Fullerene-Based Photoactive Layers for Heterojunction Solar Cells: Structure, Absorption Spectra and Charge Transfer Process

Abstract
:1. Introduction
2. Methods
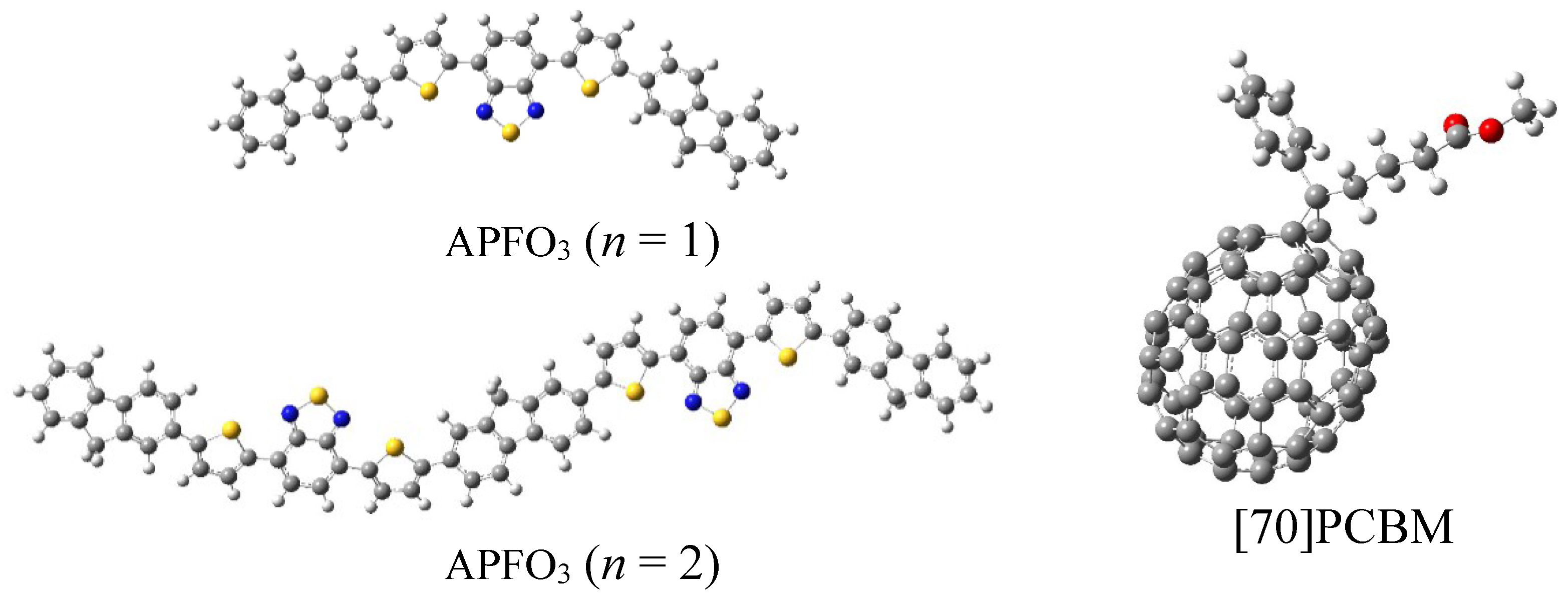
3. Results and Discussion
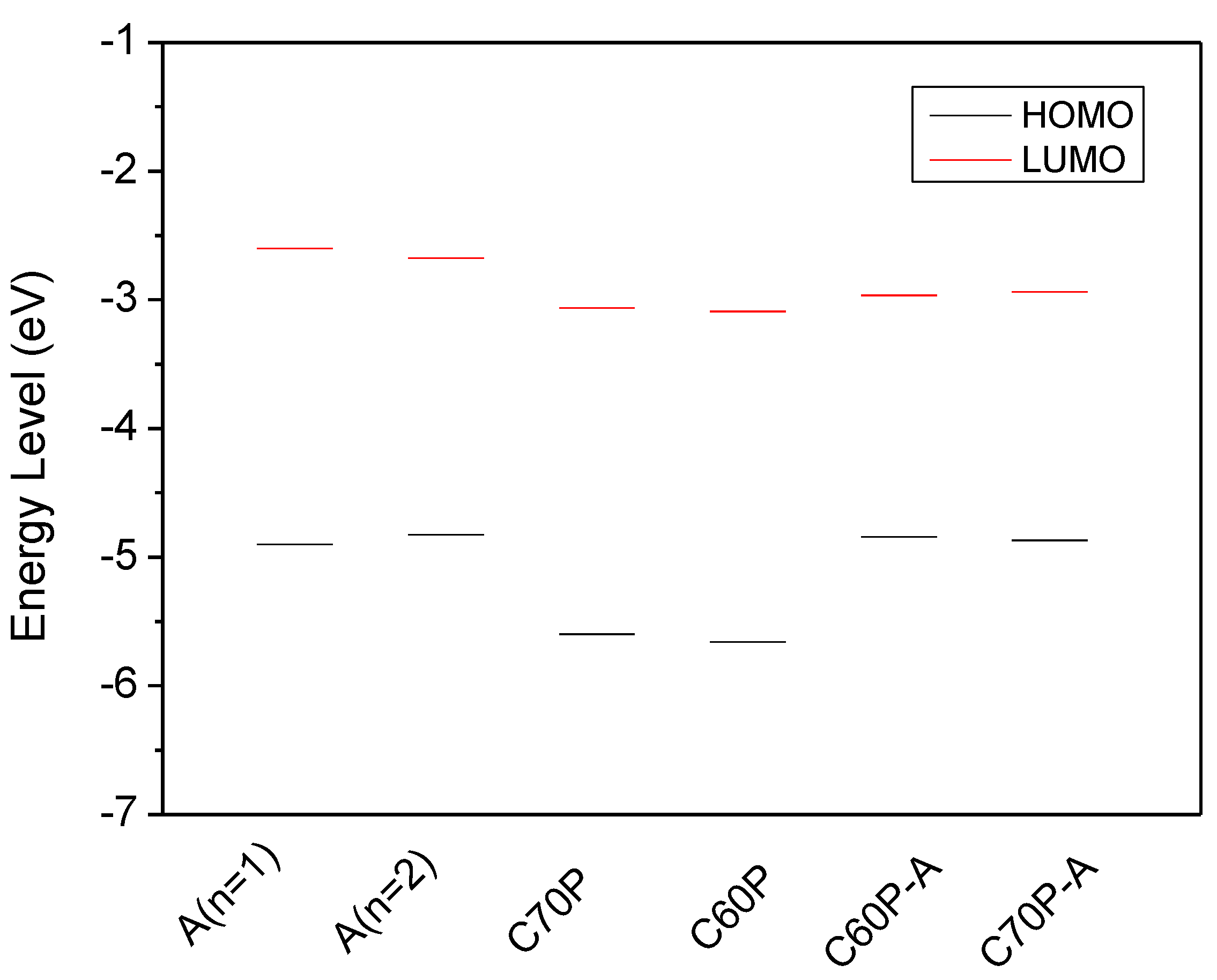
3.1. Energy Levels and Band Gap

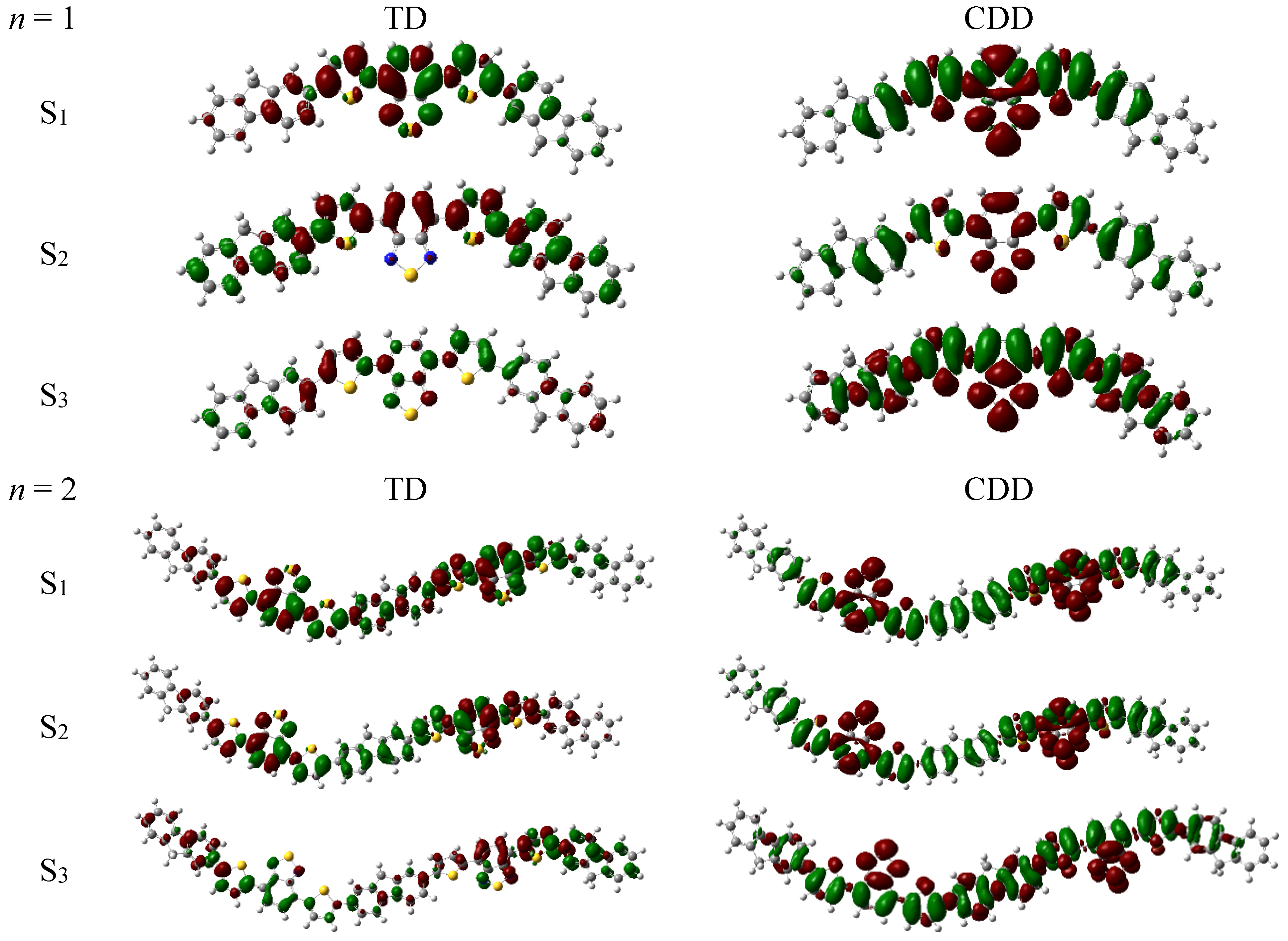
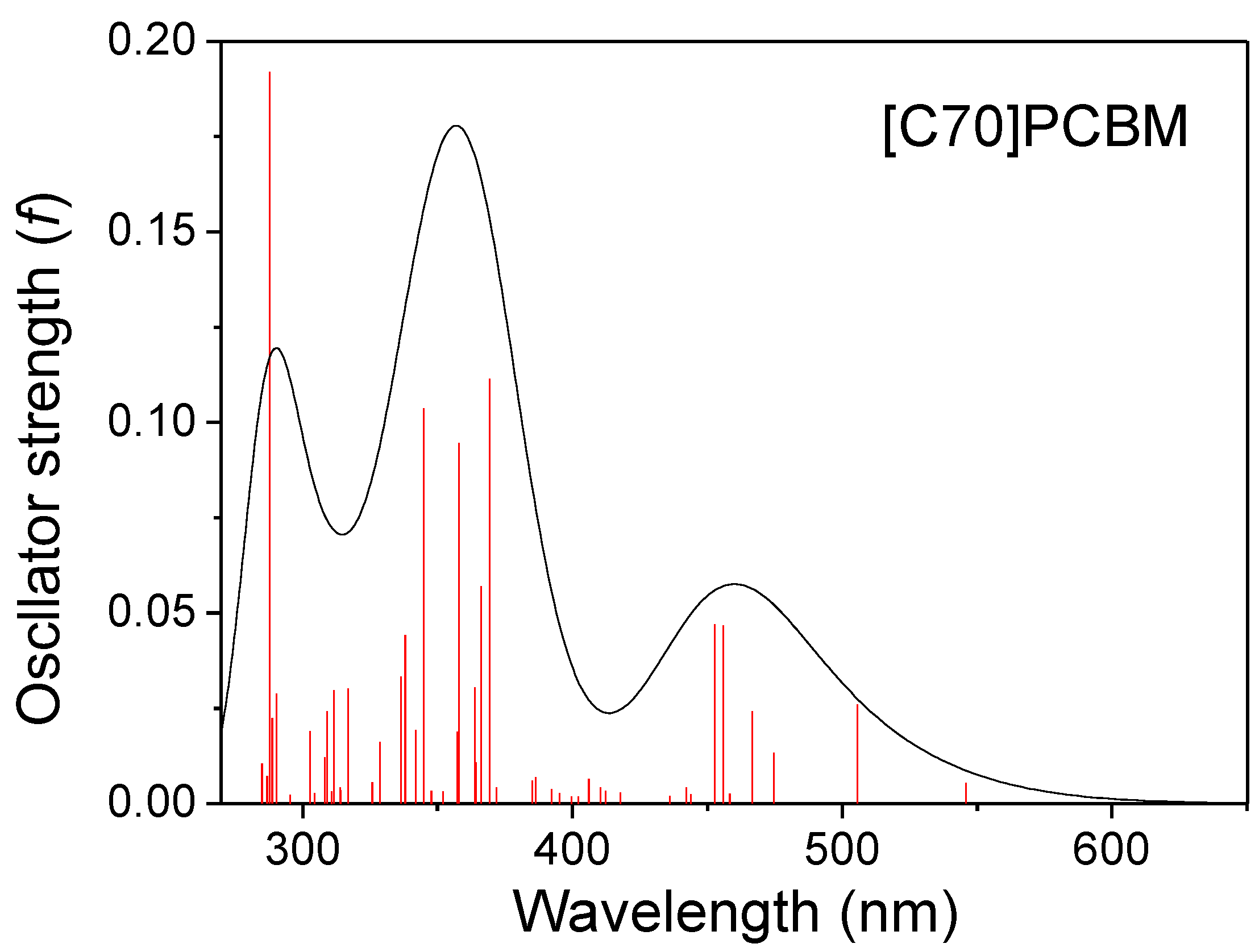
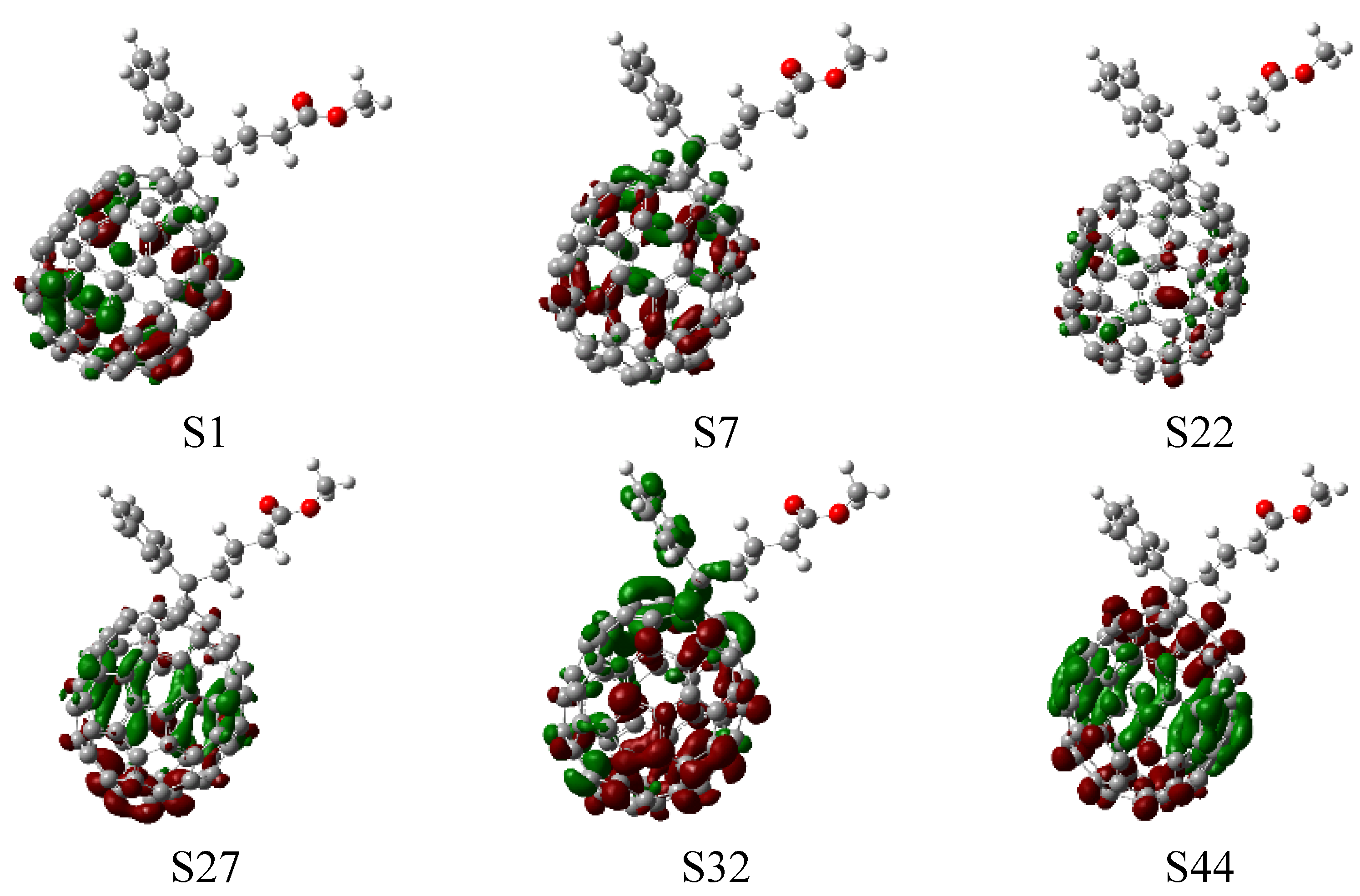
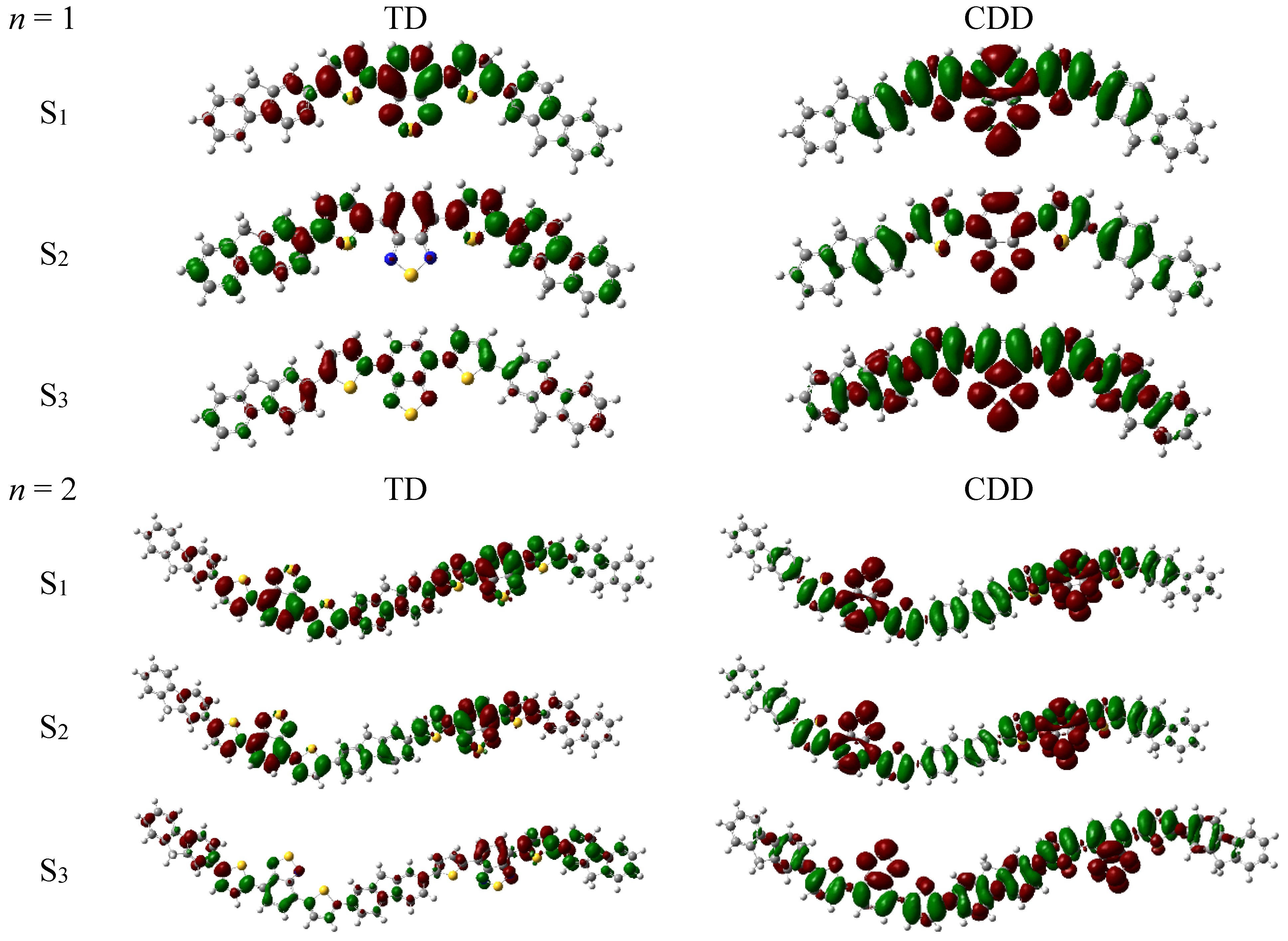
3.2. Optical Absorption of Donor, Acceptor and the Donor-Acceptor Complex

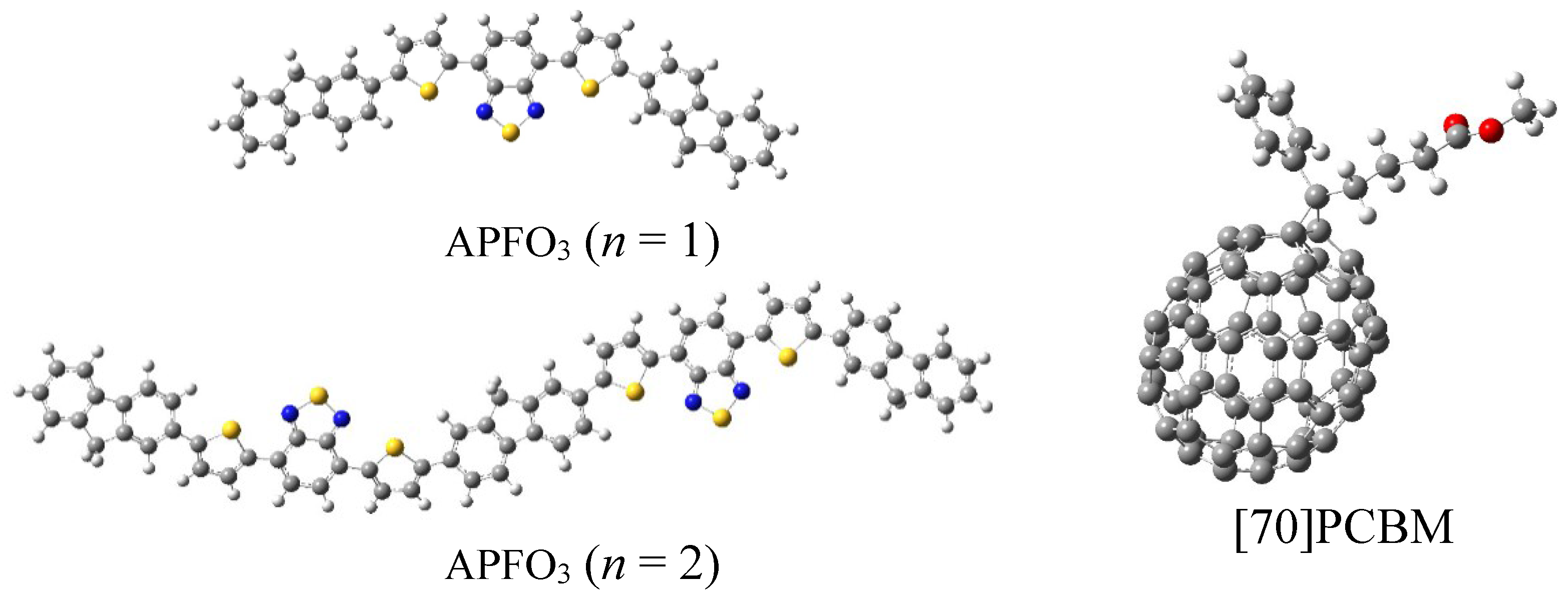{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| States | n = 1 | n = 2 | Experiment | ||
|---|---|---|---|---|---|
| eV (nm) | f | eV (nm) | f | nm | |
| S1 | 2.48(500.84) | 1.3006 | 2.40(515.68) | 2.8379 | 540 |
| S2 | 3.51(353.23) | 0.0299 | 2.52(491.41) | 0.0625 | – |
| S3 | 3.78(328.04) | 1.3606 | 3.40(364.33) | 0.1901 | 384 |
| S4 | 4.09(302.68) | 0.0862 | 3.49(355.25) | 0.0006 | – |
| S5 | 4.31(287.55) | 0.0023 | 3.67(337.96) | 1.7567 | – |
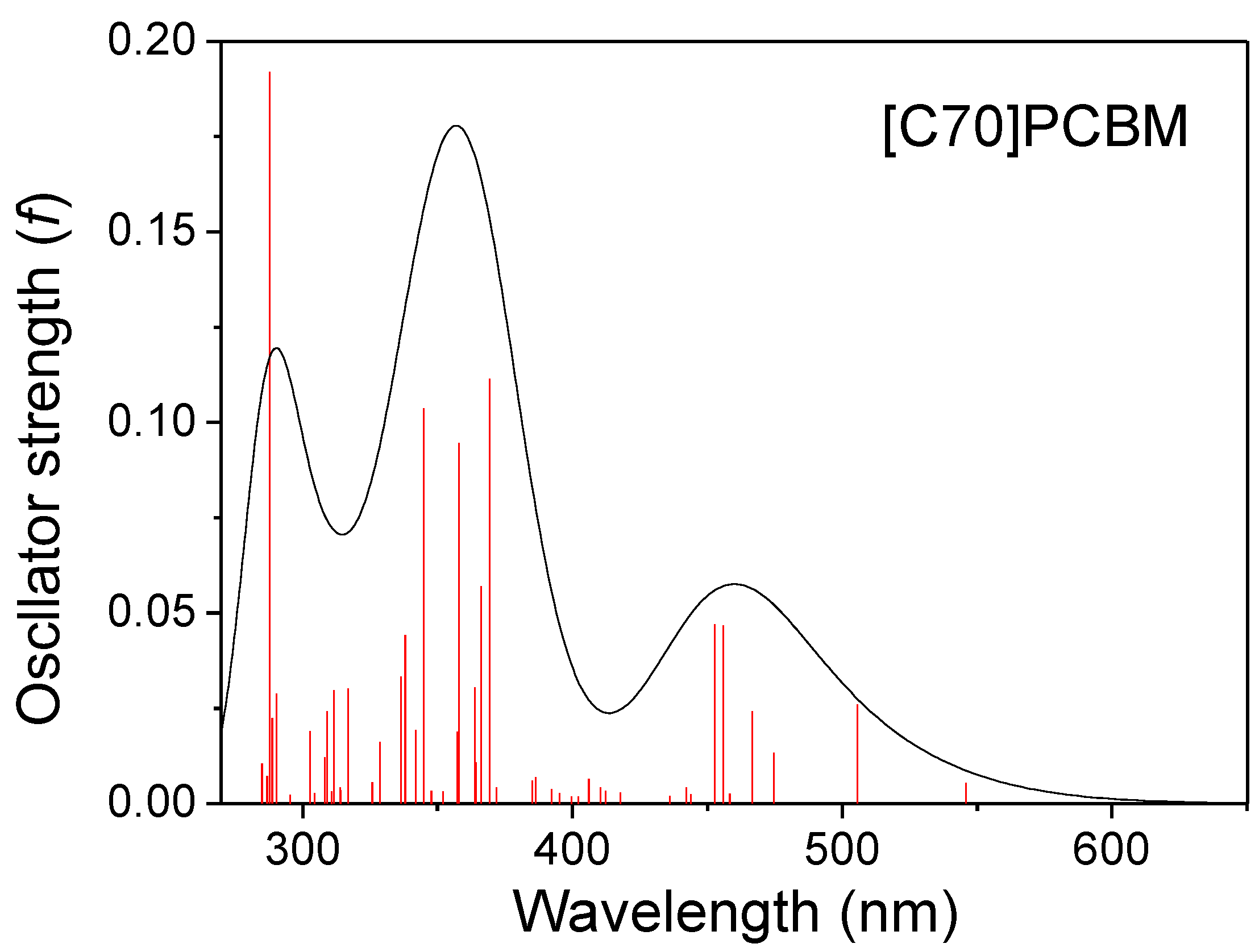
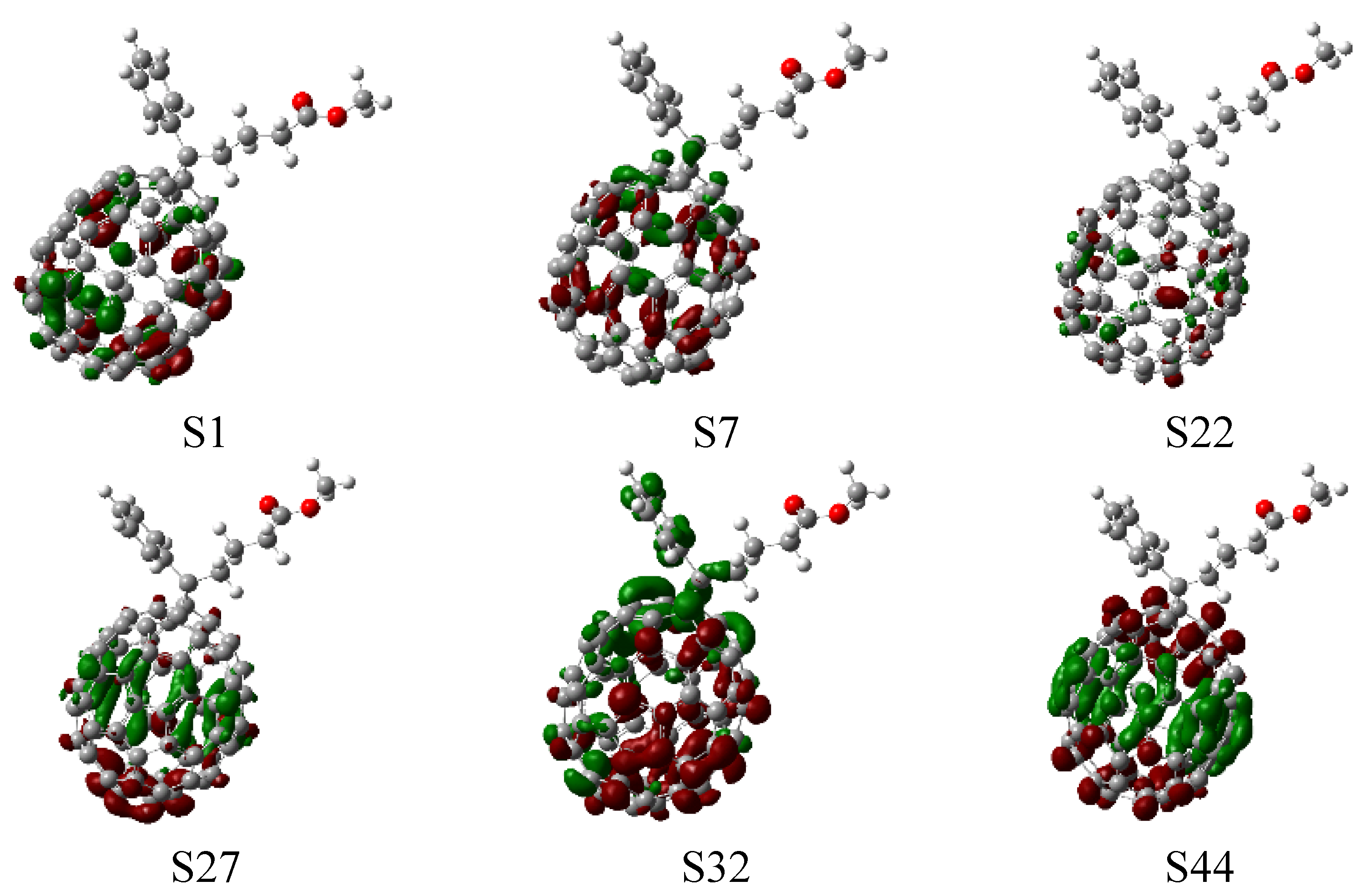
| States | [C60]PCBM & APFO3 | [C70]PCBM& APFO3 | ||
|---|---|---|---|---|
| eV (nm) | f | eV (nm) | f | |
| S1 | 2.42(511.43) | 0.0017 | 2.27(545.29) | 0.0014 |
| S2 | 2.46(504.84) | 0.0026 | 2.45(506.59) | 0.1925 |
| S3 | 2.48(500.52) | 1.1259 | 2.48(500.88) | 0.9239 |
| S4 | 2.53(490.32) | 0.0004 | 2.61(474.33) | 0.0127 |
| S5 | 2.55(486.25) | 0.0000 | 2.66(466.35) | 0.0159 |
| S6 | 2.68(463.09) | 0.0001 | 2.71(457.64) | 0.0006 |
| S7 | 2.73(454.88) | 0.0004 | 2.72(456.05) | 0.0432 |
| S8 | 2.78(445.36) | 0.0000 | 2.74(452.86) | 0.0663 |
| S9 | 2.84(437.07) | 0.0003 | 2.79(443.97) | 0.0020 |
| S10 | 2.86(433.12) | 0.0053 | 2.79(443.84) | 0.0024 |
| S11 | 2.87(431.69) | 0.0008 | 2.81(442.00) | 0.0023 |
| S12 | 2.94(421.83) | 0.0005 | 2.85(435.34) | 0.0000 |
| S13 | 2.95(419.91) | 0.0013 | 2.89(428.27) | 0.0045 |
| S14 | 2.99(415.21) | 0.0018 | 2.96(419.22) | 0.0006 |
| S15 | 3.00(412.93) | 0.0001 | 2.97(416.85) | 0.0009 |
| S16 | 3.09(401.58) | 0.0005 | 3.01(411.92) | 0.0022 |
| S17 | 3.10(400.60) | 0.0002 | 3.02(410.38) | 0.0020 |
| S18 | 3.14(394.37) | 0.0010 | 3.05(405.93) | 0.0050 |
| S19 | 3.18(389.81) | 0.0153 | 3.08(402.16) | 0.0000 |
| S20 | 3.46(358.09) | 0.0029 | 3.10(399.78) | 0.0000 |
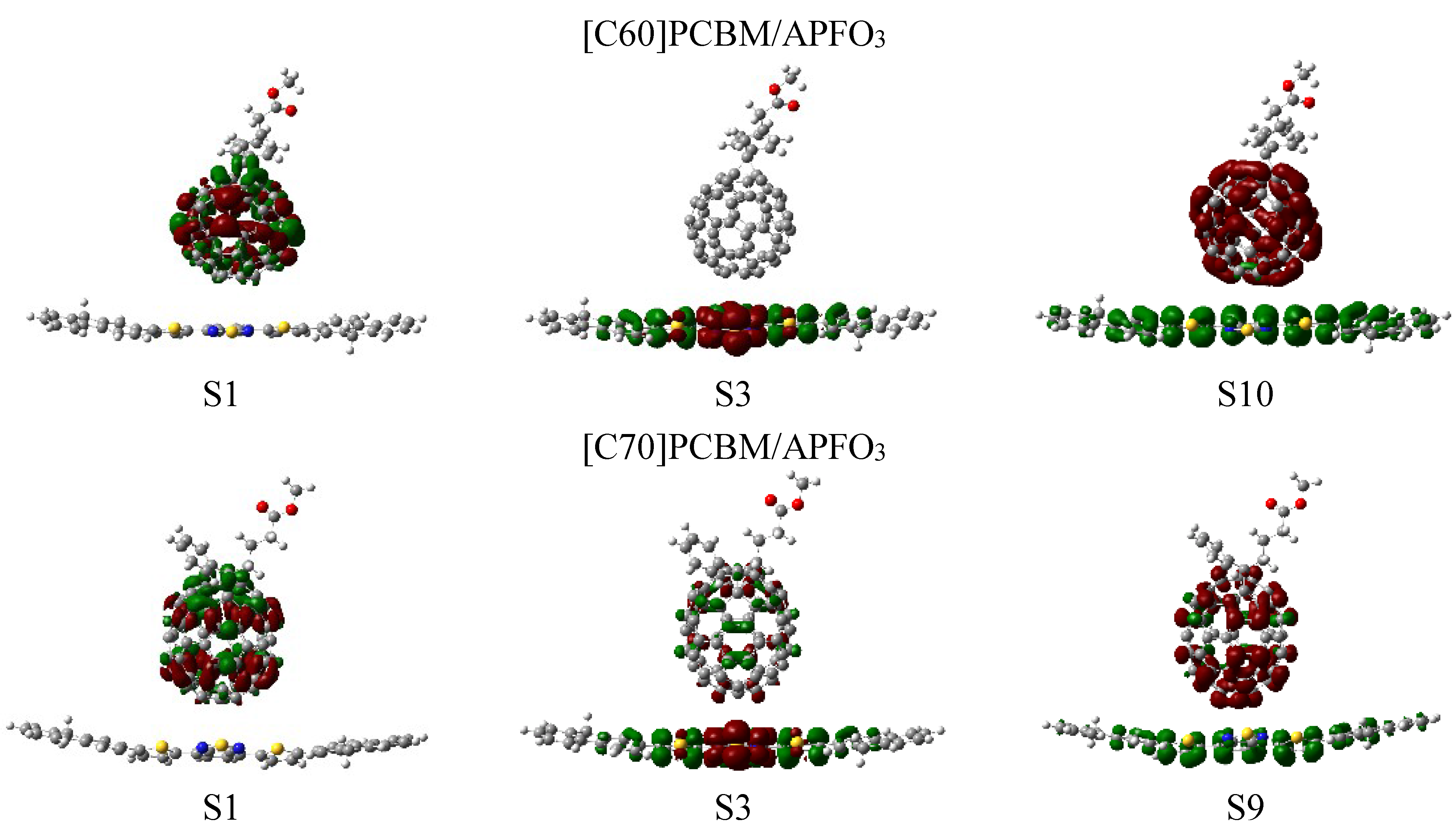
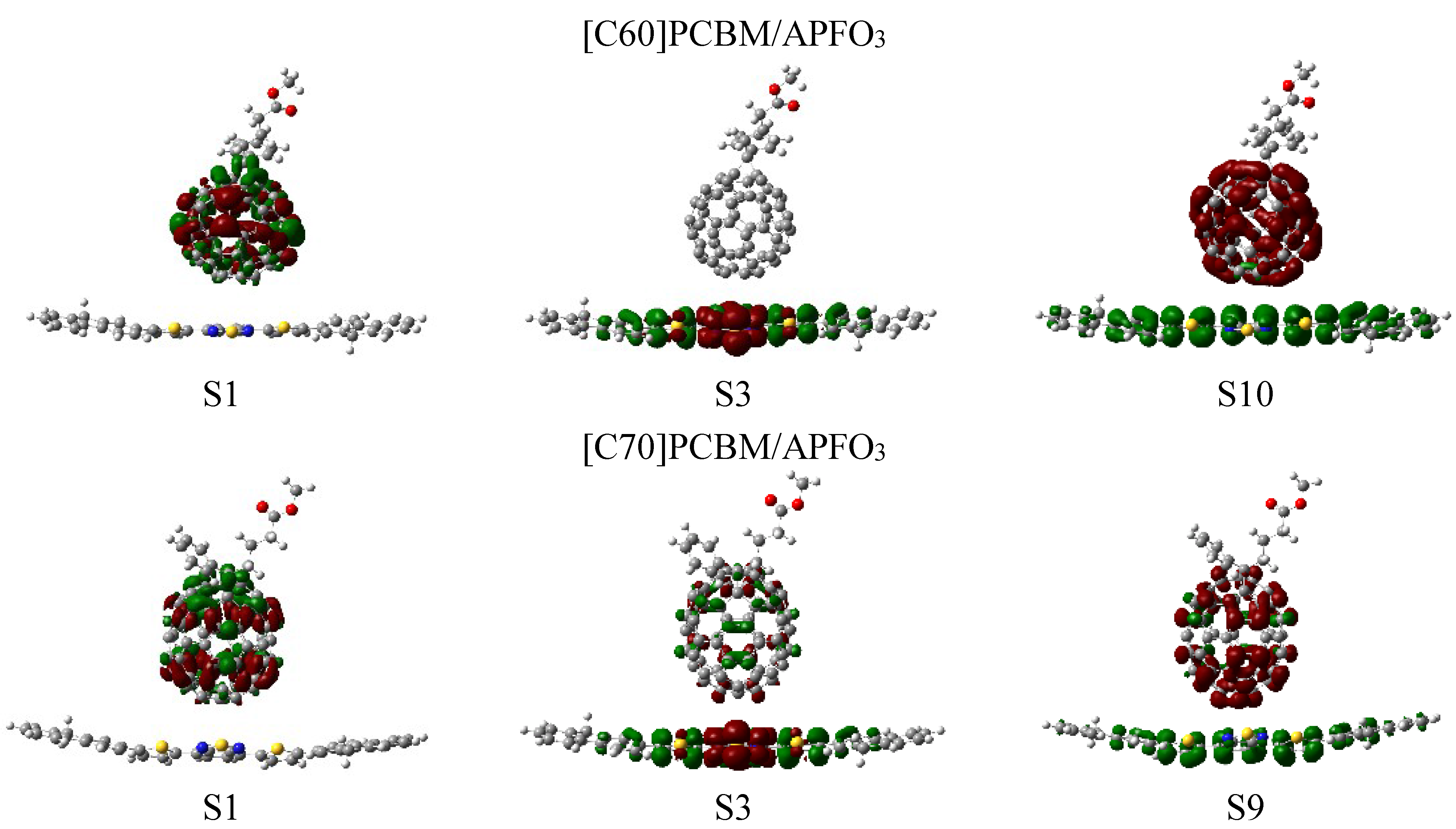
3.3. Rate of Charge Transfer in the Marcus Theory
| Complex | States | U (a.u.) | U (a.u.) | VDA (cm−1) |
|---|---|---|---|---|
| [C60]PCBM/APFO3 | S10 | 13.39286 | 0.1910 | 329.2 |
| [C70]PCBM/APFO3 | S9 | 10.41667 | 0.1204 | 260.2 |
| Complex | Δ GCR | λ | Δ GCT | VDA | KCT (×1013) | KCR (×107) |
|---|---|---|---|---|---|---|
| [C60]PCBM/APFO3 | −1.810 | 0.7 | −0.6655 | 0.04082 | 3.2811 | 0.13517 |
| [C70]PCBM/APFO3 | −1.837 | 0.7 | −0.6400 | 0.03265 | 2.0304 | 0.036515 |
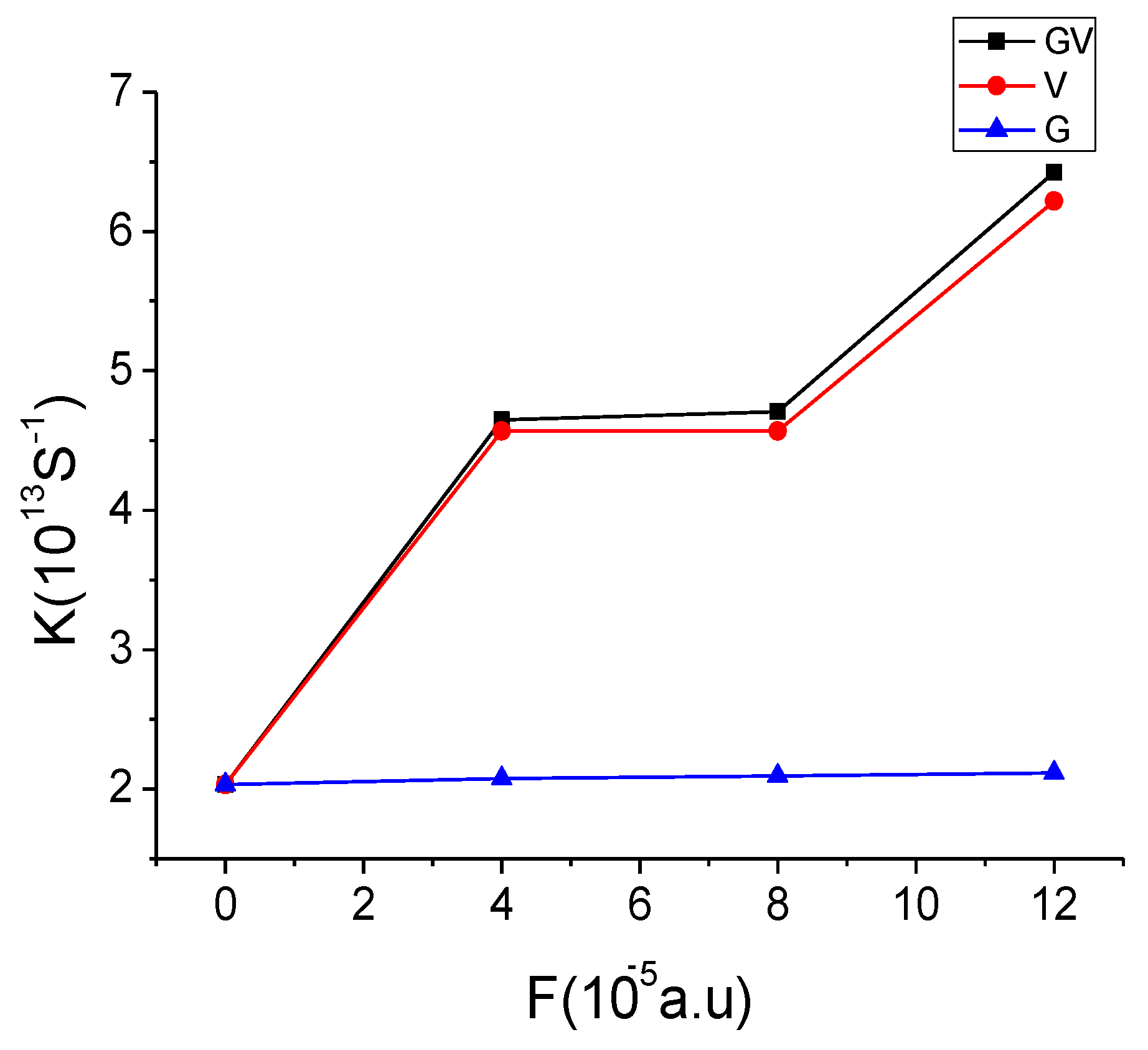
3.4. Effect of Electronic Field on CT Rate
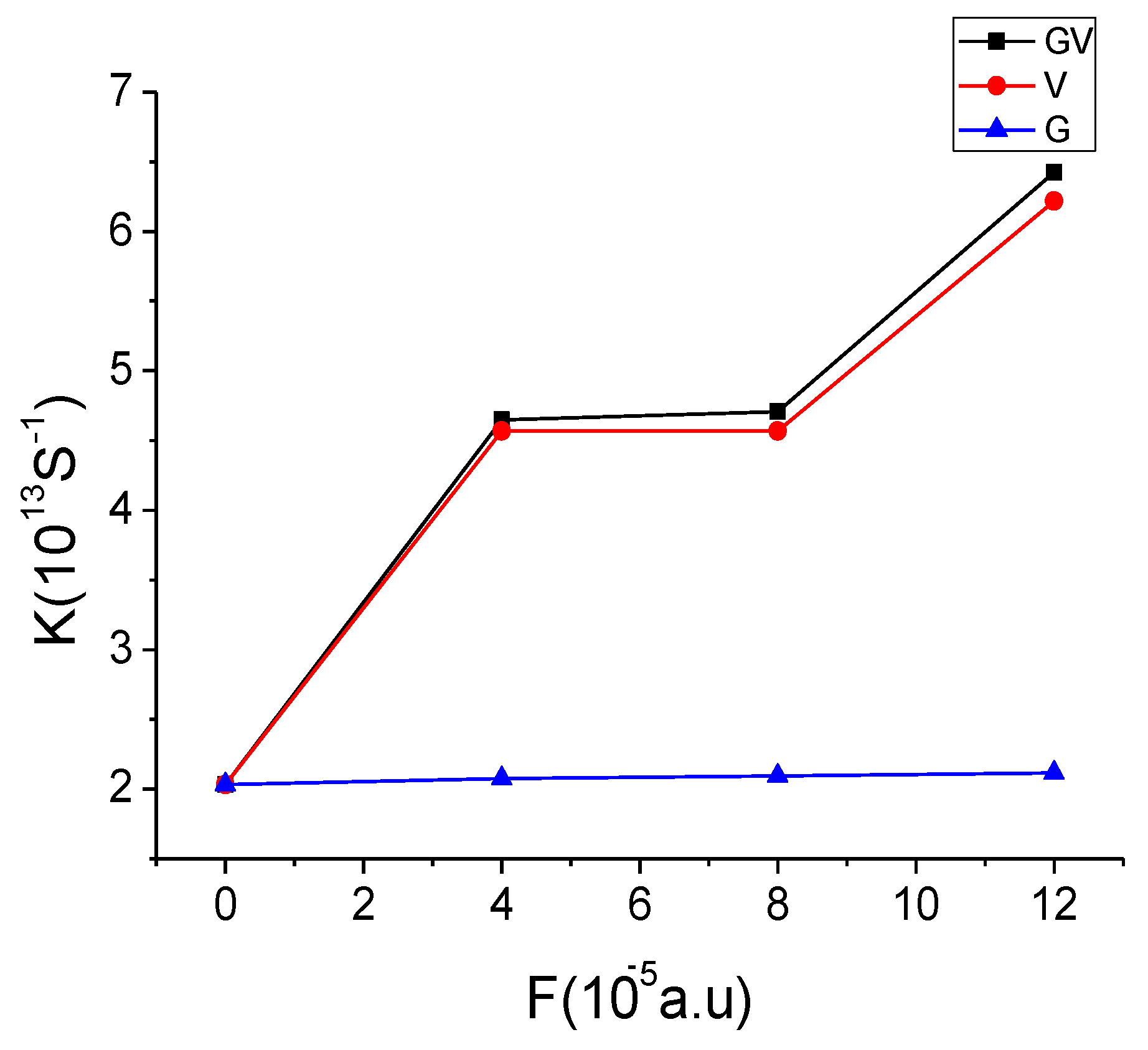
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lewis, N.S. Toward cost-effective solar energy use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.L.; Zhu, H.F.; Meng, Q.; Gong, X.; Hu, W.P. Organic photoresponse materials and devices. Chem. Soc. Rev. 2012, 41, 1754–1808. [Google Scholar] [CrossRef] [PubMed]
- Roes, A.L.; Alsema, E.A.; Blok, K.; Patel, M.K. Ex-ante environmental and economic evaluation of polymer photovoltaics. Prog. Photovolt. 2009, 17, 372–393. [Google Scholar] [CrossRef]
- Darling, S.B.; You, F.Q. The case for organic photovoltaics. RSC Adv. 2013, 3, 17633–17648. [Google Scholar] [CrossRef]
- Mihailetchi, V.D.; Xie, H.X.; Boer, B.; Koster, L.J.A.; Blom, P.W.M. Charge Transport and Photocurrent Generation in Poly(3-hexylthiophene): Methanofullerene Bulk-Heterojunction Solar Cells. Adv. Funct. Mater. 2006, 16, 699–708. [Google Scholar] [CrossRef]
- Reiss, P.; Couderc, E.; De Girolamo, J.; Pron, A. Conjugated polymers/semiconductor nanocrystals hybrid materials-preparation, electrical transport properties and applications. Nanoscale 2011, 3, 446–489. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Mohanraj, J.; Accorsi, G.; Armaroli, N.; Boyd, P.D.W. A supramolecular porphyrin-ferrocene-fullerene triad. New. J. Chem. 2011, 35, 632–639. [Google Scholar] [CrossRef]
- Ranta, J.; Kumpulainen, T.; Lemmetyinen, H.; Efimov, A. Synthesis and characterization of monoisomeric 1,8,15,22-Substituted (A3B and A2B2) phthalocyanines and phthalocyanine-fullerene dyads. J. Org. Chem. 2010, 75, 5178–5194. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Morgade, M.S.; Plonska-Brzezinska, M.E.; Athans, A.J.; Carbonell, E.; Miguel, G.D.; Guldi, D.M.; Echegoyen, L.; Torres, T. Synthesis, characterization, and photoinduced electron transfer processes of orthogonal ruthenium phthalocyanine−fullerene assemblies. J. Am. Chem. Soc. 2009, 131, 10484–10496. [Google Scholar] [CrossRef] [PubMed]
- Sakai, J.; Kawano, K.; Yamanari, T.; Taima, T.; Yoshida, Y.; Fujii, A.; Ozaki, M. Efficient organic photovoltaic tandem cells with novel transparent conductive oxide interlayer and poly (3-hexylthiophene): Fullerene active layers. Solar Energy Mater. Solar Cells. 2010, 94, 376–380. [Google Scholar] [CrossRef]
- Holmes, N.P.; Uluma, S.; Sista, P.; Burkea, K.B.; Wilsona, M.G.; Stefanb, M.C.; Zhou, X.J.; Dastoor, P.C.; Belcher, W.J. The effect of polymer molecular weight on P3HT:PCBM nanoparticulate organic photovoltaic device performance. Solar Energy Mater. Solar Cells. 2014, 128, 369–377. [Google Scholar] [CrossRef]
- Zhao, G.J.; He, Y.J.; Li, Y.F. 6.5% Efficiency of polymer solar cells based on poly(3-hexylthiophene) and indene-C60 bisadduct by device optimization. Adv. Mater. 2010, 22, 4355–4358. [Google Scholar] [CrossRef] [PubMed]
- Vandenbergh, J.; Conings, B.; Bertho, S.; Kesters, J.; Spoltore, D.; Esiner, S.; Zhao, J.; van Assche, G.; Wienk, M.M.; Maes, W.; et al. Thermal stability of poly[2-methoxy-5-(2′-phenylethoxy)-1,4-phenylenevinylene](MPE-PPV):Fullerene bulk heterojunction solar cells. Macromolecules 2011, 44, 8470–8478. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kimura, M. Extension of light-harvesting area of bulk-heterojunction solar cells by cosensitization with ring-expanded metallophthalocyanines fused with fluorene skeletons. ACS Appl. Mater. Interfaces 2013, 5, 4367–4373. [Google Scholar] [PubMed]
- Petsalakis, I.D.; Tagmatarchis, N.; Theodorakopoulos, G. Theoretical study of fulleropyrrolidines by density functional and time-dependent density functional theory. J. Phys. Chem. C 2007, 111, 14139–14149. [Google Scholar] [CrossRef]
- Shalabi, A.S.; Aal, S.A.; Assem, M.M.; Soliman, K.A. Metallophthalocyanine and Metallo phthalocyanine–fullerene complexes as potential dye sensitizers for solar cells DFT and TD-DFT. Org. Electron. 2012, 13, 2063–2074. [Google Scholar] [CrossRef]
- Pelzer, K.M.; Chan, M.K.Y.; Gray, S.K.; Darling, S.B. Polaron Structure and Transport in Fullerene Materials: Insights from First-Principles Calculations. J. Phys. Chem. C 2014, 118, 21785–21797. [Google Scholar] [CrossRef]
- Barszcz, B.; Laskowska, B.; Graja, A.; Park, E.Y.; Kim, T.D.; Lee, K.S. Electronic excitations of the fullerene–thiophene-derived dyads. Synth. Metals 2011, 161, 229–234. [Google Scholar] [CrossRef]
- Li, Y.Z.; Ma, F.C.; Dong, B.; Lie, J.; Chen, M.D. Theoretical study of charge transfer mechanism in fullerene-phenyl-phenothiazine compound: A real-space analysis. Dyes Pigment. 2012, 92, 1344–1350. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.Z.; Liu, J.; Zhao, M.Y.; Han, Y.Y.; Ding, Y.; Song, P.; Ma, F.C. Theoretical evidence for the distance-dependent photoinduced electron transfer of porphyrin- ligothiophene-fullerene triads. Int. J. Photoenergy 2012. [Google Scholar] [CrossRef]
- Pal, S.K.; Kesti, T.; Maiti, M.; Zhang, F.L.; Inganas, O.; Hellstrom, S.; Andersson, M.R.; Oswald, F.; Langa, F.; Sterman, T.O.; et al. Geminate charge recombination in polymer/fullerene bulk heterojunction films and implications for solar cell function. J. Am. Chem. Soc. 2010, 132, 12440–12451. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Li, Y.Z.; Pullerits, T.; Zhao, M.Y.; Sun, M.T. Theoretical characterization of the PC60BM: PDDTT model for an organic solar cell. J. Phys. Chem. C 2011, 115, 21865–21873. [Google Scholar] [CrossRef]
- Liu, T.; Troisi, A. Absolute rate of charge separation and recombination in a molecular model of the P3HT/PCBM Interface. J. Phys. Chem. C 2011, 115, 2406–2415. [Google Scholar] [CrossRef]
- Darling, S.B.; Sternberg, M. Importance of side chains and backbone length in defect modeling of poly(3-alkylthiophenes). J. Phys. Chem. B 2009, 113, 6215–6218. [Google Scholar] [CrossRef] [PubMed]
- Dreizler, J.M.R.; Gross, E.K.U. Density Functional Theory; Springer-Verlag: Heidelberg, Germany, 1990. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry I the effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef]
- Kleinman, D.A. Nonlinear dielectric polarization in optical media. Phys. Rev. 1962, 126, 1977–1979. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Sun, M.T.; Kjellberg, P.; Beenken, W.J.D.; Pullerits, T. Comparison of the electronic structure of PPV and its derivative DIOXA-PPV. Chem. Phys. 2006, 327, 474–484. [Google Scholar] [CrossRef]
- Beenken, W.J.D.; Pullerits, T. Spectroscopic units in conjugated polymers: A quantum chemically founded concept. J. Phys. Chem. B. 2004, 108, 6164–6169. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.T.; Xu, H.X. Direct visualization of the chemical mechanism in SERRS of 4-Aminothiophenol/Metal complexes and Metal/4-Aminothiophenol/Metal junctions. ChemPhysChem 2009, 10, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.R.; Li, Y.Z.; Xu, H.X.; Sun, M.T. Ascertaining p,p′-Dimercaptoazobenzene produced from p-Aminothiophenol by selective catalytic coupling reaction on silver nanoparticles. Langmuir 2010, 26, 7737–7746. [Google Scholar] [CrossRef] [PubMed]
- Oudar, J.L.; Chemla, D.S. Hyperpolarizabilities of the nitroanilines and their relations to the excited state dipole moment. J. Chem. Phys. 1977, 66, 2664–2668. [Google Scholar] [CrossRef]
- Oudar, J.L. Optical nonlinearities of conjugated molecules. Stilbene derivatives and highly polar aromatic compounds. J. Chem. Phys. 1977, 67, 446–457. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry: Theory and experiment. Angew. Chem. Int. Ed. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Cave, R.J.; Newton, M.D. Generalization of the Mulliken-Hush treatment for the calculation of electron transfer matrix elements. Chem. Phys. Lett. 1996, 249, 15–19. [Google Scholar] [CrossRef]
- Kjellberg, P.; He, Z.; Pullerits, T. Bacteriochlorophyll in Electric Field. J. Phys. Chem. B. 2003, 107, 13737–13742. [Google Scholar] [CrossRef]
- Kavarnos, G.J.; Turro, N.J. Photosensitization by reversible electron transfer: Theories, experimental evidence, and examples. Chem. Rev. 1986, 86, 401–449. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Qi, D.; Song, P.; Ma, F. Fullerene-Based Photoactive Layers for Heterojunction Solar Cells: Structure, Absorption Spectra and Charge Transfer Process. Materials 2015, 8, 42-56. https://doi.org/10.3390/ma8010042
Li Y, Qi D, Song P, Ma F. Fullerene-Based Photoactive Layers for Heterojunction Solar Cells: Structure, Absorption Spectra and Charge Transfer Process. Materials. 2015; 8(1):42-56. https://doi.org/10.3390/ma8010042
Chicago/Turabian StyleLi, Yuanzuo, Dawei Qi, Peng Song, and Fengcai Ma. 2015. "Fullerene-Based Photoactive Layers for Heterojunction Solar Cells: Structure, Absorption Spectra and Charge Transfer Process" Materials 8, no. 1: 42-56. https://doi.org/10.3390/ma8010042
APA StyleLi, Y., Qi, D., Song, P., & Ma, F. (2015). Fullerene-Based Photoactive Layers for Heterojunction Solar Cells: Structure, Absorption Spectra and Charge Transfer Process. Materials, 8(1), 42-56. https://doi.org/10.3390/ma8010042