Co-Precipitation of YAG Powders for Transparent Materials: Effect of the Synthesis Parameters on Processing and Microstructure

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Syntheses by Co-Precipitation Route

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation | Precursors | Precipitant |
|---|---|---|
| YAGcl-AA | YCl3·6H2O and AlCl3·9H2O | Ammonium hydroxide solution, dilute |
| YAGcl-AHC0.5 | YCl3·6H2O and AlCl3·9H2O | Ammonium hydrogen carbonate 0.5 M |
| YAGcl-AHC1.5 | YCl3·6H2O and AlCl3·9H2O | Ammonium hydrogen carbonate 1.5 M |
| YAGn-AHC0.5 | Y(NO3)3·6H2O and Al(NO3)3·9H2O | Ammonium hydrogen carbonate 0.5 M |
2.3. Processing, Sintering and Characterization
3. Results and Discussion
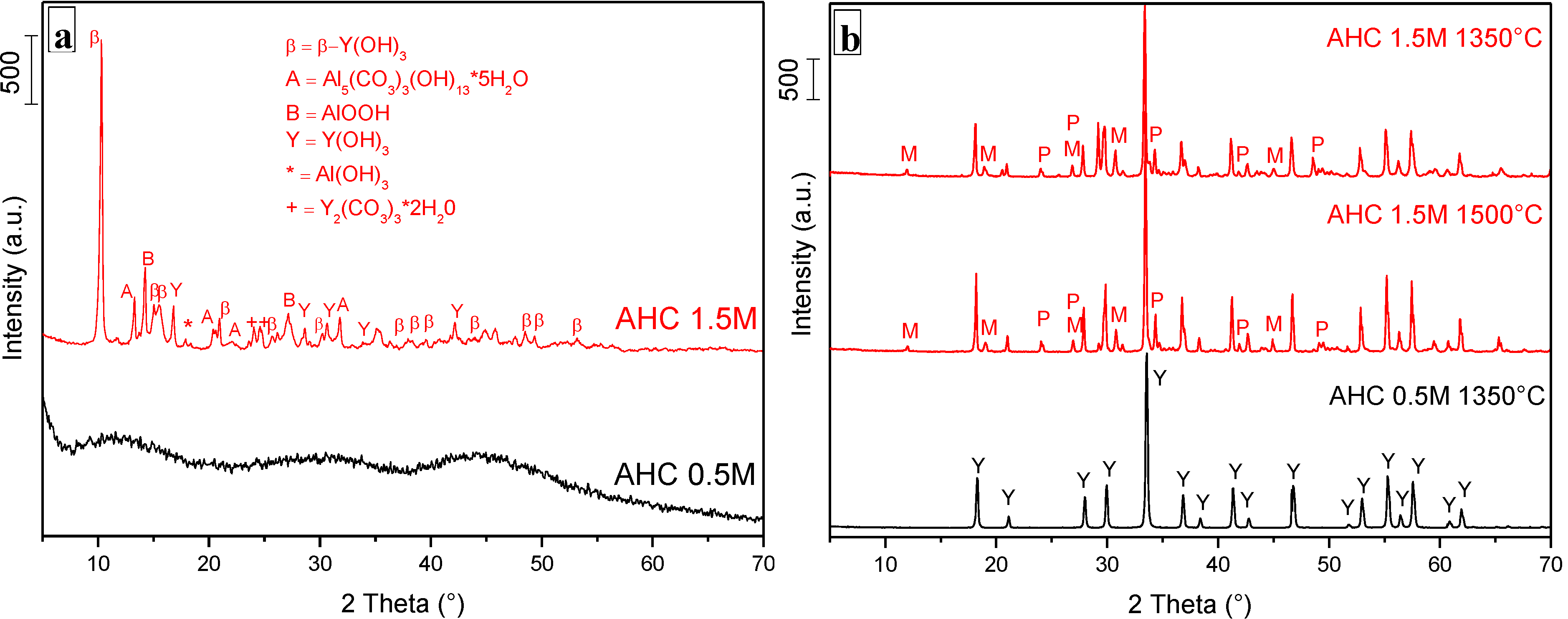
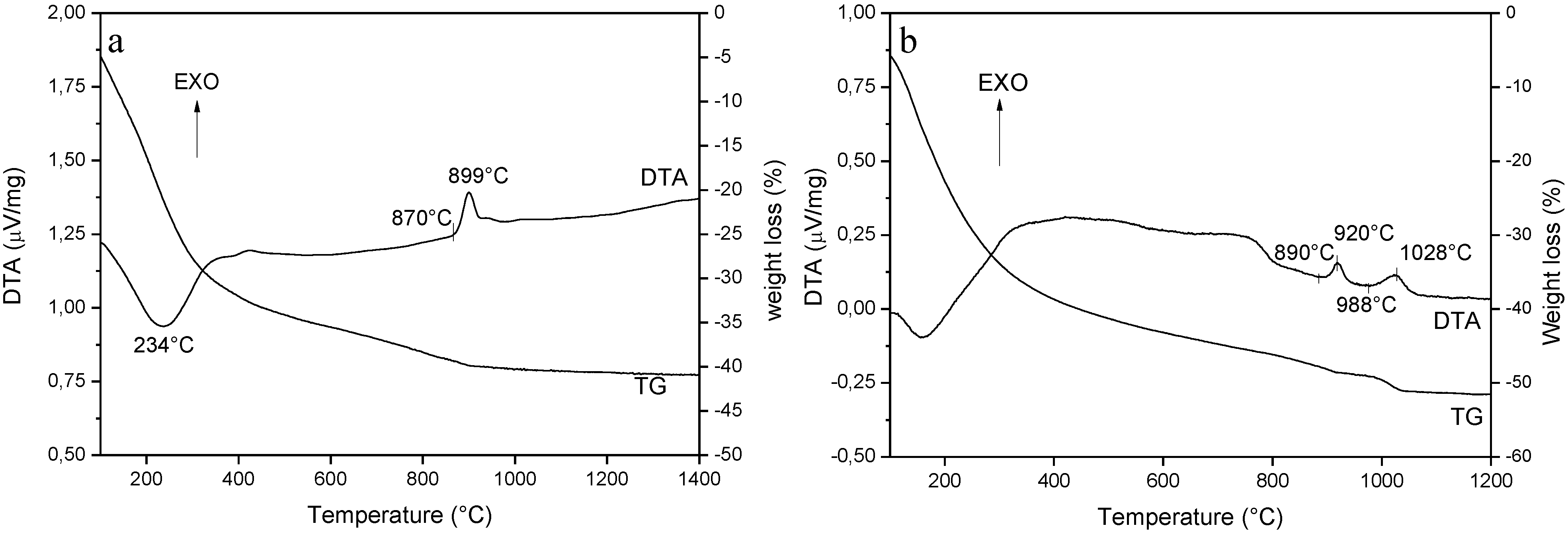
3.1. Role of the AHC Precipitant Concentration

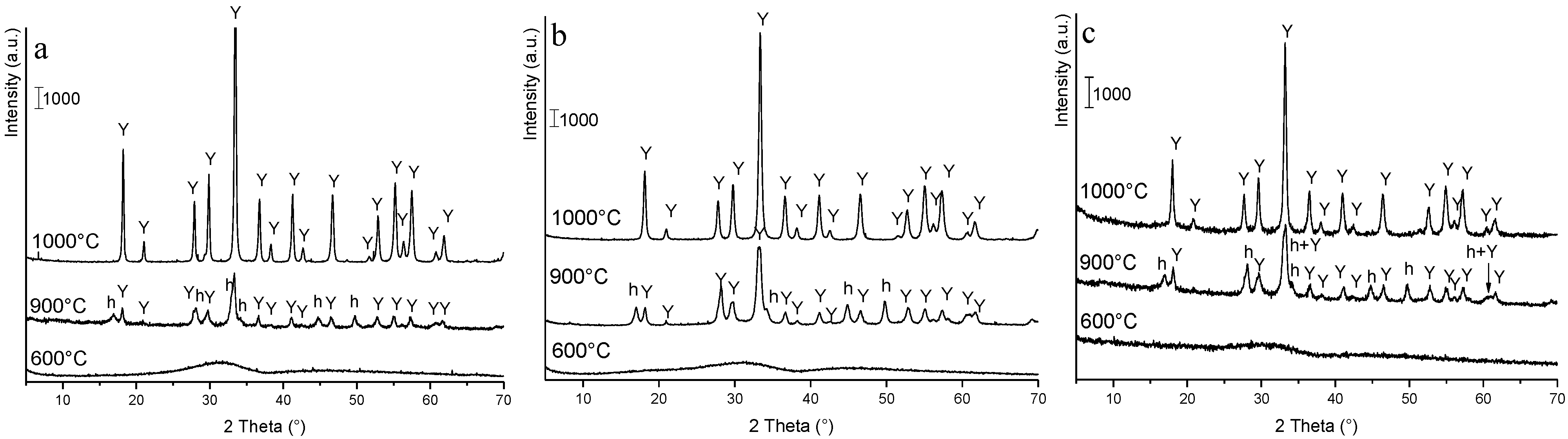
3.2. Phase Evolution: Role of the Precipitant and the Precursors
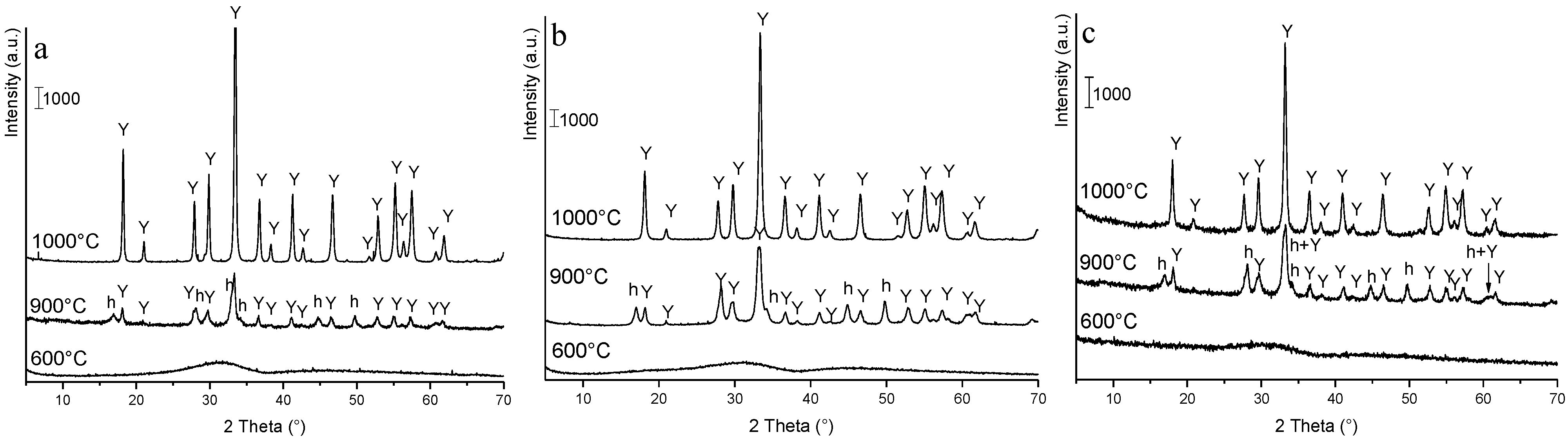
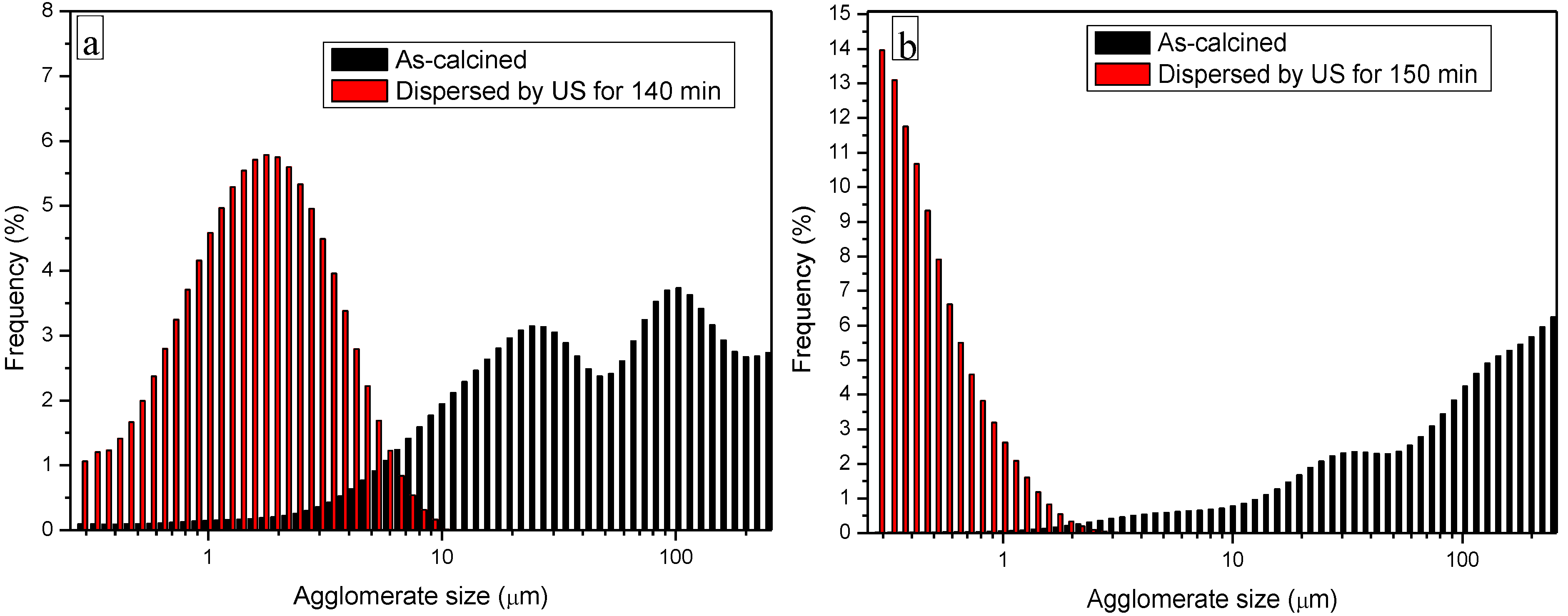
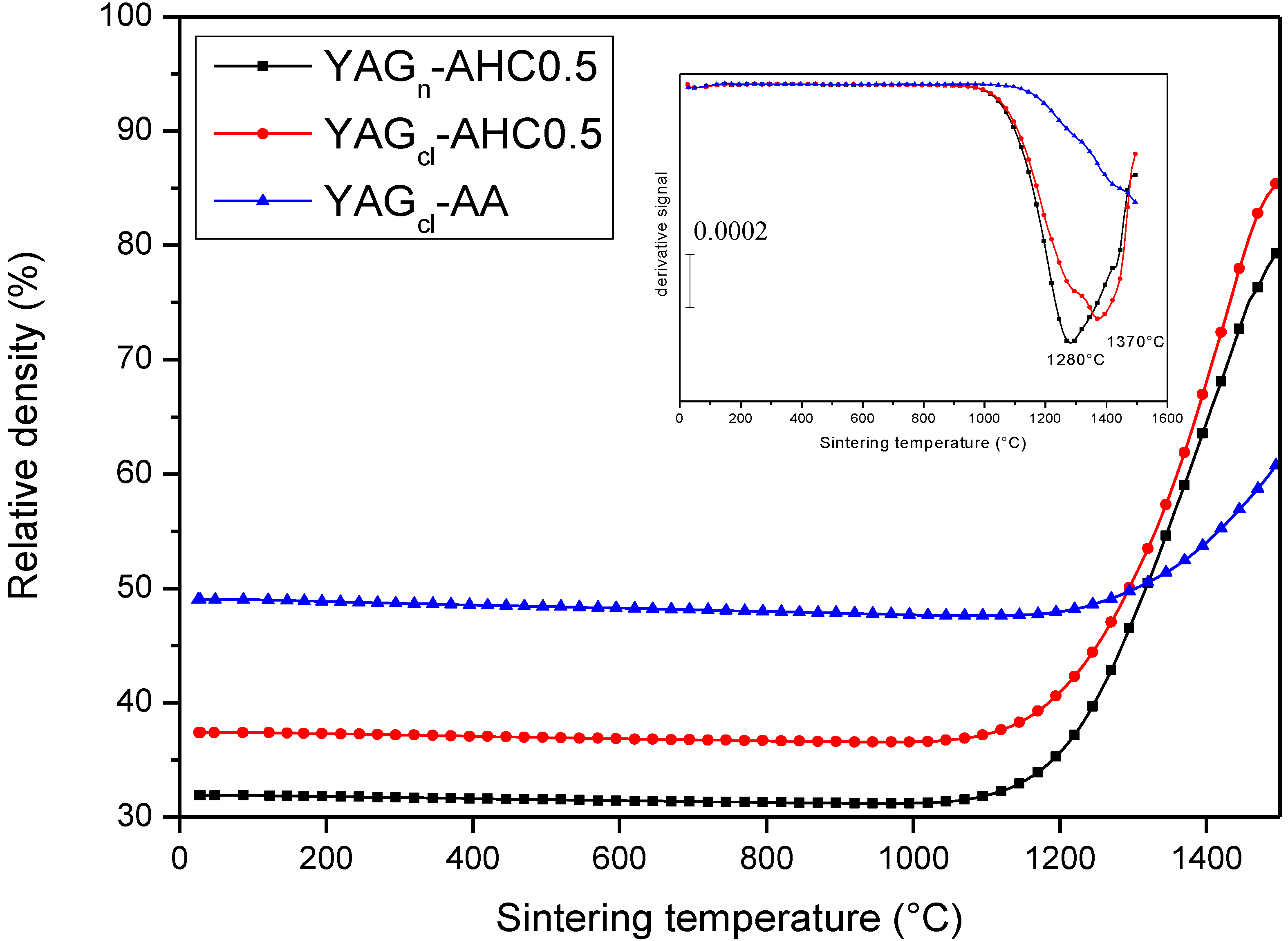
3.3. Processing and Sintering: Role of the Precipitant and the Precursors
| Sample/sintering cycle | Green density g/cm3 (%TD) | Sintered density (%TD) | Grain size (μm) | Note |
|---|---|---|---|---|
| YAGcl-AA/1500 °C-3 h | 2.23 (49.0) | 96.3 | 0.61 | Opaque |
| YAGcl-AA/1600 °C-3 h | 2.31 (50.8) | 96.3 | n.d. | Opaque |
| YAGcl-AHC0.5/1500 °C-3 h | 1.70 (37.4) | 99.0 | 0.42 | Translucent |
| YAGcl-AHC0.5/1600 °C-3 h | 1.63 (35.8) | 99.9 | 0.93 | Translucent |
| YAGn-AHC0.5/1500 °C-3 h | 1.45 (31.9) | 99.4 | 0.33 | Opaque |
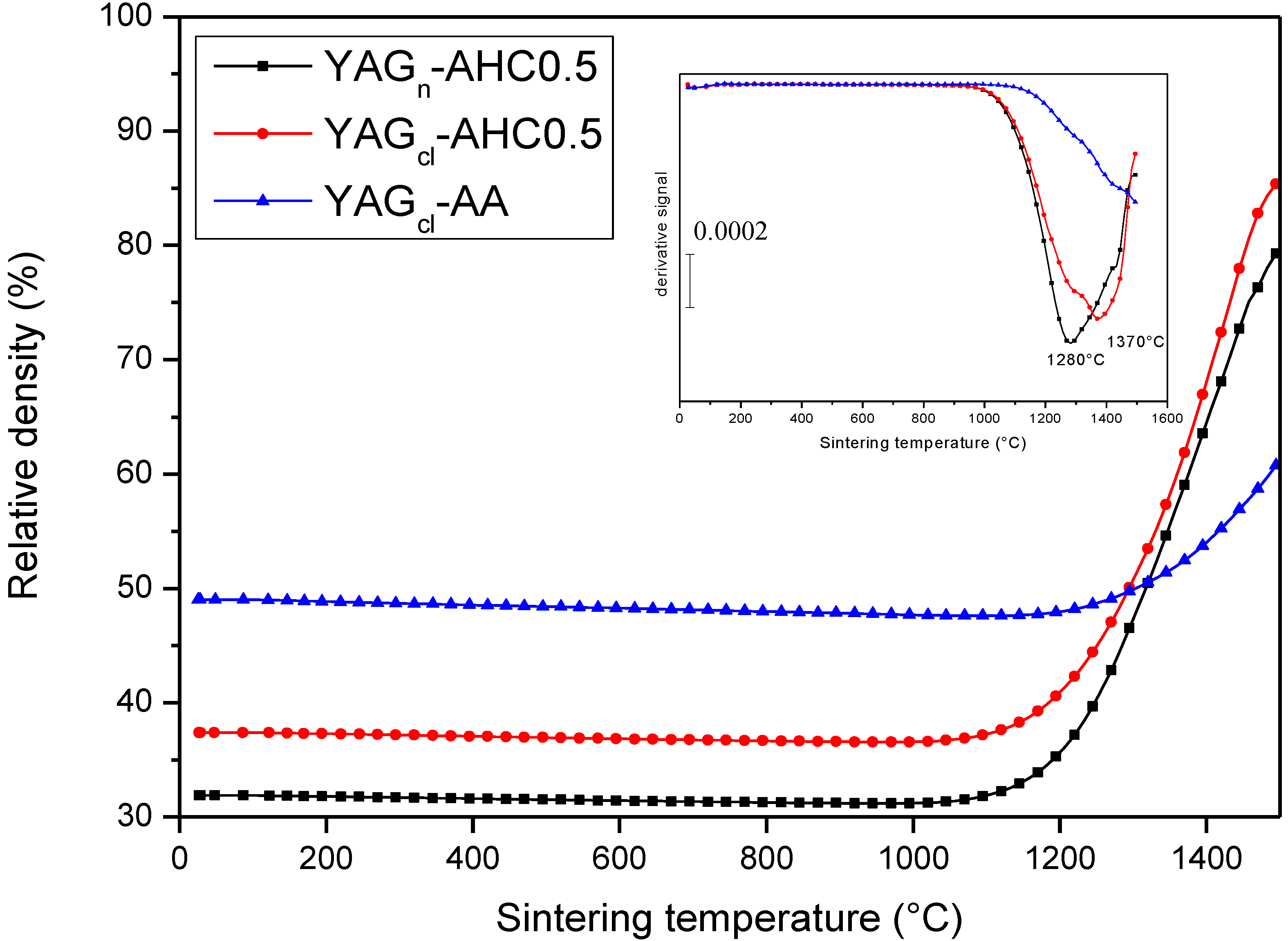
3.4. Microstructural Development: Role of the Precipitant and the Precursors

4. Conclusions
- i)
- The key role of the AHC concentration. In fact, precipitation in AHC 0.5 M gave rise to an amorphous product and to pure YAG phase after calcination at 1000 °C, for 30 min. On the opposite, the use of AHC 1.5 M produced an almost well-crystallized precipitate, made by both hydroxide and carbonate species, suggesting a non-homogeneous distribution of the cations in the as-dried product. To strengthen this hypothesis, several secondary phases beside YAG were obtained, even after high temperature treatment (1500 °C, for 30 min);
- ii)
- The key role of the precipitant. The use of AA induced a harder agglomeration in the powder calcined at 1000 °C, as compared to AHC. As a consequence, the powders derived by AHC precipitation showed better dispersibility and sinterability, giving rise to denser materials, with finer microstructures;
- iii)
- When using AHC 0.5 M as a precipitant, a minor role of the YAG precursors was observed. In fact, both chlorides and nitrates gave rise to pure YAG powders, easily dispersed under ultrasonication and showing high sinterability;
- iv)
- Finally, this work demonstrates that, when properly carried out, co-precipitation is an effective route to prepare YAG powders having the proper characteristics toward transparent ceramics. By using our best synthesis conditions (AHC at 0.5 M, by using both chlorides and nitrates as precursors), highly dense materials (99.0%–99.9%), with an ultra-fine microstructure (YAG average size of 300–400 nm) and showing translucent properties (in the case of chloride precursors) were successfully prepared.
Author Contributions
Conflicts of Interest
References
- Caslavsky, J.L.; Viechnicki, D.J. Melting behaviour and metastability of yttrium aluminium garnet (YAG) and YAlO3 determined by optical differential thermal analysis. J. Mater. Sci. 1980, 15, 1709–1718. [Google Scholar] [CrossRef]
- Waku, Y.; Nakagawa, N.; Wakamoto, T.; Ohtsubo, H.; Shimizu, K.; Kohtoku, Y. A ductile ceramic eutectic composite with high strength at 1,873 K. Nature 1997, 1873, 49–52. [Google Scholar] [CrossRef]
- Minkova, N.; Tzvetkov, G. Mechanochemical effects on yttrium-aluminum-garnet. Mater. Lett. 1998, 35, 135–138. [Google Scholar] [CrossRef]
- Parthasarathy, T.A.; Mah, T.I.; Keller, K.K. Creep mechanism of polycrystalline yttrium aluminum garnet. J. Am. Ceram. Soc. 1992, 75, 1756–1759. [Google Scholar] [CrossRef]
- Ikesue, A. Fabrication and optical properties of high performance polycrystalline Nd:YAG ceramics for solid state lasers. J. Am. Ceram. Soc. 1995, 78, 1033–1040. [Google Scholar] [CrossRef]
- Frage, N.; Kalabukhov, S.; Sverdlov, N.; Kasiyan, V.; Rothman, A.; Dariel, M.P. Effect of the spark plasma sintering (SPS) parameters and LiF doping on the mechanical properties and the transparency of polycrystalline Nd-YAG. Ceram. Int. 2012, 38, 5513–5519. [Google Scholar] [CrossRef]
- Palmero, P.; Bonelli, B.; Fantozzi, G.; Spina, G.; Bonnefont, G.; Montanaro, L.; Chevalier, J. Surface and mechanical properties of transparent polycrystalline YAG fabricated by SPS. Mater. Res. Bull. 2013, 48, 2589–2597. [Google Scholar] [CrossRef]
- Chaim, R.; Marder-Jaeckel, R.; Shen, J.Z. Transparent YAG ceramics by surface softening of nanoparticles in spark plasma sintering. Mater. Sci. Eng. A 2006, 429, 74–78. [Google Scholar] [CrossRef]
- Wang, S.F.; Zhang, J.; Luo, D.W.; Gu, F.; Tang, D.Y.; Dong, Z.L.; Tan, G.E.B.; Que, W.X.; Zhang, T.S.; Li, S.; et al. Transparent ceramics: Processing, materials and applications. Prog. Solid State Chem. 2013, 41, 20–54. [Google Scholar] [CrossRef]
- Krell, A.; Klimke, J.; Hutzler, T. Transparent compact ceramics: Inherent physical issues. Opt. Mater. 2009, 31, 1144–1150. [Google Scholar] [CrossRef]
- Li, C.; Zuo, H.; Zhang, M.; Han, J.; Meng, S. Fabrication of transparent YAG ceramics by traditional solid-state-reaction method. Trans. Nonferrous Met. Soc. China 2007, 17, 148–153. [Google Scholar] [CrossRef]
- Boukerika, A.; Guerbous, L.; Brihi, N. Ce-doped YAG phosphors prepared via sol–gel method: Effect of some modular parameters. J. Alloy Compd. 2014, 614, 383–388. [Google Scholar] [CrossRef]
- Huang, B.; Ma, Y.; Qian, S.; Zou, D.; Zheng, G.; Dai, Z. Luminescent properties of low-temperature-hydrothermally-synthesized and post-treated YAG:Ce (5%) phosphors. Opt. Mater. 2014, 36, 1561–1565. [Google Scholar] [CrossRef]
- Inoue, M. Glycothermal synthesis of metal oxides. J. Phys. Condens. Matter. 2004, 16, S1291–S1303. [Google Scholar]
- Li, J.G.; Ikegami, T.; Lee, J.H.; Mori, T.; Yajima, Y. Co-precipitation synthesis and sintering of yttrium aluminum garnet (YAG) powders: The effect of precipitant. J. Eur. Ceram. Soc. 2000, 20, 2395–2405. [Google Scholar] [CrossRef]
- Palmero, P.; Esnouf, C.; Montanaro, L.; Fantozzi, G. Influence of the co-precipitation temperature on phase evolution in yttrium-aluminum oxide materials. J. Eur. Ceram. Soc. 2005, 25, 1565–1573. [Google Scholar] [CrossRef]
- Sang, Y.; Lv, Y.; Qin, H.; Zhang, X.; Liu, H.; Wang, J.; Sun, X.; Boughton, R.I. Chemical composition evolution of YAG co-precipitate determined by pH during aging period and its effect on precursor properties. Ceram. Int. 2012, 38, 1635–1641. [Google Scholar] [CrossRef]
- Azar, M.; Palmero, P.; Lombardi, M.; Garnier, V.; Montanaro, L.; Fantozzi, G.; Chevalier, J. Effect of initial particle packing on the sintering of nanostructured transition alumina. J. Eur. Ceram. Soc. 2008, 28, 1121–1128. [Google Scholar] [CrossRef]
- Wang, L.; Kou, H.; Zeng, Y.; Li, J.; Pan, Y.; Sun, X.; Guo, J. The effect of precipitant concentration on the formation procedure of yttrium aluminum garnet (YAG) phase. Ceram. Int. 2012, 38, 3763–3771. [Google Scholar] [CrossRef]
- Palmero, P.; Di Nunzio, S.; Montanaro, L. YAG wet-chemical synthesis from chlorides and nitrates precursors: Effect on phase evolution and powder sinterability. Int. J. Mater. Prod. Technol. 2009, 35, 374–391. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmero, P.; Traverso, R. Co-Precipitation of YAG Powders for Transparent Materials: Effect of the Synthesis Parameters on Processing and Microstructure. Materials 2014, 7, 7145-7156. https://doi.org/10.3390/ma7107145
Palmero P, Traverso R. Co-Precipitation of YAG Powders for Transparent Materials: Effect of the Synthesis Parameters on Processing and Microstructure. Materials. 2014; 7(10):7145-7156. https://doi.org/10.3390/ma7107145
Chicago/Turabian StylePalmero, Paola, and Rebecca Traverso. 2014. "Co-Precipitation of YAG Powders for Transparent Materials: Effect of the Synthesis Parameters on Processing and Microstructure" Materials 7, no. 10: 7145-7156. https://doi.org/10.3390/ma7107145
APA StylePalmero, P., & Traverso, R. (2014). Co-Precipitation of YAG Powders for Transparent Materials: Effect of the Synthesis Parameters on Processing and Microstructure. Materials, 7(10), 7145-7156. https://doi.org/10.3390/ma7107145