
A New Homogeneous Catalyst for the Dehydrogenation of Dimethylamine Borane Starting with Ruthenium(III) Acetylacetonate

Abstract
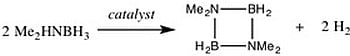
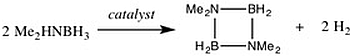
:
1. Introduction
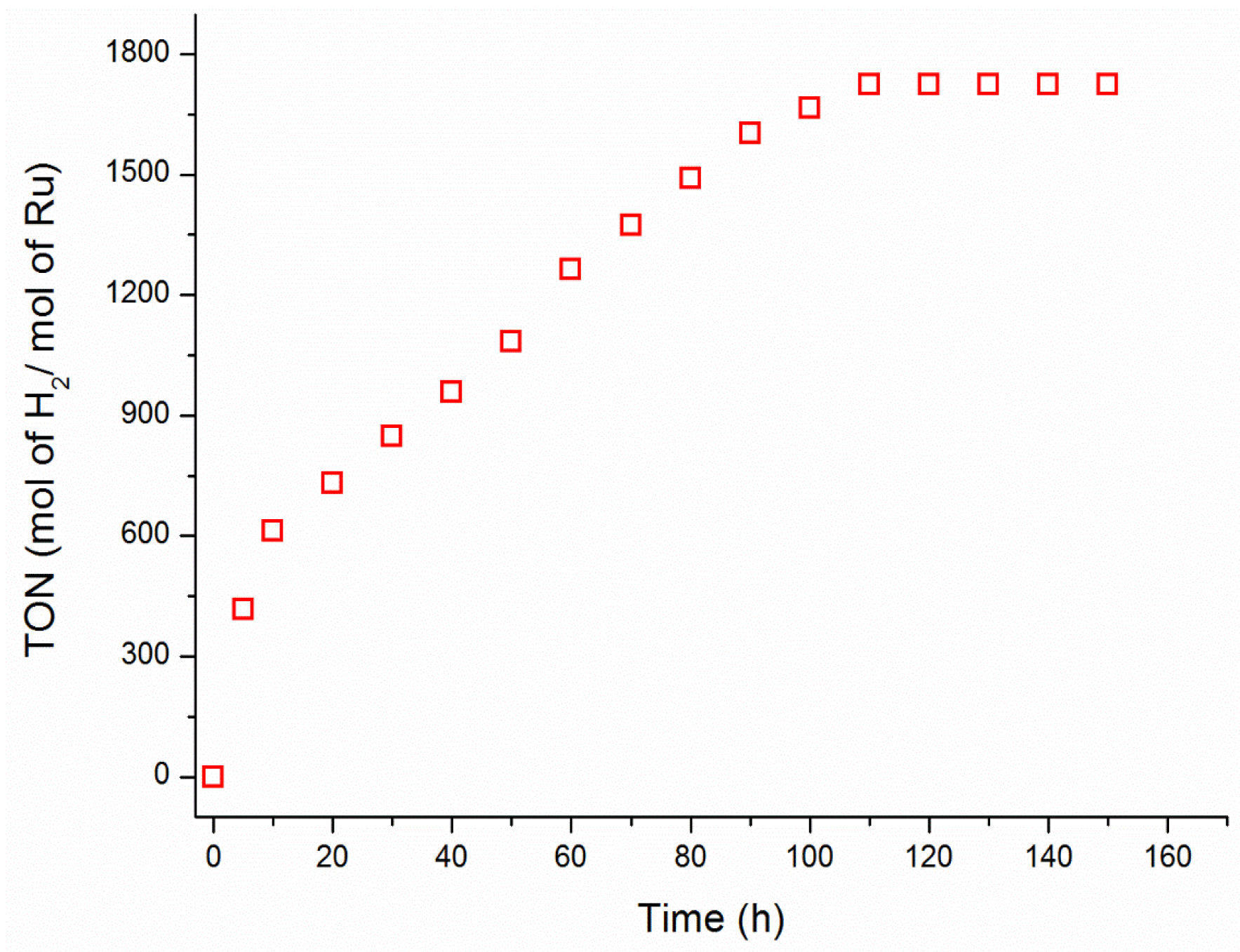
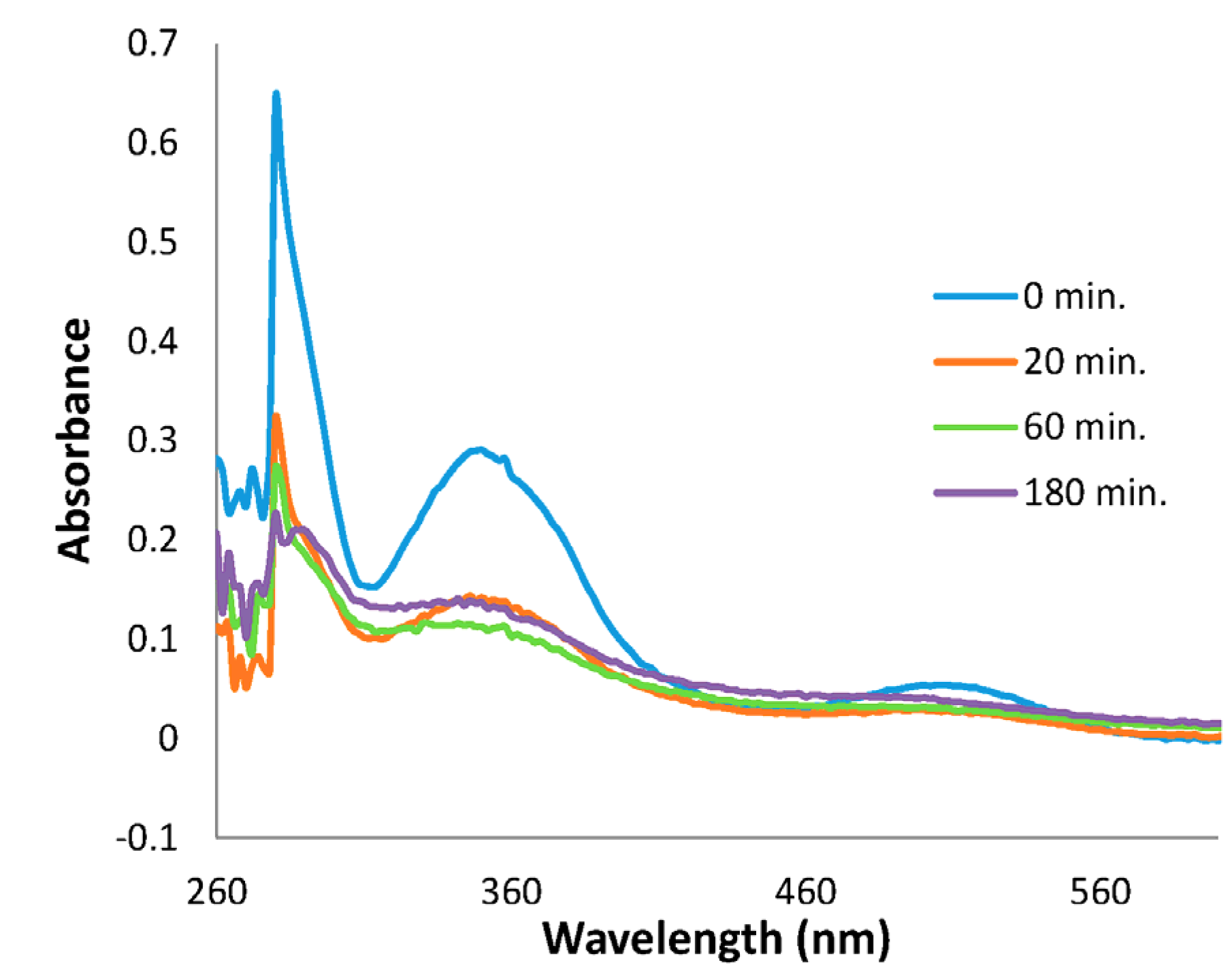
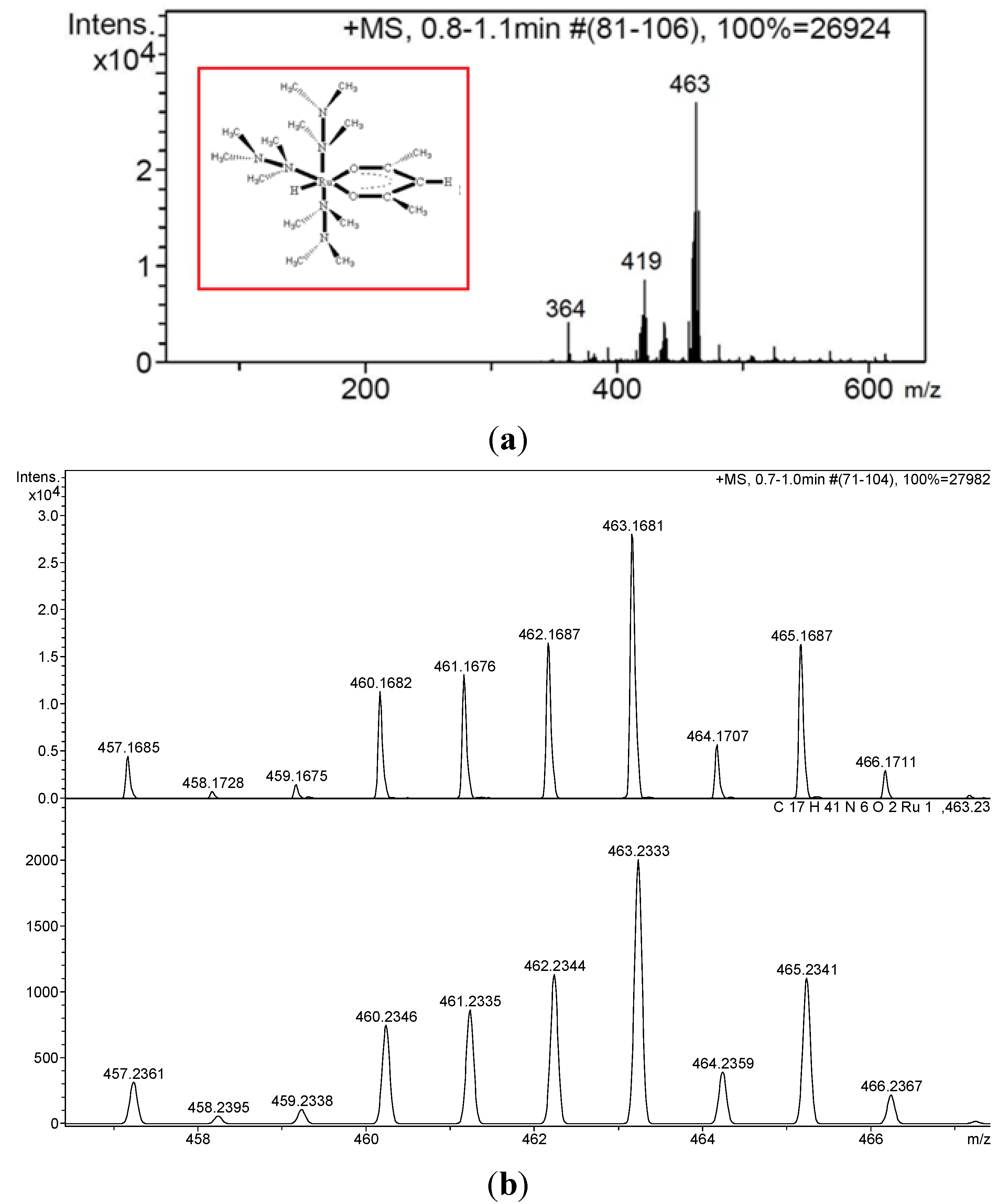
2. Results and Discussion

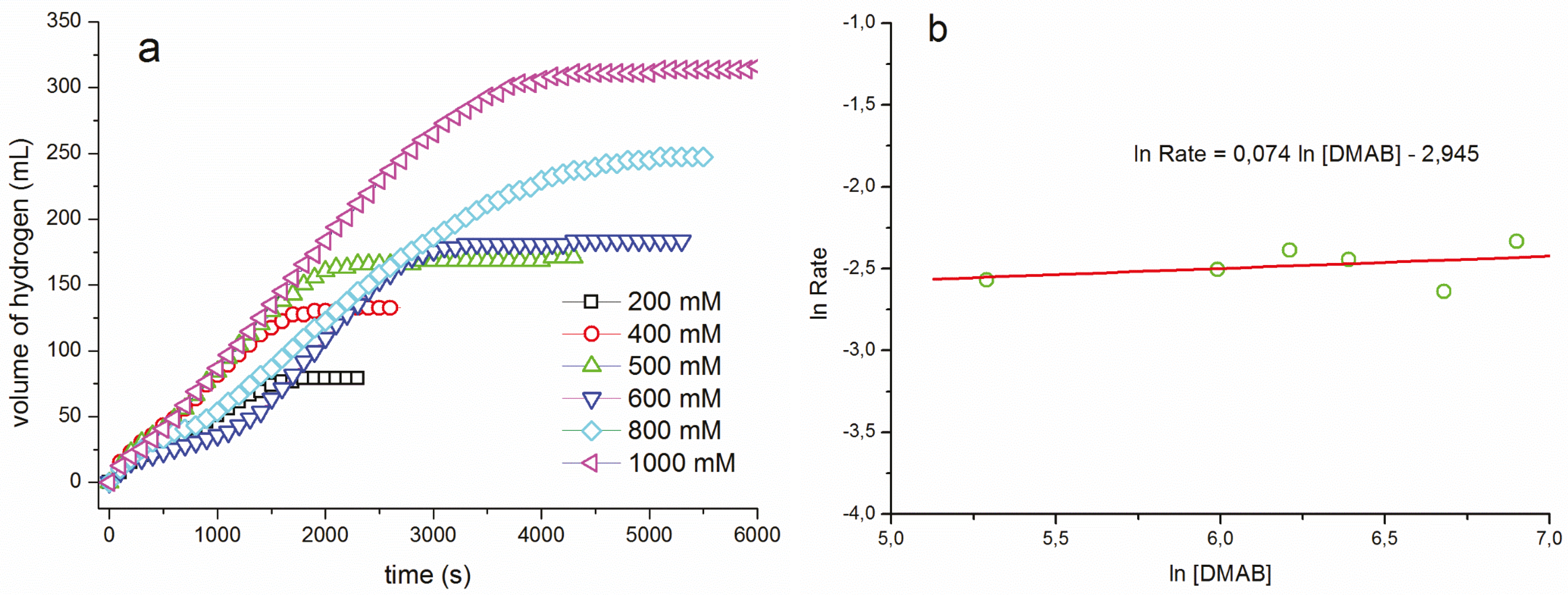
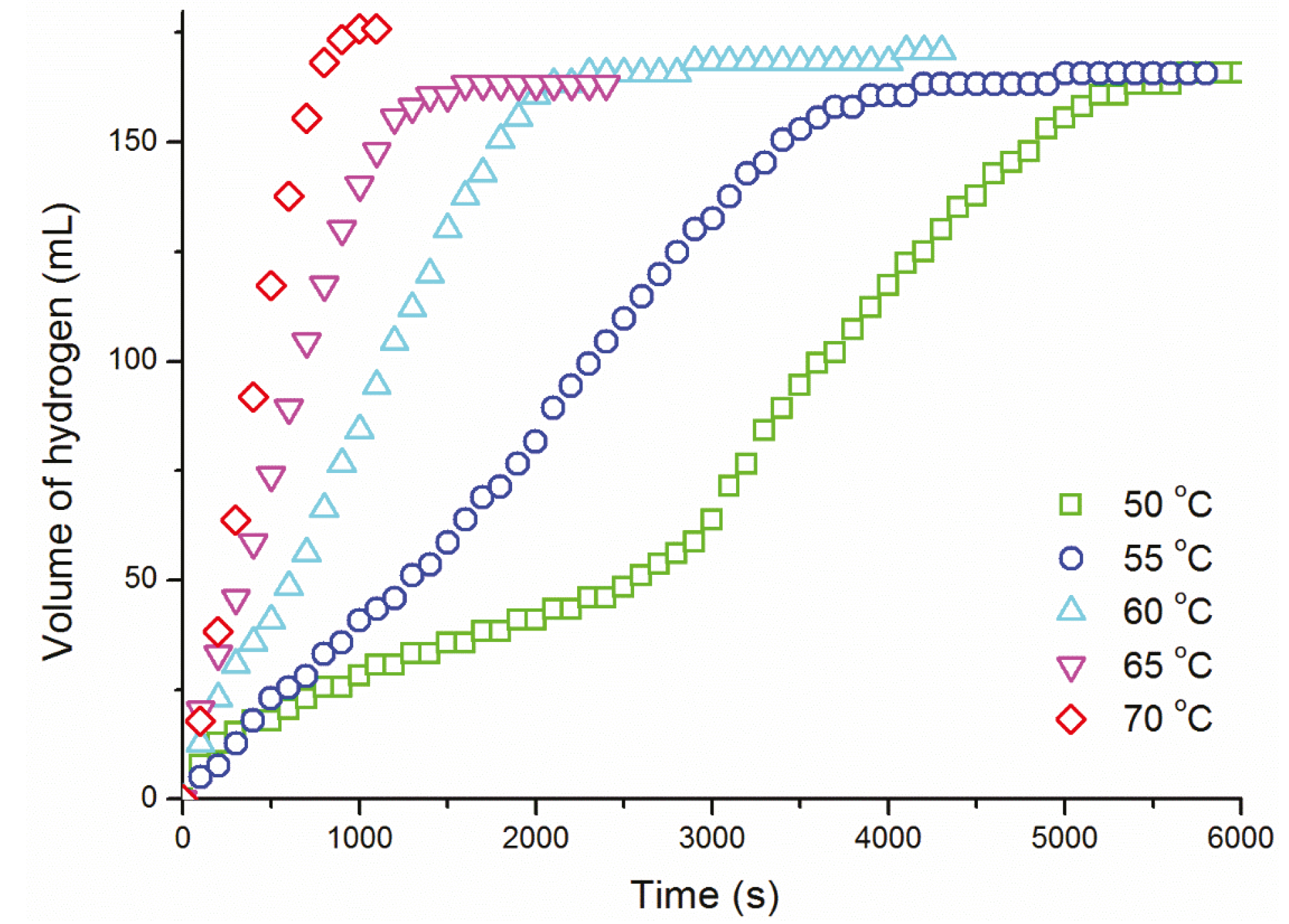

{kind=link}
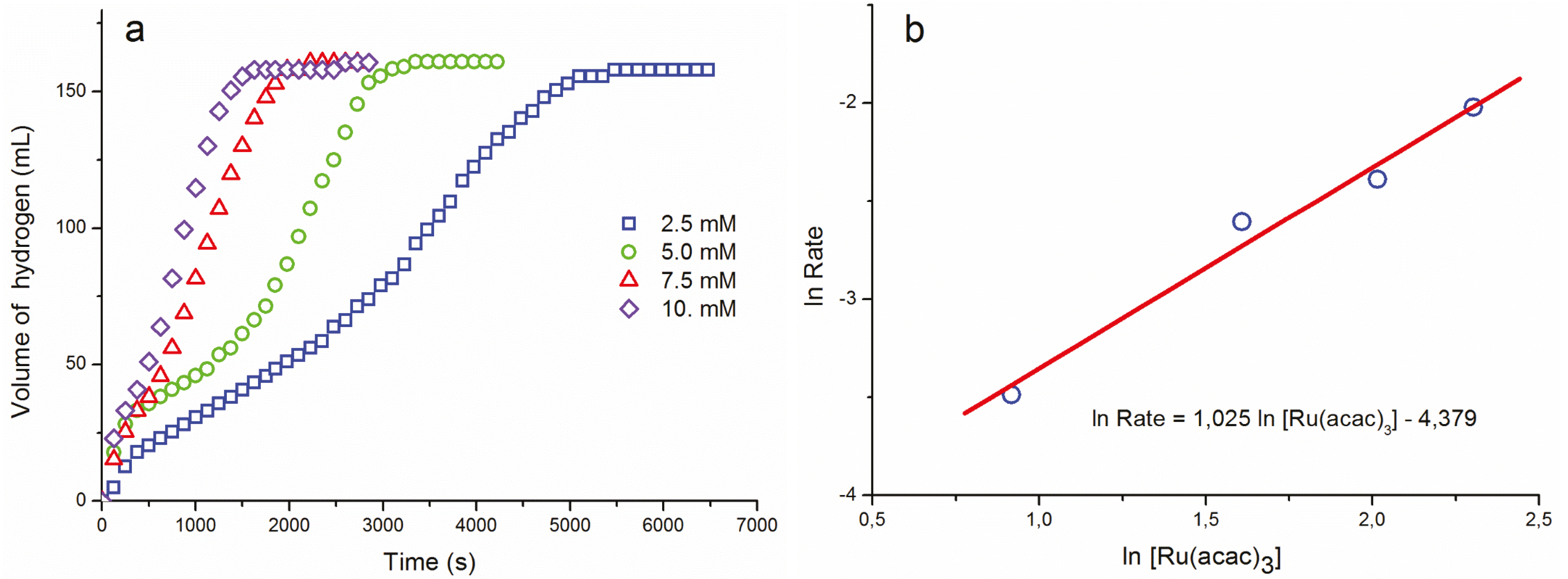{kind=link}
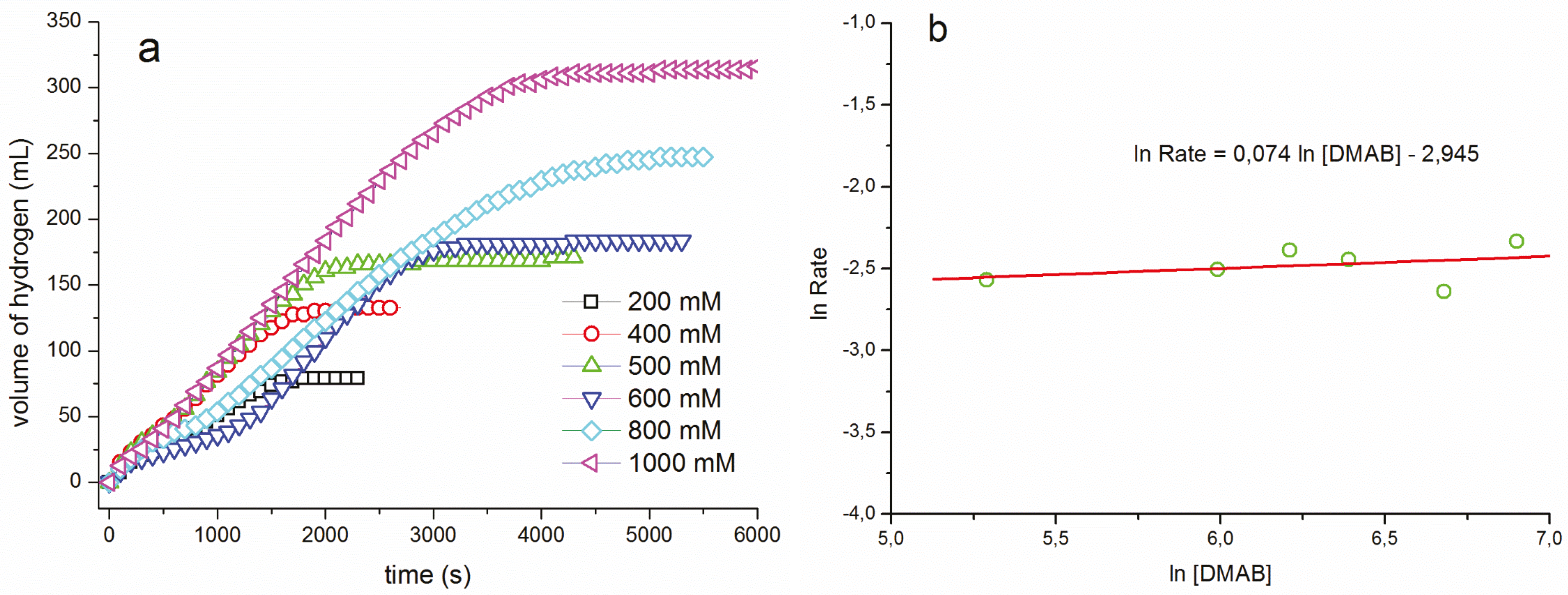{kind=link}
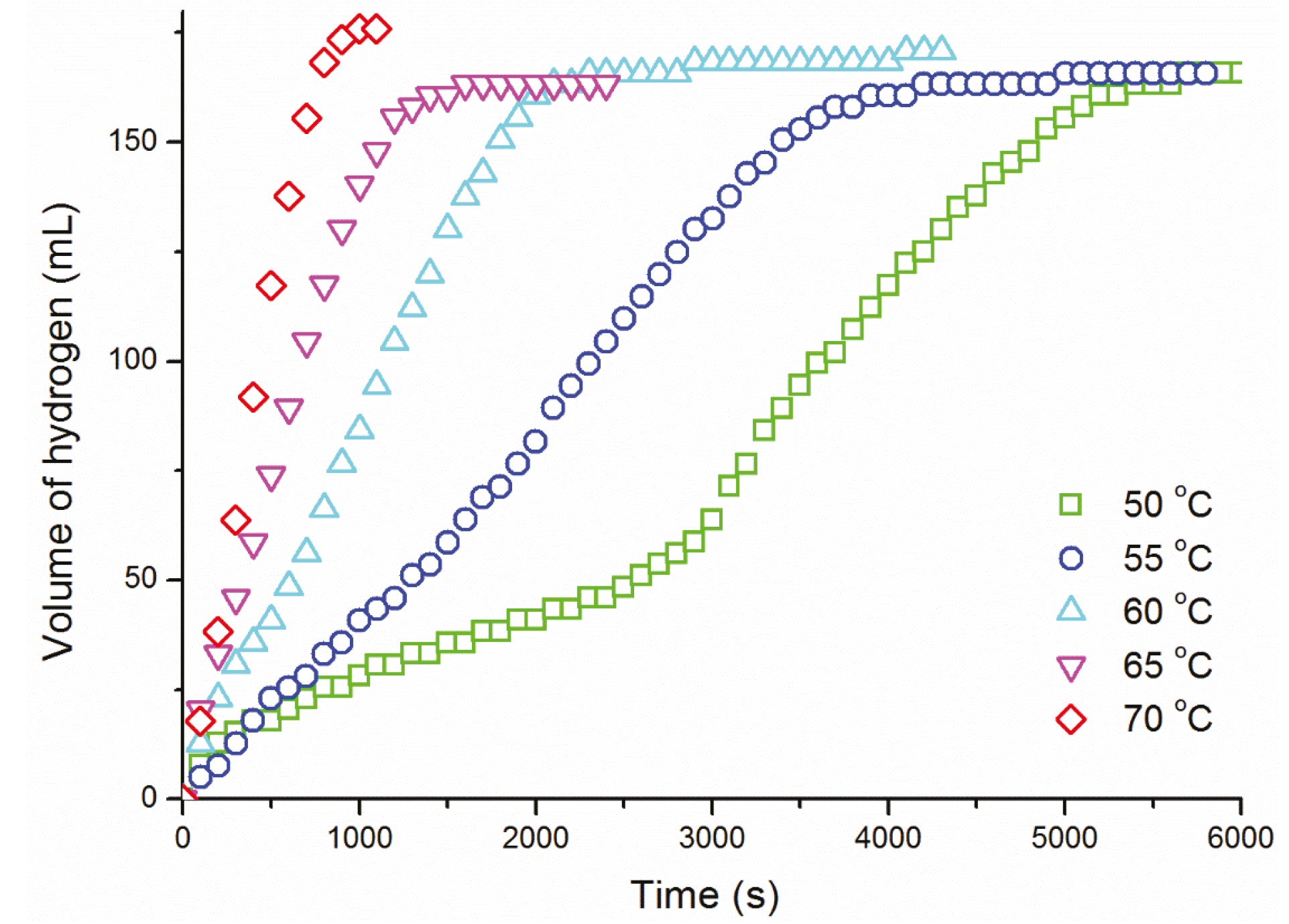{kind=link}
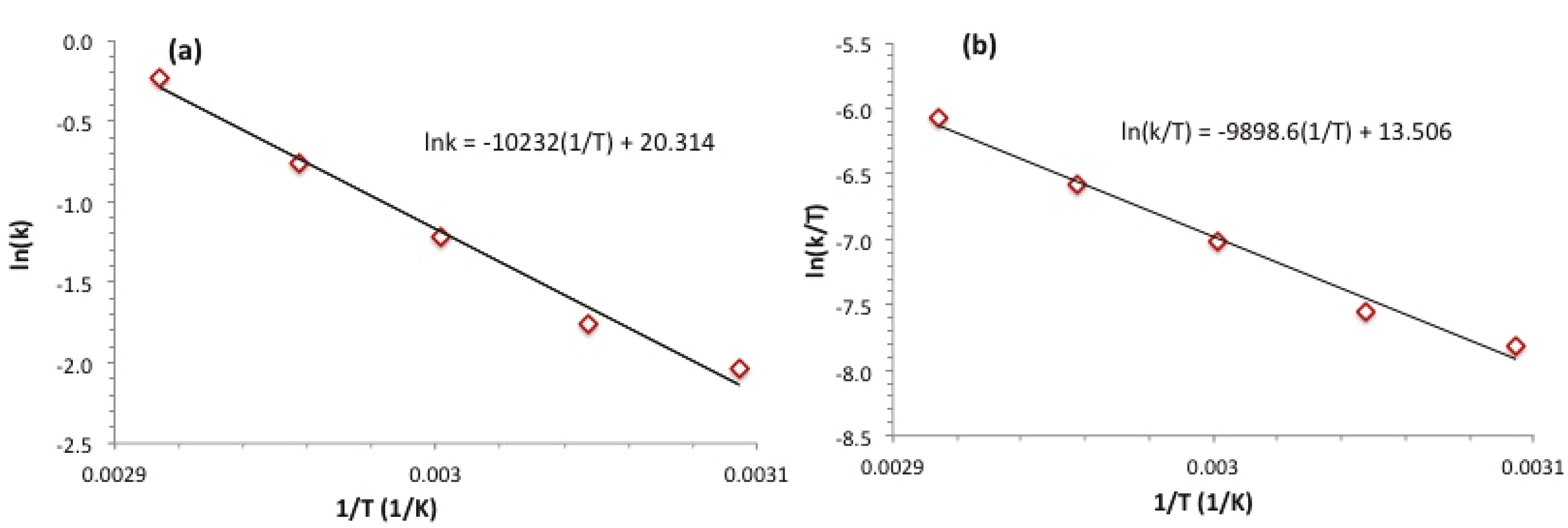{kind=link}
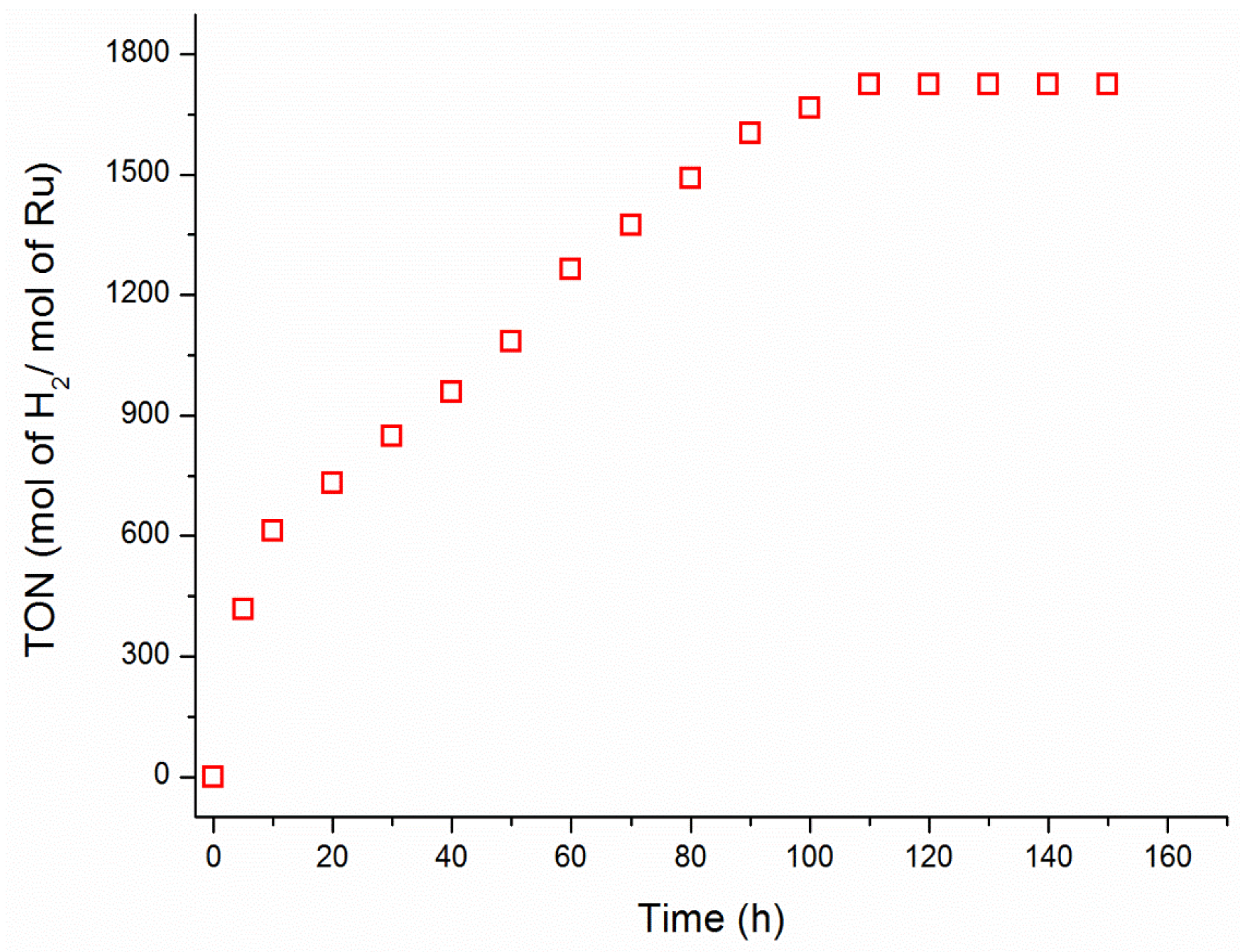{kind=link}
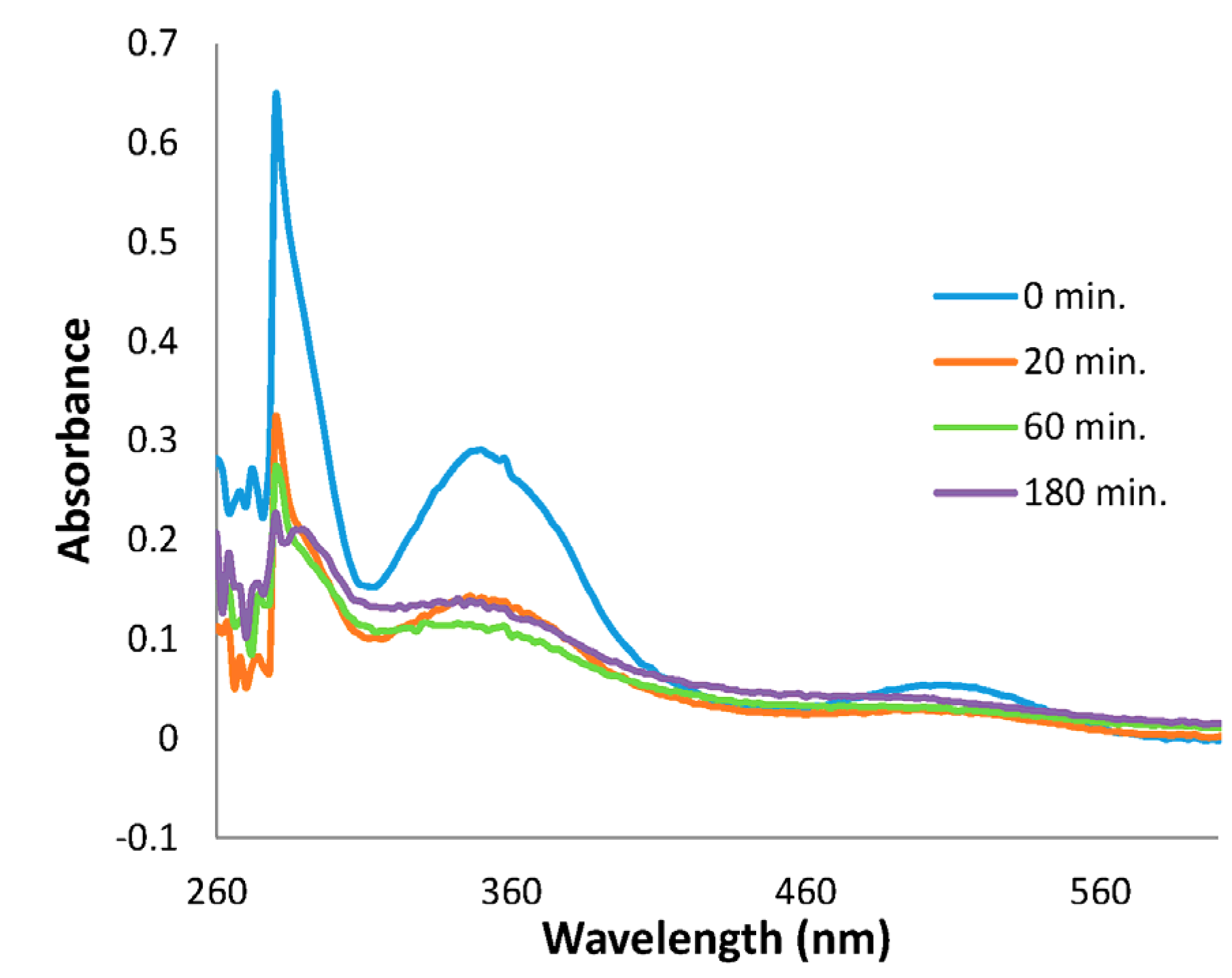{kind=link}
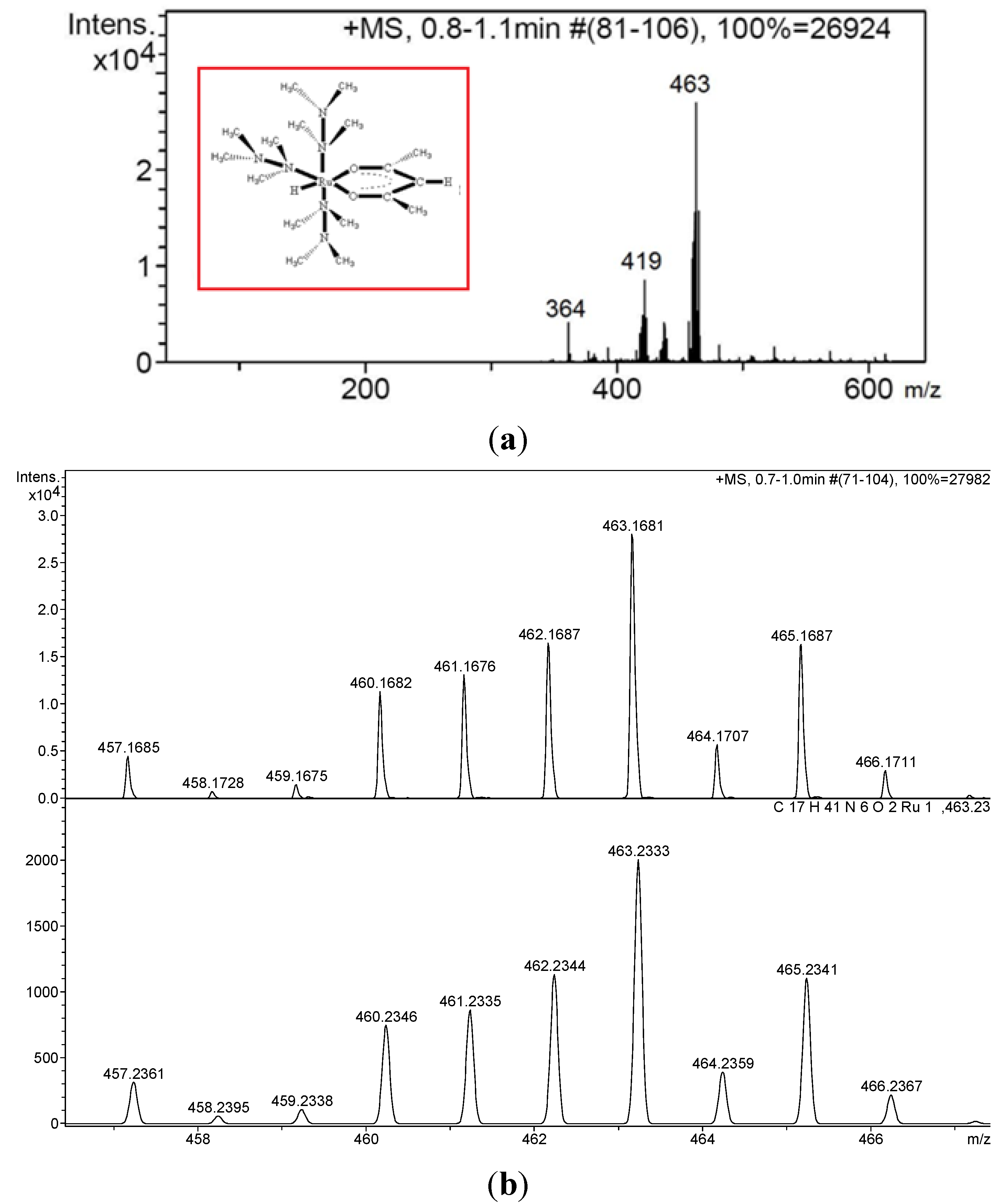{kind=link}
| T (°C) | k ((mol H2)·(mol Ru)−1·s−1) |
|---|---|
| 50 | 1.30 ± 0.07 ×10−1 |
| 55 | 1.72 ± 0.09 ×10−1 |
| 60 | 2.97 ± 0.15 ×10−1 |
| 65 | 4.67 ± 0.23 ×10−1 |
| 70 | 7.95 ± 0.40 ×10−1 |
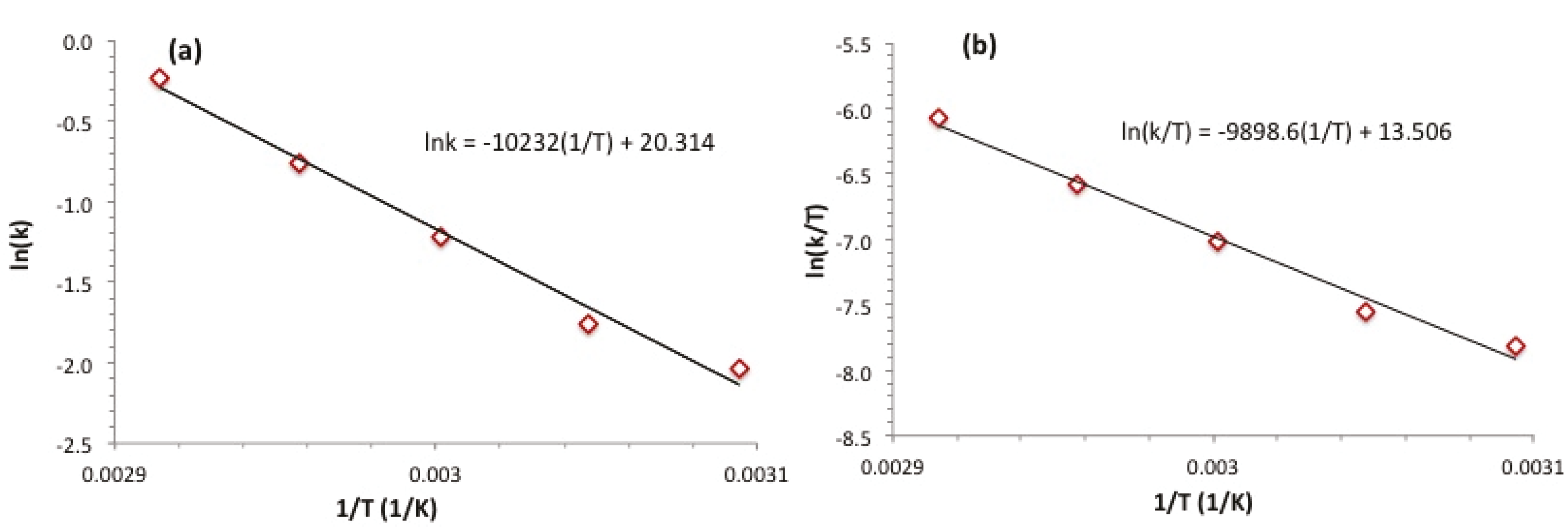
3. Experimental Section
3.1. Materials
3.2. Equipment
3.3. Catalytic Dehydrogenation of Dimethylamine Borane Starting with Ruthenium(III) Acetylacetonate
3.4. Catalytic Lifetime of Ruthenium(III) Acetylacetonate
3.5. Poisoning Experiment
3.6. Isolation and Characterization of Rutneium(II) Species, [Ru(N2Me4)3(acac)H]
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bluhm, M.E.; Bradley, M.G.; Butterick, R.; Kusari, U.; Sneddon, L.G. Amineborane-Based Chemical Hydrogen Storage: Enhanced Ammonia Borane Dehydrogenation in Ionic Liquids. J. Am. Chem. Soc. 2006, 128, 7748–7749. [Google Scholar] [CrossRef] [PubMed]
- Blaquiere, N.; Diallo-Garcia, S.; Gorelsky, S.I.; Black, D.A.; Fagnou, K. Ruthenium-Catalyzed Dehydrogenation of Ammonia Boranes. J. Am. Chem. Soc. 2008, 130, 14034–14035. [Google Scholar] [CrossRef] [PubMed]
- Käß, M.; Friedrich, A.; Dress, M.; Schneider, S. Ruthenium Complexes with Cooperative PNP Ligands: Bifunctional Catalysts for the Dehydrogenation of Ammonia-Borane. Angew. Chem. Int. Ed. 2009, 48, 905–907. [Google Scholar] [CrossRef] [PubMed]
- US Department of Energy (DOE). Basic Research Needs for the Hydrogen Economy. Available online: http://science.energy.gov/~/media/bes/pdf/reports/files/nhe_rpt.pdf (accessed on 27 May 2015).
- Inter Academy Council. Lighting the Way: Towards a Sustainable Energy Futures; Inter Academy Council: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Van den Berg, A.W.C.; Areán, C.O. Materials for Hydrogen Storage: Current Research Trends and Perspectives. Chem. Commun. 2008. [Google Scholar] [CrossRef]
- T-Raissi, A. Technoeconomic Analysis of Area II Hydrogen Production-PART II: Hydrogen from Ammonia and Ammonia-Borane Complex for Fuel Cell Applications. Available online: http://www.eere.energy.gov/hydrogenandfuelcells/pdfs/32405b15.pdf (accessed on 27 May 2015).
- Annual Energy Outlook 2005. With Projections To 2025. Available online: http://www.eia.gov/oiaf/archive/aeo05/pdf/0383(2005).pdf (accessed on 27 May 2015).
- Turner, J.; Sverdrup, G.; Mann, K.; Maness, P.G.; Kroposki, B.; Ghirardi, M.; Evans, R.J.; Blake, D. Renewable Hydrogen Production. Int. J. Energy Res. 2007, 32, 379–407. [Google Scholar] [CrossRef]
- Jaska, C.A.; Temple, K.; Lough, A.J.; Manners, I. Rhodium-Catalyzed Formation of Boron–Nitrogen Bonds: A Mild Route to Cyclic Aminoboranes and Borazines. Chem. Commun. 2001. [Google Scholar] [CrossRef]
- Jaska, C.A.; Temple, K.; Lough, A.J.; Manners, I. Transition Metal-Catalyzed Formation of Boron-Nitrogen Bonds: Catalytic Dehydrocoupling of Amine-Borane Adducts to Form Aminoboranes and Borazines. J. Am. Chem. Soc. 2003, 125, 9424–9434. [Google Scholar] [CrossRef] [PubMed]
- Friederich, A.; Drees, M.; Schneider, S. Ruthenium-Catalyzed Dimethylamineborane Dehydrogenation: Stepwise Metal-Centered Dehydrocyclization. Chem. Eur. J. 2009, 15, 10339–10342. [Google Scholar] [CrossRef] [PubMed]
- Jaska, C.A.; Manners, I. Heterogeneous or Homogeneous Catalysis? Mechanistic Studies of the Rhodium-Catalyzed Dehydrocoupling of Amine-Borane and Phosphine-Borane Adducts. J. Am. Chem. Soc. 2004, 126, 9776–9785. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fulton, J.L.; Linehan, J.C.; Autrey, T. In-situ Spectroscopic Studies of Rhodium Catalyzed Production of Hydrogen from Dimethylamine Borane: Homogeneous or Heterogeneous Catalysis? Prepr. Pap. Am. Chem. Soc. Div. Fuel Chem. 2004, 49, 972–973. [Google Scholar]
- Chen, Y.; Fulton, J.L.; Linehan, J.C.; Autrey, T. In Situ XAFS and NMR Study of Rhodium-Catalyzed Dehydrogenation of Dimethylamine Borane. J. Am. Chem. Soc. 2005, 127, 3254–3255. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.J.; Russell, C.A.; Manners, I. Homogeneous, Titanocene-Catalyzed Dehydrocoupling of Amine–Borane Adducts. J. Am. Chem. Soc. 2006, 128, 9582–9583. [Google Scholar] [CrossRef] [PubMed]
- Sloan, M.E.; Staubitz, A.; Clark, T.J.; Russell, C.A.; Lloyd-Jones, G.C.; Manners, I. Homogeneous Catalytic Dehydrocoupling/Dehydrogenation of Amine-Borane Adducts by Early Transition Metal, Group 4 Metallocene Complexes. J. Am. Chem. Soc. 2010, 132, 3831–3841. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, G.; Vendier, L.; Clot, E.; Sabo-Etienne, S. Ruthenium Bis(σ-B H) Aminoborane Complexes from Dehydrogenation of Amine–Boranes: Trapping of H2BNH2. Angew. Chem. Int. Ed. 2010, 49, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Jaska, C.A.; Manners, I. Ambient Temperature, Tandem Catalytic Dehydrocoupling-Hydrogenation Reactions Using Rh Colloids and Me2NH·BH3 as a Stoichiometric H2 Source. J. Am. Chem. Soc. 2004, 126, 2698–2699. [Google Scholar] [CrossRef] [PubMed]
- Durap, F.; Zahmakıran, M.; Özkar, S. Water Soluble Laurate-Stabilized Rhodium(0) Nanoclusters Catalyst with Unprecedented Catalytic Lifetime in the Hydrolytic Dehydrogenation of Ammonia-Borane. Appl. Cat. A Gen. 2009, 369, 53–59. [Google Scholar] [CrossRef]
- Zahmakıran, M.; Özkar, S. Dimethylammonium Hexanoate Stabilized Rhodium(0) Nanoclusters Identified as True Heterogeneous Catalysts with the Highest Observed Activity in the Dehydrogenation of Dimethylamine-Borane. Inorg. Chem. 2009, 48, 8955–8964. [Google Scholar] [CrossRef] [PubMed]
- Zahmakıran, M.; Tristany, M.; Philippot, K.; Fajerweg, K.; Özkar, S.; Chaudret, B. Aminopropyltriethoxysilane Stabilized Ruthenium(0) Nanoclusters as an Isolable and Reusable Heterogeneous Catalyst for the Dehydrogenation of Dimethylamine–Borane. Chem. Commun. 2010, 46, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Berke, H. Dehydrocoupling of Dimethylamine-Borane Catalysed by Rhenium Complexes and its Application in Olefin Transfer-Hydrogenations. Chem. Commun. 2007, 34, 3571–3573. [Google Scholar] [CrossRef]
- Fulton, J.L.; Linehan, J.C.; Autrey, T.; Balasubramanian, M.; Chen, Y.; Szymczak, N.K. When is a Nanoparticle a Cluster? An Operando EXAFS Study of Amine Borane Dehydrocoupling by Rh4-6 Clusters. J. Am. Chem. Soc. 2007, 129, 11936–11949. [Google Scholar] [CrossRef] [PubMed]
- Sloan, M.E.; Clark, T.J.; Manners, I. Homogeneous Catalytic Dehydrogenation/Dehydrocoupling of Amine-Borane Adducts by the Rh(I) Wilkinson’s Complex Analogue RhCl(PHCy2)3 (Cy = cyclohexyl). Inorg. Chem. 2009, 48, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Yurderi, M.; Bulut, A.; Zahmakiran, M.; Gulcan, M.; Özkar, S. Ruthenium(0) Nanoparticles Stabilized by Metal-Organic Framework (ZIF-8): Highly Efficient Catalyst for the Dehydrogenation of Dimethylamine-Borane and Transfer Hydrogenation of Unsaturated Hydrocarbons using Dimethylamine-Borane as Hydrogen Source. Appl. Catal. B Environ. 2014, 160–161, 534–541. [Google Scholar] [CrossRef]
- Munoz-Olasagasti, M.; Telleria, A.; Perez-Miqueo, J.; Garralda, M.A.; Freixa, Z. A Readily Accessible Ruthenium Catalyst for the Solvolytic Dehydrogenation of Amine–Borane Adducts. Dalton Trans. 2014, 43, 11404–11409. [Google Scholar] [CrossRef] [PubMed]
- Gulcan, M.; Zahmakiran, M.; Özkar, S. Palladium(0) Nanoparticles Supported on Metal Organic Framework as Highly Active and Reusable Nanocatalyst in Dehydrogenation of Dimethylamine-Borane. Appl. Catal. B Environ. 2014, 147, 394–401. [Google Scholar] [CrossRef]
- Sen, F.; Karatas, Y.; Gulcan, M.; Zahmakiran, M. Amylamine Stabilized Platinum(0) Nanoparticles: Active and Reusable Nanocatalyst in the Room Temperature Dehydrogenation of Dimethylamine-Borane. RSC Adv. 2014, 4, 1526–1531. [Google Scholar] [CrossRef]
- Keceli, E.; Özkar, S. Ruthenium(III) Acetylacetonate: A Homogeneous Catalyst in the Hydrolysis of Sodium Borohydride. J. Mol. Catal. A Chem. 2008, 286, 87–91. [Google Scholar] [CrossRef]
- Masjedi, M.; Demiralp, T.; Özkar, S. Testing Catalytic Activity of Ruthenium(III) Acetylacetonate in the Presence of Trialkylphosphite or Trialkylphosphine in Hydrogen Generation from the Hydrolysis of Sodium Borohydride. J. Mol. Catal. A Chem. 2009, 310, 59–63. [Google Scholar] [CrossRef]
- Masjedi, M.; Yildirim, L.T.; Özkar, S. Novel Homogeneous Catalyst Comprising Ruthenium and Trimethylphosphite for the Hydrolysis of Sodium Borohydride. J. Mol. Catal. A Chem. 2012, 355, 186–191. [Google Scholar] [CrossRef]
- Connors, K.A. Chemical Kinetics: The Study of Reaction Rates in Solution; VCH Publishers: New York, NY, USA, 1990. [Google Scholar]
- Twigg, M.V. Mechanism of Inorganic and Organometallic Reactions; Plenum Press: New York, NY, USA, 1994. [Google Scholar]
- Hornstein, B.J.; Aiken, J.D.; Finke, R.G. Nanoclusters in Catalysis: A Comparison of CS2 Catalyst Poisoning of Polyoxoanion- and Tetrabutylammonium-Stabilized 40 ± 6 Å Rh(0) Nanoclusters to 5 Rh/Al2O3, Including an Analysis of the Literature Related to the CS2 to Metal Stoichiometry Issue. Inorg. Chem. 2002, 41, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, P.; Latha, J.N.L.; Satyanarayana, S. DNA-Binding Studies of Mixed-Ligand (Ethylenediamine)Ruthenium(II) Complexes. Chem. Biodivers. 2006, 3, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Masjedi, M.; Yildirim, L.T.; Özkar, S. Crystal Structure of Trans and Cis-Bis(Acetylacetonato)Bis(Trimethylphosphite)Ruthenium(II) Complexes and Testing their Catalytic Activity in Hydrogen Generation from the Hydrolysis of Sodium Borohydride. Inorg. Chim. Acta 2010, 363, 1713–1718. [Google Scholar] [CrossRef]
- Barth, M.; Kaestele, X.; Kluefers, P. Polyol Metal Complexes. 46. Nitrosyl Ruthenium Diolato Complexes. Eur. J. Inorg. Chem. 2005, 7, 1353–1359. [Google Scholar] [CrossRef]
- Diamantis, A.A.; Moritz, P.S.; Snow, M.R.; Tiekink, E.R.T. Substituted Ruthenium Triammine Complexes: the Crystal Structure of Triammineaqua(Pentane-2,4-Dionato)Ruthenium(III) Dithionate Dihydrate, [Ru(NH3)3(acac)(OH2)][S2O6]·2H2O. Aust. J. Chem. 1988, 41, 1251–1256. [Google Scholar] [CrossRef]
- King, B.T.; Michl, J. The Explosive “Inert” Anion CB11(CF3)12−. J. Am. Chem. Soc. 2000, 122, 10255–10256. [Google Scholar] [CrossRef]
- Hancock, K.G.; Dickinson, D.A. Photochemistry of Dimethylamine in Hydrocarbon Solvents. Striking Differences between Solution- and Gas-Phase Photochemical Reactivity. J. Org. Chem. 1975, 40, 969–970. [Google Scholar] [CrossRef]
- Prakash, H. Ureas as Amine Substrates for Hydrazine and Alkylhydrazine Synthesis-Base-Assisted Chlorination of Urea by Dimethylchloramine as a Route for Hydrazine and Tetramethylhydrazine Synthesis. Indian J. Chem. A Inorg. Phys. Theo. Anal. 1986, 25A, 764–767. [Google Scholar]
- Bouman, M.; Zaera, F. Reductive Eliminations from Amido Metal Complexes: Implications for Metal Film Deposition. J. Electrochem. Soc. 2011, 158, D524–D526. [Google Scholar] [CrossRef]
- Widegren, J.A.; Finke, R.G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A Chem. 2003, 198, 317–341. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barın, E.Ü.; Masjedi, M.; Özkar, S. A New Homogeneous Catalyst for the Dehydrogenation of Dimethylamine Borane Starting with Ruthenium(III) Acetylacetonate. Materials 2015, 8, 3155-3167. https://doi.org/10.3390/ma8063155
Barın EÜ, Masjedi M, Özkar S. A New Homogeneous Catalyst for the Dehydrogenation of Dimethylamine Borane Starting with Ruthenium(III) Acetylacetonate. Materials. 2015; 8(6):3155-3167. https://doi.org/10.3390/ma8063155
Chicago/Turabian StyleBarın, Ebru Ünel, Mehdi Masjedi, and Saim Özkar. 2015. "A New Homogeneous Catalyst for the Dehydrogenation of Dimethylamine Borane Starting with Ruthenium(III) Acetylacetonate" Materials 8, no. 6: 3155-3167. https://doi.org/10.3390/ma8063155
APA StyleBarın, E. Ü., Masjedi, M., & Özkar, S. (2015). A New Homogeneous Catalyst for the Dehydrogenation of Dimethylamine Borane Starting with Ruthenium(III) Acetylacetonate. Materials, 8(6), 3155-3167. https://doi.org/10.3390/ma8063155