic alloy nanoparticles. ChemCatChem 2013, 5, 652–676. [Google Scholar] [CrossRef]
- Zhu, Q.L.; Li, J.; Xu, Q. Immobilizing metal nanoparticles to metal-organic frameworks with size and location control for optimizing catalytic performance. J. Am. Chem. Soc. 2013, 135, 10210–10213. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, Q.L.; Xu, Q. Highly active AuCo alloy nanoparticles encapsulated in the pore of metal-organic frameworks for hydrolytic dehydrogenation of ammonia borane. Chem. Commun. 2014, 5899–5901. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.Y.; Su, B.L. Insights into hierarchically meso-macroporous structured materials. J. Mater. Chem. 2006, 16, 663–677. [Google Scholar] [CrossRef]
- Elias, J.; Bechelany, M.; Utke, I.; Erni, R.; Hosseini, D.; Michler, J.; Philippe, L. Urchin-inspired zinc oxide as building blocks for nanostructured solar cells. Nano Energy 2012, 1, 696–705. [Google Scholar] [CrossRef]
- Marichy, C.; Bechelany, M.; Pinna, N. Atomic layer deposition of nanostructured materials for energy and environmental applications. Adv. Mater. 2012, 24, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.H.; Wang, C.C.; Chen, C.Y. Synthesis of mono-disperse hollow nickel spheres with a double shell structure using poly(methyl methacrylate) as a template. Scr. Mater. 2008, 58, 37–40. [Google Scholar] [CrossRef]
- Xu, S.; Hessel, C.M.; Ren, H.; Yu, R.; Jin, Q.; Yang, M.; Zhao, H.; Wang, D. α-Fe2O3 multi-shelled hollow microspheres for lithium ion battery anodes with superior capacity and charge retention. Energy Environ. Sci. 2014, 7, 632–637. [Google Scholar] [CrossRef]
- Dong, Z.; Lai, X.; Halpert, J.E.; Yang, N.; Yi, L.; Zhai, J.; Wang, D.; Tang, Z.; Jiang, L. Accurate control of multishelled ZnO hollow microspheres for dye-sensitized solar cells with high efficiency. Adv. Mater. 2012, 24, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.W.; Archer, L.A.; Yang, Z. Hollow micro-/nanostructures: Synthesis and applications. Adv. Mater. 2008, 20, 3987–4019. [Google Scholar] [CrossRef]
- Jiao, W.; Hu, X.; Ren, H.; Xu, P.; Yu, R.; Chen, J.; Xing, X. Magnetic Ni and Ni/Pt hollow nanospheres and their catalytic activities for hydrolysis of ammonia borane. J. Mater. Chem. A 2014, 2, 18171–18176. [Google Scholar] [CrossRef]
- Umegaki, T.; Takei, C.; Xu, Q.; Kojima, Y. Fabrication of hollow metal oxide-nickel composite spheres and their catalytic activity for hydrolytic dehydrogenation of ammonia borane. Int. J. Hydrogen Energy 2013, 38, 1397–1404. [Google Scholar] [CrossRef]
- Umegaki, T.; Takei, C.; Watanuki, Y.; Xu, Q.; Kojima, Y. Fabrication of hollow nickel-silica composite spheres using L(+)-arginine and their catalytic performance for hydrolytic dehydrogenation of ammonia borane. J. Mol. Catal. A 2013, 371, 1–7. [Google Scholar] [CrossRef]
- Umegaki, T.; Seki, A.; Xu, Q.; Kojima, Y. Influence of preparation conditions of hollow silica-nickel composite spheres on their catalytic activity for hydrolytic dehydrogenation of ammonia borane. J. Alloy. Compd. 2014, 588, 615–621. [Google Scholar] [CrossRef]
- Umegaki, T.; Watanuki, Y.; Xu, Q.; Kojima, Y. Effect of solvents on morphology of hollow nickel-silica composite spheres and their catalytic performance for hydrolytic dehydrogenation of ammonia borane. J. Jpn. Inst. Energy 2014, 93, 703–709. [Google Scholar] [CrossRef]
- Umegaki, T.; Hosoya, T.; Toyama, N.; Xu, Q.; Kojima, Y. Fabrication of hollow silica-zirconia composite spheres and their activity for hydrolytic dehydrogenation of ammonia borane. J. Alloy. Compd. 2014, 608, 261–265. [Google Scholar] [CrossRef]
- Toyama, N.; Umegaki, T.; Kojima, Y. Fabrication of hollow silica-alumina composite spheres and their activity for hydrolytic dehydrogenation of ammonia borane. Int. J. Hydrogen Energy 2014, 39, 17136–17143. [Google Scholar] [CrossRef]
- Toyama, N.; Umegaki, T.; Xu, Q.; Kojima, Y. Control of particle size of hollow silica-alumina composite spheres and their activity for hydrolytic dehydrogenation of ammonia borane. J. Jpn. Inst. Energy 2014, 93, 511–516. [Google Scholar] [CrossRef]
- Umegaki, T.; Imamura, S.; Toyama, N.; Kojima, Y. Influence of preparation conditions on the morphology of hollow silica-alumina composite spheres and their activity for hydrolytic dehydrogenation of ammonia borane. Microporous Mesoporous Mater. 2014, 196, 349–353. [Google Scholar] [CrossRef]
- Bechelany, M.; Chaaya, A.A.; Frances, F.; Akdim, O.; Cot, D.; Demirci, U.B.; Miele, P. Nanowires with controlled porosity for hydrogen production. J. Mater. Chem. A 2013, 1, 2133–2138. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umegaki, T.; Xu, Q.; Kojima, Y. Porous Materials for Hydrolytic Dehydrogenation of Ammonia Borane. Materials 2015, 8, 4512-4534. https://doi.org/10.3390/ma8074512
Umegaki T, Xu Q, Kojima Y. Porous Materials for Hydrolytic Dehydrogenation of Ammonia Borane. Materials. 2015; 8(7):4512-4534. https://doi.org/10.3390/ma8074512
Chicago/Turabian StyleUmegaki, Tetsuo, Qiang Xu, and Yoshiyuki Kojima. 2015. "Porous Materials for Hydrolytic Dehydrogenation of Ammonia Borane" Materials 8, no. 7: 4512-4534. https://doi.org/10.3390/ma8074512
APA StyleUmegaki, T., Xu, Q., & Kojima, Y. (2015). Porous Materials for Hydrolytic Dehydrogenation of Ammonia Borane. Materials, 8(7), 4512-4534. https://doi.org/10.3390/ma8074512