Challenges in the Development of Functional Assays of Membrane Proteins

Abstract
:
1. Introduction
2. Lipid Bilayers with Integrated Functional Membrane Proteins on Surfaces

{kind=link}
{kind=link}
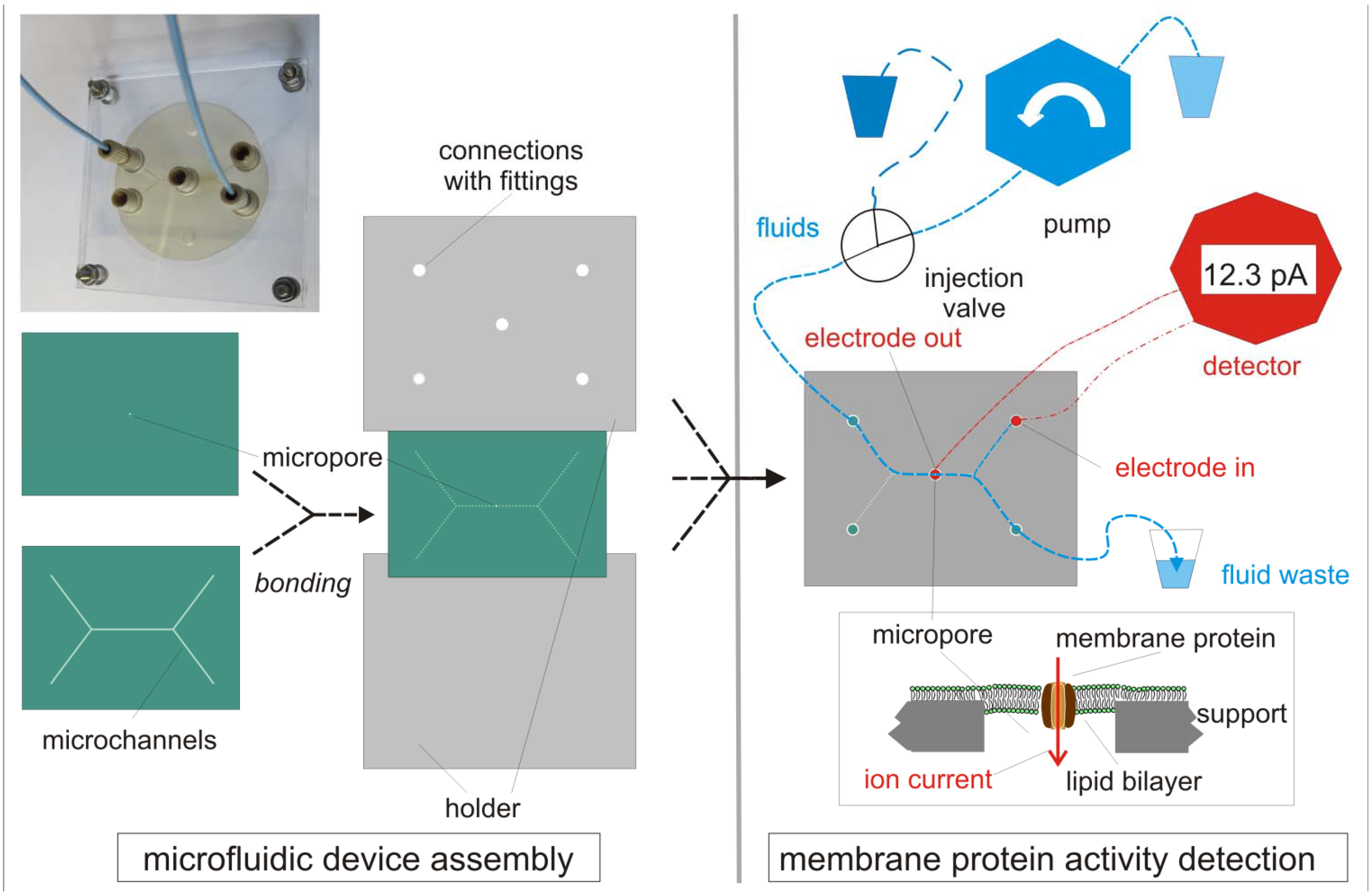{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Abbreviation | Material used for | Remarks | ||
|---|---|---|---|---|---|
| Bilayer support | MFS | Detector | |||
| anodic aluminum oxide | AAO | porous [19] | – | – | nanopores |
| gold | Au | tethered [15,20,21,22] | – | P-L 1 [23] | thiol-compounds |
| indium tin oxide | ITO | – | – | glass [24,25] | conducting plates |
| mercury | Hg | tethered [26] | – | drop [27] | – |
| platinum | Pt | – | – | P-L [28]; wire [29,30] | – |
| polycarbonate | PC | pores, las 2 [31,32] pores, ion 3 [33] | mec 4 [34]; emb 5 [35] | – | – |
| polylactic acid | PLLA | pores, las [32] | – | – | – |
| polyethyleneterephthalate | PETE | pores, las [32] | – | – | – |
| polymetylmetacrylate | PMMA | pore(s), mec [36,37] pore(s), emb [38] | mec [39,40] emb [38,41] | – | fitting layers [39] |
| polydimethylsiloxane | PDMS | pore, emb [42] | cas 6 [43] | – | – |
| polyvinylidenechloride | PVDC | pore, mec [34,35] | – | – | – |
| polytetrafluoroethylene | PTFE Teflon | pore P-L [30] | mec [31] | – | fitting layers [44] |
| pore(s) emb [38], las [44] | |||||
| poly(p-xylylene) | parylene | pores, P-L [39,40] | – | – | nanopore [39] |
| photoresist, epoxy-based | SU-8 | P-L [28,45] | – | – | – |
| quartz or glass | – | pore, elc 7 [46]; ion [47] | – | – | – |
| silicon | Si | pore(s), P-L + dry 8 [48] porous [49,50] pore(s), ion [51] | wet 9 + dry [48] | – | gold coated [49] |
| silicon nitride | Si3N4 | pore(s), e-b 10 [52] P-L [53,54,55] pores, C-L 11+ dry [55,56] | – | – | polyimide [54] on glass [43] |
| silver/silver chlorid | Ag/AgCl | – | – | cvd 12 [35] wire [34,37,42,57] | |
2.1. Materials Used for Artificial Bilayers and Microfluidic Systems
2.2. Silicon-Based Micro- and Nanofabrication Technologies
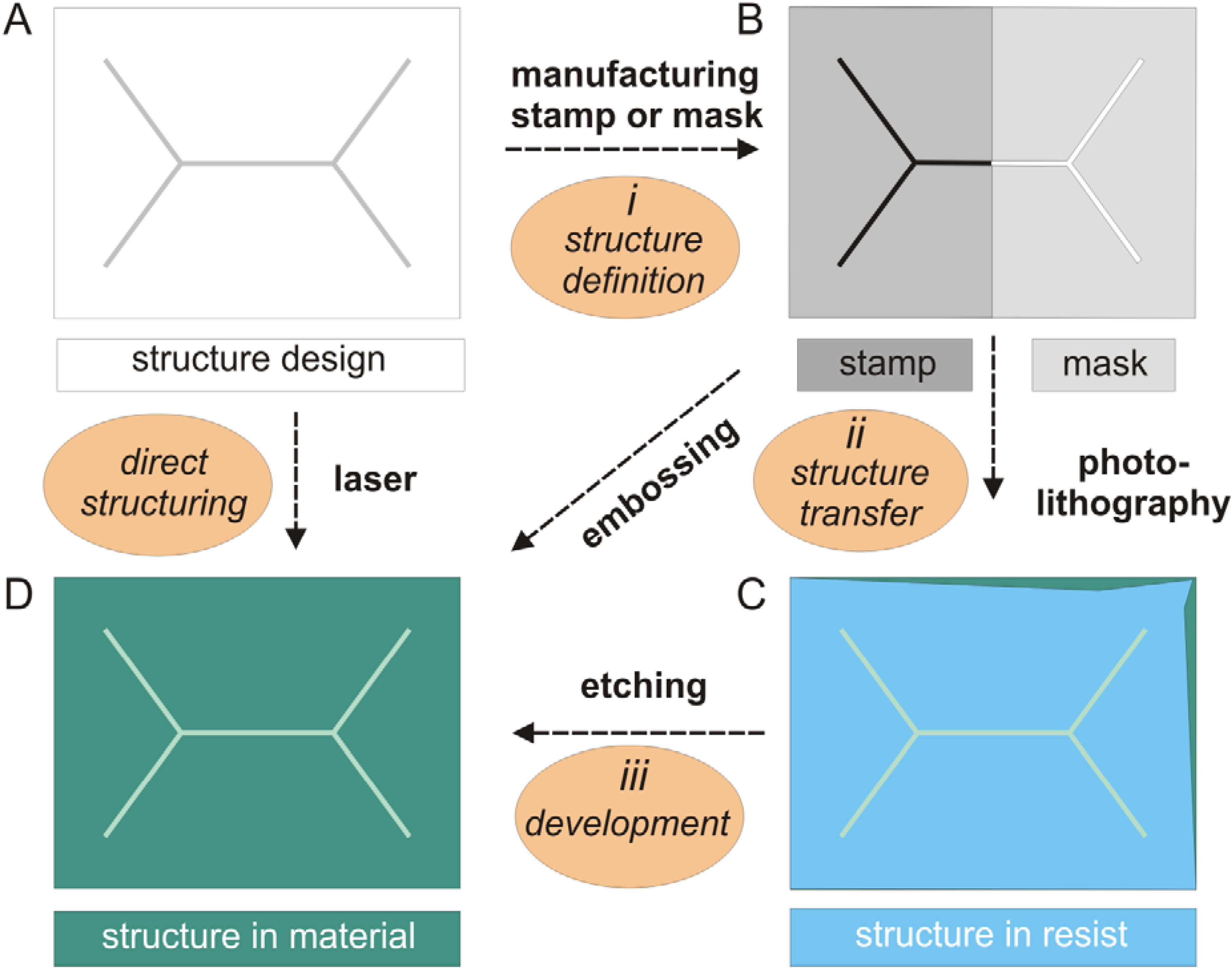
2.3. Micro-Pore and Microfluidic Channels in Polymers

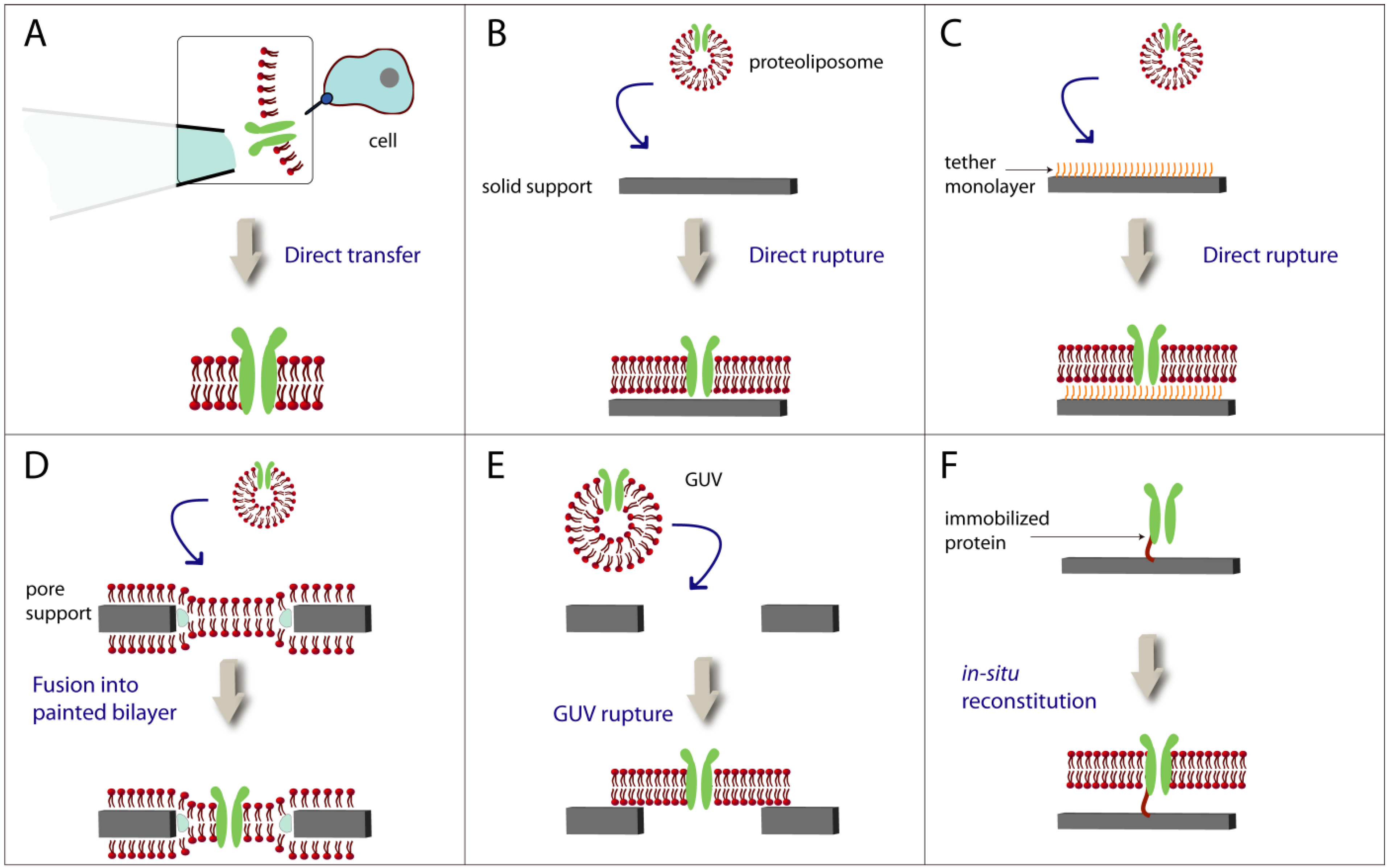
3. Lipid Bilayers with Functional Membrane Proteins
3.1. Formation of Lipid Bilayers on Solid Supports and Pores
3.2. Integration of Peptides in Lipid Bilayers
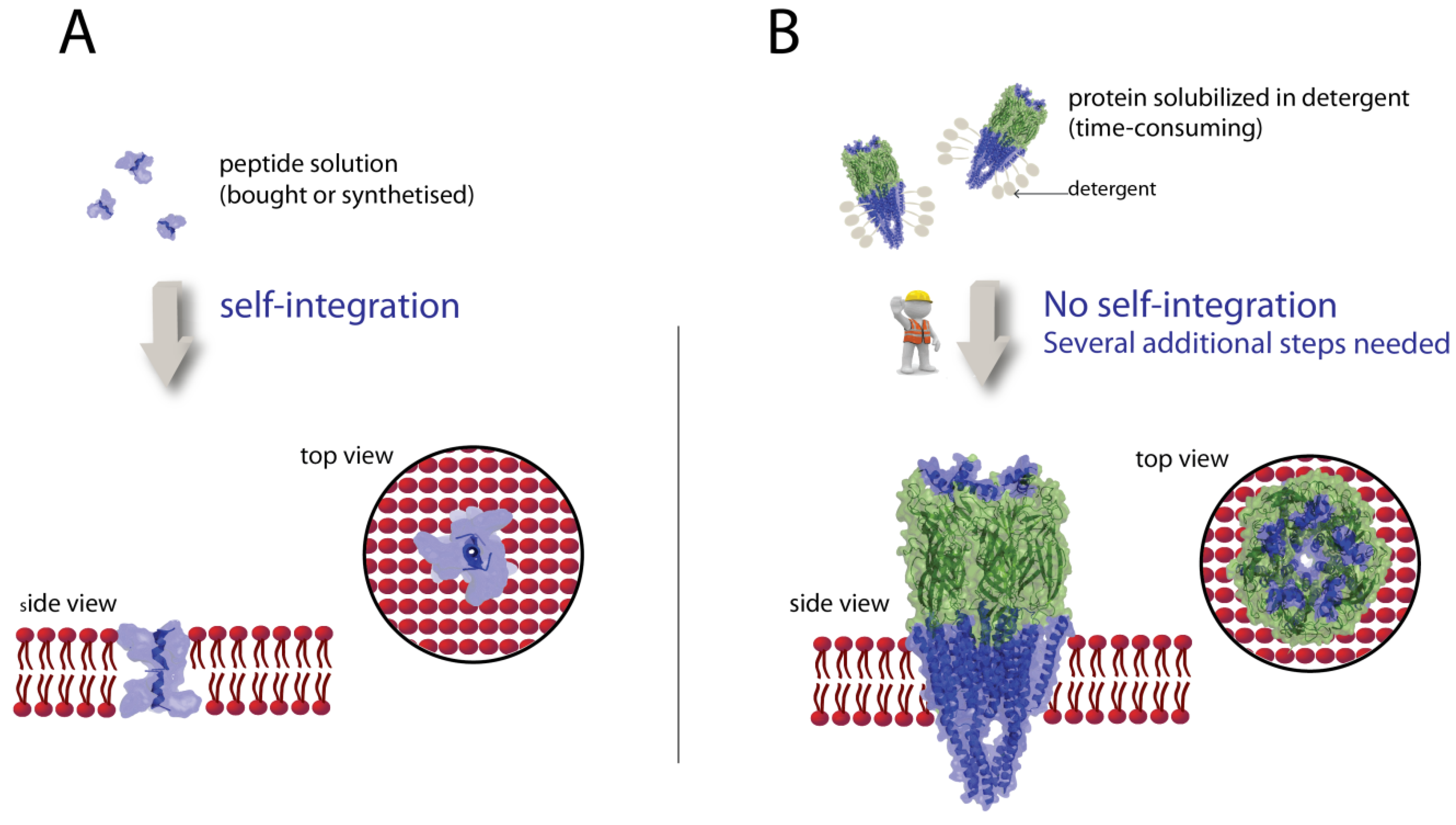
3.3. Integration of Proteins in Lipid Bilayers
3.4. The Impact of Lipid Composition on Lipid Bilayer Stability and Protein Function
4. Importance of Membrane Proteins in Biology and as Drug Targets
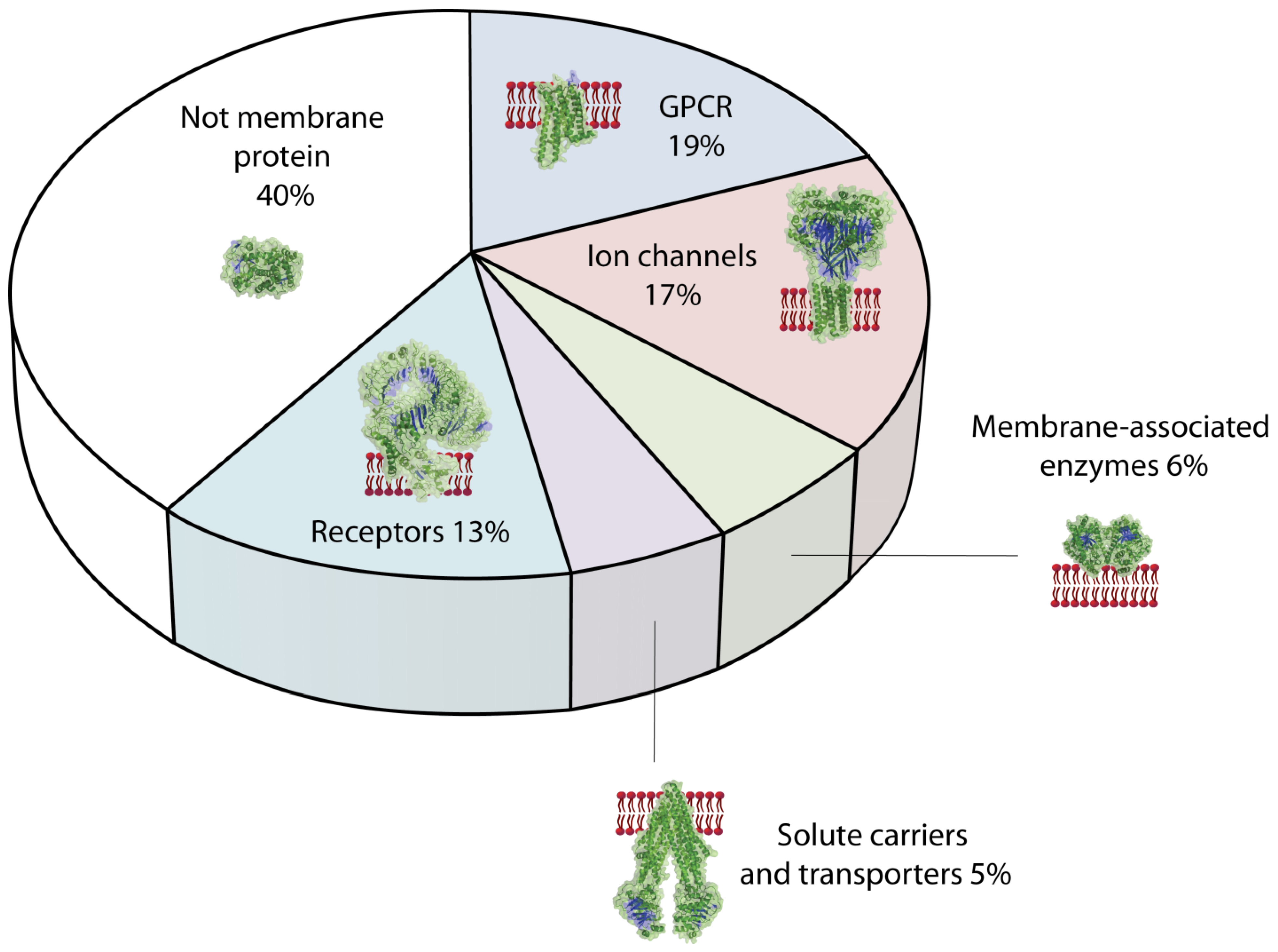
4.1. Classification of Membrane Proteins Relevant as Drug Targets
4.2. Prokaryotic vs. Eukaryotic Protein Structure
4.3. Study of Protein—Protein Interactions at the Membrane
4.4. Biosensor for Drug Discovery
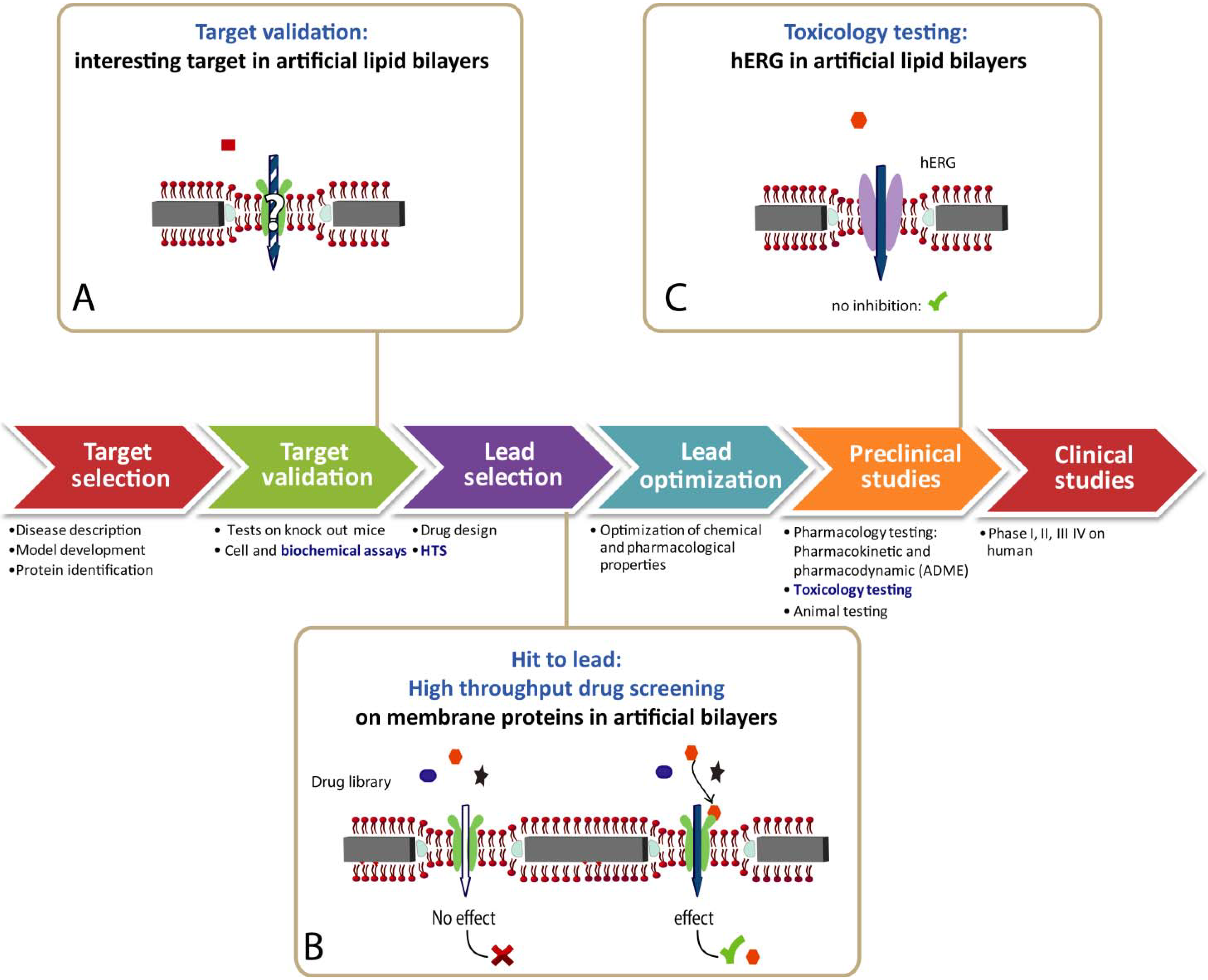
5. Information Expected from Functional Devices
5.1. Working Mechanism of GPCRs
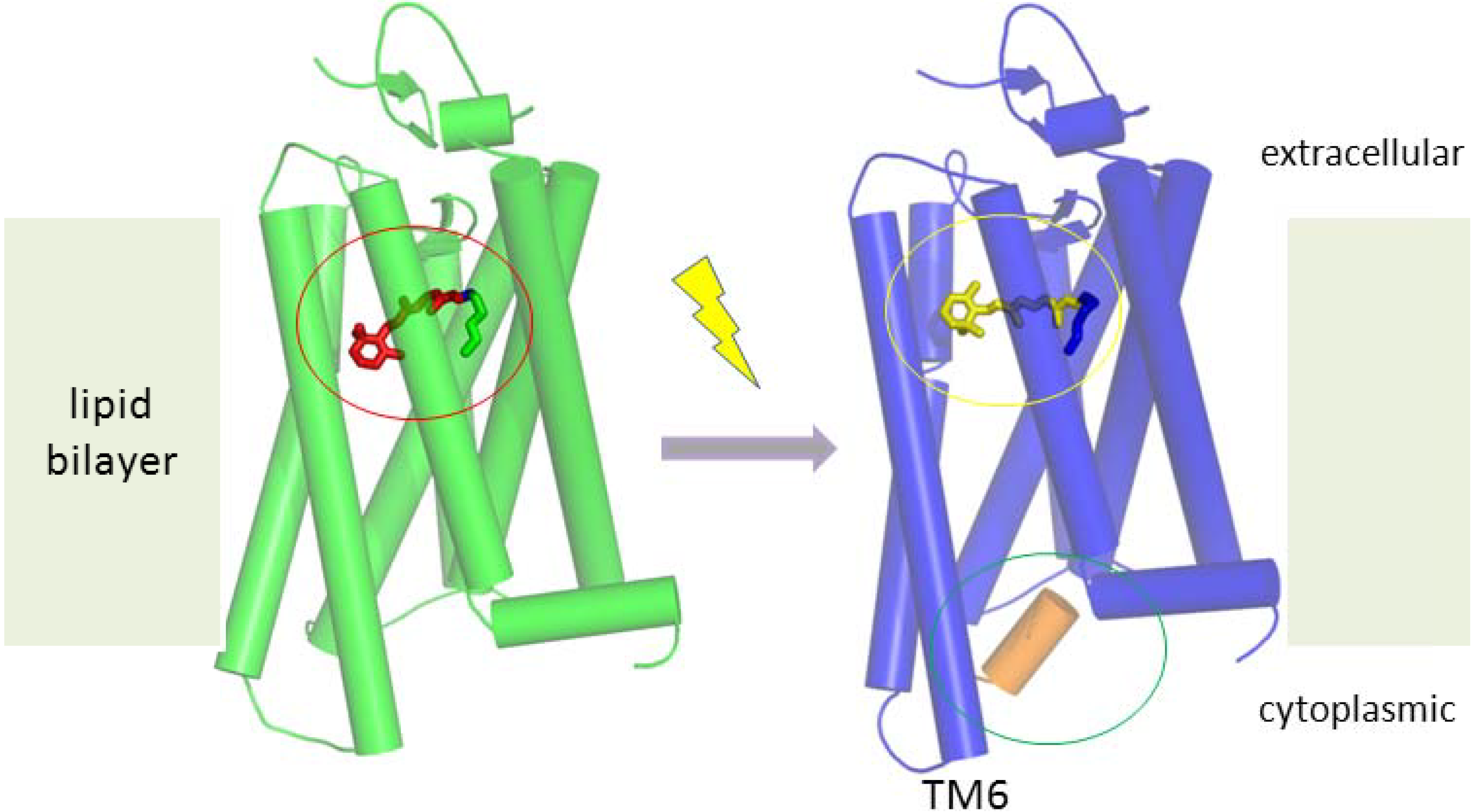
5.2. Detection Methods for Membrane Protein Activities
5.3. Contribution of Artificial Systems to the Understanding of Cell Functions
6. Outlook
Acknowledgments
References
- Fagerberg, L.; Jonasson, K.; von Heijne, G.; Uhlen, M.; Berglund, L. Prediction of the human membrane proteome. Proteomics 2010, 10, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- White, S. Membrane proteins of known structures. Available online: http://blanco.biomol.uci.edu/mpstruc/listAll/list (accessed on 1 November 2012).
- Nielsen, C.H. Biomimetic membranes for sensor and separation applications. Anal. Bioanal. Chem. 2009, 395, 697–718. [Google Scholar] [CrossRef] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids, and detergents: Not just a soap opera. Biochem. Biophys. Acta 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Demarche, S.; Sugihara, K.; Zambelli, T.; Tiefenauer, L.; Vörös, J. Techniques for recording reconstituted ion channels. Analyst 2011, 136, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Vareiro, M.M.L.; Jenkins, A.T. Fluorophore-encapsulated solid-supported bilayer vesicles: A method for studying membrane permeation processes. Langmuir 2006, 22, 6473–6476. [Google Scholar] [CrossRef] [PubMed]
- Pick, H.; Schmid, E.L.; Tairi, A.P.; Ilegems, E.; Hovius, R.; Vogel, H. Investigating cellular signaling reactions in single attoliter vesicles. J. Am. Chem. Soc. 2004, 127, 2908–2912. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Iwamoto, M.; Kato, A.; Yoshikawa, K.; Oiki, S. Oriented reconstitution of a membrane protein in a giant unilamellar vesicle: Experimental verification with the potassium channel KcsA. J. Am. Chem. Soc. 2011, 133, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Aimon, S.; Manzi, J.; Schmidt, D.; Larrosa, J.A.P.; Bassereau, P.; Toombes, G.E.S. Functional reconstitution of a voltage-gated potassium channel in giant unilamellar vesicles. PLoS One 2011, 6, e25529. [Google Scholar] [CrossRef]
- Lu, P.H.; Liu, R.H.; Sharom, F.J. Drug transport by reconstituted P-glycoprotein in proteoliposomes—Effect of substrates and modulators, and dependence on bilayer phase state. Eur. J. Biochem. 2001, 268, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Heron, A.J.; Thompson, J.R.; Cronin, B.; Bayley, H.; Wallace, M.I. Simultaneous measurement of ionic current and fluorescence from single protein pores. J. Am. Chem. Soc. 2009, 131, 1652–1653. [Google Scholar] [CrossRef] [PubMed]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981, 391, 85–100. [Google Scholar]
- Sackmann, E. Supported membranes: Scientific and practial applications. Science 1996, 271, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Vockenroth, I.K.; Fine, D.; Dodobalapur, A.; Toby, A.; Jenkins, A.; Köper, I. Tethered bilayer lipid membranes with giga-ohm resistance. Electrochem. Commun. 2008, 10, 323–328. [Google Scholar] [CrossRef]
- Köper, I. Insulating tethered bilayer lipid membranes to study membrane proteins. Mol. Biosyst. 2007, 3, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Anrather, D.; Smetazko, M.; Saba, M.; Alguel, Y.; Schalkhammer, T. Supported membrane nanodevices. J. Nanosci. Nanotechnol. 2004, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Janshoff, A.; Steinem, C. Transport across artificial membranes—An analytical perspective. Anal. Bioanal. Chem. 2006, 385, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Takeuchi, S. Microtechnologies for membrane protein studies. Anal. Bioanal. Chem. 2008, 391, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, T.D.; Kliesch, T.T.; Janshoff, A.; Steinem, C. Orthogonal functionalization of nanoporous substrates: Control of 3D surface functionality. ACS Appl. Mater. Interfaces 2011, 3, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Chopineau, J. Biomimetic tethered lipid membranes designed for membrane-protein interaction studies. Eur. Biophys. J. Biophys. Lett. 2007, 36, 955–965. [Google Scholar] [CrossRef]
- He, L.H.; Robertson, J.W.F.; Li, J.; Karcher, I.; Schiller, S.M.; Knoll, W.; Naumann, R. Tethered bilayer lipid membranes based on monolayers of thiolipids mixed with a complementary dilution molecule. 1. Incorporation of channel peptides. Langmuir 2005, 21, 11666–11672. [Google Scholar] [CrossRef] [PubMed]
- Cornell, B.A.; Braach-Maksvytis, L.B.; King, L.G.; Osman, P.D.J.; Raguse, B.; Wieczorek, L.; Pace, R.J. A biosensor that uses ion-channel switches. Nature 1997, 387, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Critchley, K.; Zhang, L.; Pradeep, S.N.D.; Bushby, R.J.; Evans, S.D. A novel method to fabricate patterned bilayer lipid membranes. Langmuir 2007, 23, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Estes, D.J.; Lopez, S.R.; Fuller, A.O.; Mayer, M. Triggering and visualizing the aggregation and fusion of lipid membranes in microfluidic chambers. Biophys. J. 2006, 91, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.K.; Yen, C.W.; El-Sayed, M.A. Bacteriorhodopsin-based photo-electrochemical cell. Biosens. Bioelectron. 2010, 26, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Becucci, L.; Moncelli, M.R.; Guidelli, R. Impedance spectroscopy of OmpF porin reconstituted into a mercury-supported lipid bilayer. Langmuir 2006, 22, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Becucci, L.; Guidelli, R. Kinetics of channel formation in bilayer lipid membranes (BLMs) and tethered BLMs: Monazomycin and melittin. Langmuir 2007, 23, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Aghdaei, S.; Sandison, M.E.; Zagnoni, M.; Green, N.G.; Morgan, H. Formation of artificial lipid bilayers using droplet dielectrophoresis. Lab Chip 2008, 8, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.P.; Chand, A.; Ramachandran, S.; Daraio, C.; Jin, S.; Lal, R. Atomic force microscopy imaging and electrical recording of lipid bilayers supported over microfabricated silicon chip nanopores: Lab-on-a-chip system for lipid membranes and ion channels. Langmuir 2007, 23, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Kriebel, J.K.; Tosteson, M.T.; Whitesides, G.M. Microfabricated teflon membranes for low-noise recordings of ion channels in planar lipid bilayers. Biophys. J. 2003, 85, 2684–2695. [Google Scholar] [CrossRef] [PubMed]
- O'Shaughnessy, T.J.; Hu, J.E.; Kulp, J.L.; Daly, S.M.; Ligler, F.S. Laser ablation of micropores for formation of artificial planar lipid bilayers. Biomed. Microdevices 2007, 9, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Dhoke, M.A.; Ladha, P.J.; Boerio, F.J.; Lessard, L.B.; Malinowska, D.H.; Cuppoletti, J.; Wieczorek, D.S. Porous membranes for reconstitution of ion channels. Biochim. Biophys. Acta 2005, 1716, 117–125. [Google Scholar] [PubMed]
- Favero, G.; D'Annibale, A.; Campanella, L.; Santucci, R.; Ferri, T. Membrane supported bilayer lipid membranes array: Preparation, stability and ion-channel insertion. Anal. Chim. Acta 2002, 460, 23–34. [Google Scholar] [CrossRef]
- Shao, C.R.; Sun, B.; Colombini, M.; DeVoe, D.L. Rapid microfluidic perfusion enabling kinetic studies of lipid ion channels in a bilayer lipid membrane chip. Ann. Biomed. Eng. 2011, 39, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Hromada, L.P.; Nablo, B.J.; Kasianowicz, J.J.; Gaitan, M.A.; DeVoe, D.L. Single molecule measurements within individual membrane-bound ion channels using a polymer-based bilayer lipid membrane chip. Lab Chip 2008, 8, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tabata, K.V.; Noji, H.; Takeuchi, S. Highly reproducible method of planar lipid bilayer reconstitution in polymethyl methacrylate microfluidic chip. Langmuir 2006, 22, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, K.; Suzuki, H.; Takeuchi, S. Lipid bilayer formation by contacting monolayers in a microfluidic device for membrane protein analysis. Anal. Chem. 2006, 78, 8169–8174. [Google Scholar] [CrossRef] [PubMed]
- Sandison, M.E.; Morgan, H. Rapid fabrication of polymer microfluidic systems for the production of artificial lipid bilayers. J. Micromech. Microeng. 2005, 15, S139–S144. [Google Scholar] [CrossRef]
- Kawano, R.; Osaki, T.; Sasaki, H.; Takeuchi, S. A polymer-based nanopore-integrated microfluidic device for generating stable bilayer lipid membranes. Small 2010, 6, 2100–2104. [Google Scholar] [CrossRef] [PubMed]
- Le Pioufle, B.; Suzuki, H.; Tabata, K.V.; Noji, H.; Takeuchi, S. Lipid bilayer microarray for parallel recording of transmembrane ion currents. Anal. Chem. 2008, 80, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Sandison, M.E.; Zagnoni, M.; Morgan, H. Air-exposure technique for the formation of artificial lipid bilayers in microsystems. Langmuir 2007, 23, 8277–8284. [Google Scholar] [CrossRef] [PubMed]
- Malmstadt, N.; Nash, M.A.; Purnell, R.F.; Schmidt, J.J. Automated formation of lipid-bilayer membranes in a microfluidic device. Nano Lett. 2006, 6, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Ota, S.; Suzuki, H.; Takeuchi, S. Microfluidic lipid membrane formation on microchamber arrays. Lab Chip 2011, 11, 2485–2487. [Google Scholar] [CrossRef] [PubMed]
- Zagnoni, M.; Sandison, M.E.; Morgan, H. Microfluidic array platform for simultaneous lipid bilayer membrane formation. Biosens. Bioelectron. 2009, 24, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.W.; Rieck, D.; van Wie, B.J.; Cheng, G.J.; Moffett, D.F.; Kidwell, D.A. Bilayer lipid membrane (BLM) based ion selective electrodes at the meso-, micro-, and nano-scales. Biosens. Bioelectron. 2009, 24, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Schibel, A.E.P.; Edwards, T.; Kawano, R.; Lan, W.J.; White, H.S. Quartz nanopore membranes for suspended bilayer ion channel recordings. Anal. Chem. 2010, 82, 7259–7266. [Google Scholar] [CrossRef] [PubMed]
- Fertig, N.; Klau, M.; George, M.; Blick, R.H.; Behrends, J.C. Activity of single ion channel proteins detected with a planar microstructure. Appl. Phys. Lett. 2002, 81, 4865–4867. [Google Scholar] [CrossRef]
- Suzuki, H.; Tabata, K.; Kato-Yamada, Y.; Noji, H.; Takeuchi, S. Planar lipid bilayer reconstitution with a micro-fluidic system. Lab Chip 2004, 4, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Orth, A.; Johannes, L.; Römer, W.; Steinem, C. Creating and modulating microdomains in pore-spanning membranes. ChemPhysChem 2012, 13, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schmitt, E.K.; Klühr, M.H.; Dertinger, S.K.; Steinem, C. Micro-BLMs on highly ordered porous silicon substrate: rupture process and lateral mobility. Langmuir 2007, 23, 9134–9139. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Lee, J.R.I.; Ratto, T.V.; Letant, S.E. Localized functionalization of single nanopores. Adv. Mater. 2006, 18, 427–431. [Google Scholar] [CrossRef]
- Heyderman, L.J.; Ketterer, B.; Bächle, D.; Glaus, F.; Haas, B.; Schift, H.; Vogelsang, K.; Gobrecht, J.; Tiefenauer, L.; Dubochet, O.; Surbled, P.; Hessler, T. High volume fabrication of customised nanopore membrane chips. Microelectron. Eng. 2003, 67–68, 208–213. [Google Scholar] [CrossRef]
- Hirano-Iwata, A.; Aoto, K.; Oshima, A.; Taira, T.; Yamaguchi, R.T.; Kimura, Y.; Niwano, M. Free-standing lipid bilayers in silicon chips—Membrane stabilization based on microfabricated apertures with a nanometer-scale smoothness. Langmuir 2010, 26, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Peterman, M.C.; Ziebarth, J.M.; Braha, O.; Bayley, H.; Fishman, H.A.; Bloom, D.M. Ion channels and lipid bilayer membranes under high potentials using microfabricated apertures. Biochem. Microdevices 2002, 4, 231–236. [Google Scholar] [CrossRef]
- Tiefenauer, L.; Studer, A. Nano for bio: Nanopore arrays for stable and functionnal lipid bilayer membranes (Mini Review). Biointerphases 2008, 3, 74–78. [Google Scholar] [CrossRef]
- Kumar, K.; Isa, L.; Egner, A.; Schmidt, R.; Textor, M.; Reimhult, E. Formation of nanopore-spanning lipid bilayers through liposome fusion. Langmuir 2011, 27, 10920–10928. [Google Scholar] [CrossRef] [PubMed]
- Hutter, I.; Müller, E.; Kristiansen, P.M.; Kresak, S.; Tiefenauer, L. Polymer-based microfluidic device for measuring membrane protein activities. Microfluid. Nanofluid. 2012. [Google Scholar] [CrossRef]
- Reimhult, E.; Kumar, K. Membrane biosensor platforms using nano- and microporous supports. Trends Biotechnol. 2008, 26, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Worsfold, O.; Voelcker, N.H.; Nishiya, T. Biosensing using lipid bilayers suspended on porous silicon. Langmuir 2006, 22, 7078–7083. [Google Scholar] [CrossRef] [PubMed]
- Hennesthal, C.; Drexler, J.; Steinem, C. Membrane-suspended nanocompartments based on ordered pores in alumina. ChemPhysChem 2002, 3, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Danelon, C.; Santschi, C.; Brugger, J.; Vogel, H. Fabrication and functionalizuation of nanochannels by electron-beam-induced silicon oxide deposition. Langmuir 2006, 22, 10711–10715. [Google Scholar] [CrossRef] [PubMed]
- Studer, A.; Han, X.J.; Winkler, F.K.; Tiefenauer, L.X. Formation of individual protein channels in lipid bilayers suspended in nanopores. Colloids Surfaces B 2009, 73, 325–331. [Google Scholar] [CrossRef]
- Ervin, E.N.; Kawano, R.; White, R.J.; White, H.S. Simultaneous alternating and direct current readout of protein ion channel blocking events using glass nanopore membranes. Anal. Chem. 2008, 80, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Studer, A.; Sehr, H.; Geissbühler, I.; DiBerardino, M.; Winkler, F.K.; Tiefenauer, L. Nanopore arrays for stable and functional free-standing lipid bilayers. Adv. Mater. 2007, 19, 4466–4470. [Google Scholar] [CrossRef]
- Schmitt, E.K.; Vrouenraets, M.; Steinem, C. Channel activity of OmpF monitored in nano-BLMs. Biophys. J. 2006, 91, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Römer, W.; Steinem, C. Impedance analysis and single-channel recordings on nano-black lipid membranes based on porous alumina. Biophys. J. 2004, 86, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, D.C. Ion transport—Spot the difference. Nature 2004, 427, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Studer, A.; Demarche, S.; Langenegger, D.; Tiefenauer, L. Integration and recording of a reconstituted voltage-gated sodium channel in planar lipid bilayers. Biosens. Bioelectron. 2011, 26, 1924–1928. [Google Scholar] [CrossRef] [PubMed]
- Fologea, D.; Gershow, M.; Ledden, B.; McNabb, D.S.; Golovchenko, J.A.; Li, J.L. Detecting single stranded DNA with a solid state nanopore. Nano Lett. 2005, 5, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.W.; Flores, A. Nanoporous microbead supported bilayers: stability, physical characterization, and incorporation of functional transmembrane proteins. Langmuir 2005, 21, 11666–11672. [Google Scholar] [CrossRef] [PubMed]
- Sandison, M.E.; Zagnoni, M.; Abu-Hantash, M.; Morgan, H. Micromachined glass apertures for artificial lipid bilayer formation in a microfluidic system. J. Micromech. Microeng. 2007, 17, S189–S196. [Google Scholar] [CrossRef]
- Kim, P.; Lee, S.E.; Jung, H.S.; Lee, H.Y.; Kawai, T.; Jeong, H.E.; Suh, K.Y. Supported lipid bilayers microarrays onto a surface and inside microfluidic channels. In Proceeding of 2006 International Conference on Microtechnologies in Medicine and Biology, Okinawa, Japan, 9–12 May 2006.
- Allbritton, N.L. Micro total analyis systems for cell biology and biochemical assays. Anal. Chem. 2012, 212, 516–540. [Google Scholar]
- Gervais, L.; de Rooij, N.; Delamarche, E. Microfluidic chips for point-of-care immunodiagnostics. Adv. Mater. 2011, 23, H151–H176. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Simone, G.; Salieb-Beugelaar, G.B.; Kim, J.T.; Manz, A. Latest developments in micro total analysis aystems. Anal. Chem. 2010, 82, 4830–4847. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Frutos, A.G.; Lahiri, J. Membrane protein microarrays. J. Am. Chem. Soc. 2002, 124, 2394–2395. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Furukawa, K.; Kashimura, Y.; Sumitomo, K.; Shinozaki, Y.; Torimitsu, K. Pattern formation and molecular transport of histidine-tagged GFPs using supported lipid bilayers. Langmuir 2010, 26, 12716–12721. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Hennig, M.; Wixforth, A.; Manus, S.; Radler, J.O.; Schneider, M.F. Transport, separation, and Accumulation of Proteins on Supported Lipid Bilayers. Nano Lett. 2010, 10, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Bailey, K.; Sugihara, K.; Grieshaber, D.; Vörös, J.; Städler, B. Liposome and lipid bilayer arrays towards biosensing applications. Small 2010, 6, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Aiba, T. Supported lipid bilayer composition microarray fabricated by pattern-guided self-spreading. Langmuir 2011, 27, 7341–7344. [Google Scholar] [CrossRef] [PubMed]
- Vinchurkar, M.S.; Bricarello, D.A.; Lagerstedt, J.O.; Buban, J.P.; Stahlberg, H.; Oda, N.M.; Voss, J.C.; Parikh, A.N. Bridging across length scales: multi-scale ordering of supported lipid bilayers via lipoprotein self-assembly and surface patterning. J. Am. Chem. Soc. 2008, 130, 11164–11169. [Google Scholar] [CrossRef] [PubMed]
- Schift, H. Nanoimprint lithography: An old story in modern times? A review. J. Vac. Sci. Technol. B 2008, 26, 458–480. [Google Scholar] [CrossRef]
- Zema, L.; Loreti, G.; Melocchi, A.; Maroni, A.; Gazzaniga, A. Injection molding and its application to drug delivery. J. Control. Release 2012, 159, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Gornall, J.L.; Mahendran, K.R.; Pambos, O.J.; Steinbock, L.J.; Otto, O.; Chimerel, C.; Winterhalter, M.; Keyser, U.F. Simple reconstitution of protein pores in nano lipid bilayers. Nano Lett. 2011, 11, 3334–3340. [Google Scholar] [CrossRef] [PubMed]
- Trietsch, S.J.; Hankemeier, T.; van der Linden, H.J. Lab-on-a-chip technologies for massive parallel data generation in the life sciences: A review. Chemometr. Intell. Lab. Syst. 2011, 108, 64–75. [Google Scholar] [CrossRef]
- Suzuki, H.; Le, P.B.; Takeuchi, S. Ninety-six-well planar lipid bilayer chip for ion channel recording fabricated by hybrid stereolithography. Biomed. Microdevices 2009, 11, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ekins, R.P. Ligand assays: From electrophoresis to miniaturized microarrays. Clin. Chem. 1998, 44, 2015–2030. [Google Scholar] [PubMed]
- Komolov, K.E.; Senin, I.I.; Philippov, P.P.; Koch, K.W. Surface plasmon resonance study of G protein/receptor coupling in a lipid bilayer-free system. Anal. Chem. 2006, 78, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Engel, A. Strategies to prepare and characterize native membrane proteins and protein membranes by AFM. Curr. Opin. Colloid Interface Sci. 2008, 13, 338–350. [Google Scholar] [CrossRef]
- Müller, P.; Rudin, D.O.; Tien, H.T.; Wescott, W.C. Reconstitution of cell membrane structure in vitro and its transformation into an excitable system. Nature 1962, 194, 979–980. [Google Scholar] [CrossRef]
- Montal, M.; Müller, P. Formation of biomolecular membranes from lipid monolayers and a study of their electrical properties. Proc . Natl Acad. Sci. USA 1972, 69, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Läuger, P. Carrier-mediated ion transport. Science 1972, 178, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, J.; Steinem, C.; Hildebrandt, P.; Millo, D. Combined electrochemistry and surface-enhanced infrared absorption spectroscopy of gramicidin A incorporated into thethered bilayer lipid membranes. Angew. Chem. Int. Ed. 2012, 51, 8114–8117. [Google Scholar] [CrossRef]
- Ovchinnikov, Y.A. Membrane active complexones—Chemistry and biological function. FEBS Lett. 1974, 44, 1–21. [Google Scholar]
- Lingler, S.; Rubenstein, I.; Knoll, W.; Offenhäuser, A. Fusion of small unilamellar lipid vesicles to alkanethiol and thiolipid self-assembled monolayers on gold. Langmuir 1997, 13, 7085–7091. [Google Scholar] [CrossRef]
- Puu, G.; Gustafson, I. Planar lipid bilayers on solid supports from liposomes—Factors of importance for kinetics and stability. Biochim. Biophys. Acta 1997, 1327, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.B.; Plant, A.L. Self assembly driven by hydrophobic interactions at alkanethiol monolayers: Mechanism of formation of hybrid bilayer membranes. Biophys. Chem. 1998, 75, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.P.; Brisson, A. Characterization of lipid bilayers and protein assemblies supported on rough surfaces by atomic force microscopy. Langmuir 2003, 19, 1632–1640. [Google Scholar] [CrossRef]
- Reimhult, E.; Zäch, M.; Höök, F.; Kasemo, B. A multitechnique study of liposome adsorption on Au and lipid bilayer formation on SiO2. Langmuir 2006, 22, 3313–3319. [Google Scholar] [CrossRef] [PubMed]
- Ratnayaka, S.N.; Wysockki, R.J.; Saavedra, S.S. Preparation and characterization of asymmetric planar supported bilayers composed of poly(bis-sorbylphosphatidylcholine) on n-octadecyltrichlorosilane SAMs. J. Colloid Interface Sci. 2008, 327, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, R.; Last, J.A.; Slade, A.L.; Ista, L.K.; Bisong, P.; O'Brien, M.J.; Brueck, S.R.J.; Sasaki, D.Y.; Lopez, G.P. Using bicellar mixtures to form supported and suspended lipid bilayers on silicon chips. Langmuir 2006, 22, 8163–8168. [Google Scholar] [CrossRef] [PubMed]
- Stamou, D.; Duschl, C.; Delamarche, E.; Vogel, H. Self-assembled microarrays of attoliter molecular vessels. Angew. Chem. Int. Ed. 2003, 42, 5580–5583. [Google Scholar] [CrossRef]
- Kim, Y.H.; Rahman, M.M.; Zhang, Z.L.; Misawa, N.; Tero, R.; Urisu, T. Supported lipid bilayer formation by the giant vesicle fusion induced by vesicle-surface electrostatic attractive interaction. Chem. Phys. Lett. 2006, 420, 569–573. [Google Scholar] [CrossRef]
- Rapuano, R.; Carmona-Ribeiro, A.M. Supported bilayers on silica. J. Colloid Interface Sci. 2000, 226, 299–307. [Google Scholar] [CrossRef]
- Bucak, S.; Wang, C.; Laibinis, P.E.; Hatton, T.A. Dynamics of supported lipid bilayer deposition from vesicle suspensions. J. Colloid Interface Sci. 2010, 348, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.; Steinberger, A.; Cottin-Bizonne, C.; Rieu, J.P.; Charlaix, E. Boundary flow of water on supported phospholipid films. Europhys. Lett. 2006, 73, 390–395. [Google Scholar] [CrossRef]
- Cho, N.J.; Frank, C.W.; Kasemo, B.; Höök, F. Quartz crystal microbalance with dissipation monitoring of supported lipid bilayers on various substrates. Nat. Protoc. 2010, 5, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, L.; Gunnarsson, A.; Wallin, P.; Jönsson, P.; Höök, F. Continuous lipid bilayers derived from cell membranes for spatial molecular manipulation. J. Am. Chem. Soc. 2011, 133, 14027–14032. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, P.; Jonsson, M.P.; Tegenfeldt, J.O.; Höök, F. A method improving the accuracy of fluorescence recovery after photobleaching analysis. Biophys. J. 2008, 95, 5334–5348. [Google Scholar] [CrossRef] [PubMed]
- Mullineaux, C.W.; Kirchhoff, H. Using fluorescence recovery after photobleaching to measure lipid diffusion in membranes. In Methods in membrane lipids; Dopico, A.M., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 267–275. [Google Scholar]
- Merzlyakov, M.; Li, E.; Hristova, K. Directed assembly of surface-supported bilayers with transmembrane helixes. Langmuir 2006, 22, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Berquand, A.; Mazeran, P.E.; Pantigny, J.; Proux-Delrouyre, V.; Laval, J.M.; Bourdillon, C. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir 2003, 19, 1700–1707. [Google Scholar] [CrossRef]
- Seu, K.J.; Pandey, A.P.; Haque, F.; Proctor, E.A.; Ribbe, A.E.; Hovis, J.S. Effect of surface treatment on diffusion and domain formation in supported lipid bilayers. Biophys. J. 2007, 92, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.F.F.; Vaz, W.L.C. Lateral diffusion in membranes. In Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier: Amsterdam, the Netherland, 1995; Volume 1, pp. 306–357. [Google Scholar]
- Benz, M.; Gutsmann, T.; Chen, N.H.; Tadmor, R.; Israelachvili, J. Correlation of AFM and SFA measurements concerning the stability of supported lipid bilayers. Biophys. J. 2004, 86, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Seantier, B.; Breffa, C.; Felix, O.; Decher, G. In situ investigations of the formation of mixed supported lipid bilayers close to phase transition temperature. Nano Lett. 2004, 4, 5–10. [Google Scholar] [CrossRef]
- Jönsson, P.; Beech, J.P.; Tegenfeldt, J.O.; Höök, F. Mechanical behavior of a supported lipid bilayer under external shear forces. Langmuir 2009, 25, 6279–6286. [Google Scholar] [CrossRef] [PubMed]
- Tien, H.T.; Ottova, A.L. From self-assembled bilayer lipid membranes (BLMs) to supported BLMs on metal and gel substrates to practical applications. Colloids Surfaces A 1999, 149, 217–233. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackmann, E. Supported membranes as biofunctional interfaces and smart biosensor platforms. Phys. Status Solidi A 2006, 203, 3452–3462. [Google Scholar] [CrossRef]
- Tun, T.N.; Jenkins, A.T.A. An electrochemical impedance study of the effect of pathogenic bacterial toxins on tethered bilayer lipid membrane. Electrochem. Commun. 2010, 12, 1411–1415. [Google Scholar] [CrossRef]
- Sinner, E.K.; Knoll, W. Functional tethered membranes. Curr. Oppi. Chem. Biol. 2001, 5, 705–711. [Google Scholar] [CrossRef]
- Danelon, C.; Terrettaz, S.; Guenat, O.; Koudelka, M.; Vogel, H. Probing the function of ionotropic and G protein-coupled receptors in surface-confined membranes. Methods 2008, 46, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Terrettaz, S.; Mayer, M.; Vogel, H. Highly electrically insulating tethered lipid bilayers for probing the function of ion channel proteins. Langmuir 2003, 19, 5567–5569. [Google Scholar] [CrossRef]
- Bunjes, N.; Schmidt, E.K.; Jonczyk, A.; Rippmann, F.; Beyer, D.; Ringsdorf, H.; Gräber, P.; Knoll, W.; Naumann, R. Thiopeptide-supported lipid layers on solid substrates. Langmuir 1997, 13, 6188–6194. [Google Scholar] [CrossRef]
- Jadhav, S.R.; Sui, D.X.; Garavito, R.M.; Worden, R.M. Fabrication of highly insulating tethered bilayer lipid membrane using yeast cell membrane fractions for measuring ion channel activity. J. Colloid Interface Sci. 2008, 322, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Stora, T.; Lakey, J.H.; Vogel, H. Ion-channel gating in transmembrane receptor proteins: Functional activity in tethered lipid membranes. Angew. Chem. Int. Ed. 1999, 38, 389–391. [Google Scholar] [CrossRef]
- Atanasov, V.; Knorr, N.; Duran, R.S.; Ingebrandt, S.; Offenhäuser, A.; Knoll, W.; Köper, I. Membrane on a chip: A functional tethered lipid bilayer membrane on silicon oxide surfaces. Biophys. J. 2005, 89, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Raguse, B.; Braach-Maksvytis, V.; Cornell, B.A.; King, L.G.; Osman, P.D.J.; Pace, R.J.; Wieczorek, L. Tethered lipid bilayer membranes: Formation and ionic reservoir characterization. Langmuir 1998, 14, 648–659. [Google Scholar] [CrossRef]
- Vockenroth, I.K.; Atanasova, P.P.; Long, J.R.; Jenkins, A.T.A.; Knoll, W.; Köper, I. Functional incorporation of the pore forming segment of AChR M2 into tethered bilayer lipid membranes. Biochim. Biophys. Acta 2007, 1768, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Kügler, R.; Knoll, W. Polyelectrolyte-supported lipid membranes. Bioelectrochemistry 2002, 56, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.W.; Boxer, S.G.; Knoll, W.; Curtis, W.F. Polymer-supported bilayers on benzophenone-modified substrates. Biomacromolecules 2001, 2, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Wong, J.Y.; Park, C.K.; Alcantar, N.A.; Israelachvili, J. Formation of tethered supported bilayers via membrane-inserting reactive lipids. Thin solid films 1998, 327–329, 767–771. [Google Scholar] [CrossRef]
- Stenlund, P.; Babcock, G.J.; Sodroski, J.; Myszka, D.G. Capture and reconstitution of G protein-coupled receptors on a biosensor surface. Anal. Biochem. 2003, 316, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J. G protein-coupled receptors. In Encyclopedia of Life Sciences; Nature Publishing group: London, UK, 2001; pp. 1–9. [Google Scholar]
- Claesson, M.; Frost, R.; Svedhem, S.; Andersson, M. Pore spanning lipid bilayers on mesoporous silica having varying pore size. Langmuir 2011, 27, 8974–8982. [Google Scholar] [CrossRef] [PubMed]
- Kresak, S.; Hianik, T.; Naumann, R.L.C. Giga-seal solvent-free bilayer lipid membranes: From single nanopores to nanopore arrays. Soft Matter 2009, 5, 4021–4032. [Google Scholar] [CrossRef]
- Tiefenauer, L.X.; Studer, A. Nano for bio: Nanopore arrays for stable and functional lipid bilayer membranes (Mini Review). Biointerphases 2008, 3, FA74–FA79. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Toshima, Y.; Minamikawa, H.; Hato, M.; Suzuki, K.; Kamo, N. Formation and characterization of planar lipid bilayer membranes from synthetic phytanyl-chained glycolipids. Biochim. Biophys. Acta 1999, 1421, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Heitz, B.A.; Jones, I.W.; Hall, H.K.; Aspinwall, C.A.; Saavedra, S.S. Fractional polymerization of a suspended planar bilayer creates a fluid, highly stable membrane for ion channel recordings. J. Am. Chem. Soc. 2010, 132, 7086–7093. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.J.; Malmstadt, N.; Poulos, J.L.; Schmidt, J.J. Black lipid membranes stabilized through substrate conjugation to a hydrogel. Biointerphases 2008, 3, FA96–FA100. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Vörös, J.; Zambelli, T. A Gigaseal obtained with a self-assembled long-lifetime lipid bilayer on a single polyelectrolyte multilayer-filled manopore. ACS Nano 2010, 4, 5047–5054. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Vörös, J.; Zambelli, T. The resistance of polyelectrolyte multilayers in a free-hanging configuration. J. Phys. Chem. B 2010, 114, 13982–13987. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.; Capone, R.; Mayer, M.; Yang, J. Chemically reactive derivatives of gramicidin A for developing ion channel-based nanoprobes. Bioconjugate Chem. 2008, 19, 1614–1624. [Google Scholar] [CrossRef]
- Leonenko, Z.V.; Carini, A.; Cramb, D.T. Supported planar bilayer formation by vesicle fusion: The interaction of phospholipid vesicles with surfaces and the effect of gramicidin on bilayer properties using atomic force microscopy. Biochim. Biophys. Acta 2000, 1509, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Structure and function of channel-forming peptides: Maganinins, cecropin, melittin and alamethicin. J. Membr. Biol. 1997, 156, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Steinem, C.; Janshoff, A.; Galla, H.J. Evidence for multilayer formation of melittin on solid-supported phospholipid membranes by shear-wave resonator measurements. Chem. Phys. Lipids 1998, 95, 95–104. [Google Scholar] [CrossRef]
- Strömstedt, A.A.; Wessman, P.; Ringstad, L.; Edwards, K.; Malmsten, M. Effect of lipid headgroup composition on the interaction between melittin and lipid bilayers. J. Colloid Sci. Interfaces Sci. 2007, 311, 59–69. [Google Scholar] [CrossRef]
- Favero, G.; Campanella, L.; D'Annibale, A.; Santucci, R.; Ferri, T. Mixed hybrid bilayer lipid membrane incorporating valinomycin: Improvement in preparation and functioning. Microchem. J. 2003, 74, 141–148. [Google Scholar] [CrossRef]
- Naumann, R.; Walz, D.; Schiller, S.M.; Knoll, W. Kinetics of valinomycin-mediated K+ ion transport through tethered bilayer lipid membranes. J. Electroanal. Chem. 2003, 550, 241–252. [Google Scholar] [CrossRef]
- Constantinescu, I.; Lafleur, M. Influence of the lipid composition on the kinetics of concerted insertion and folding of melittin in bilayers. Biochim. Biophys. Acta 2004, 1667, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Shirai, O.; Yamana, H.; Ohnuki, T.; Yoshida, Y.; Kihara, S. Ion transport across a bilayer lipid membrane facilitated by valinomycin. J. Electroanal. Chem. 2004, 570, 219–226. [Google Scholar] [CrossRef]
- Iacovache, I.; Bischofberger, M.; van der Goot, F.G. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 2010, 20, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Hong, D.L.; Zhu, T.Y.; Liu, S.L.; Li, G.X. Electrochemical sensing of the ion-channel formation of OmpF. J. Appl. Electrochem. 2009, 39, 1163–1167. [Google Scholar] [CrossRef]
- Hemmler, R.; Bose, G.; Wagner, R.; Peters, R. Nanopore unitary permeability measured by electrochemical and optical single transporter recording. Biophys. J. 2005, 88, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H.; Cremer, P.S. Stochastic sensors inspired by biology. Nature 2001, 413, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Baaken, G.; Ankri, N.; Schuler, A.K.; Rühe, J.; Behrends, J.C. Nanopore-based single-molecule mass spectrometry on a lipid membrane microarray. ACS Nano 2011, 5, 8080–8088. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H.; Jayasinghe, L. Functional engineered channels and pores. Mol. Membr. Biol. 2004, 21, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Search for pore-fection. Science 2012, 336, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Gonzalez, M.R.; van der Goot, F.G. Membrane injury by pore-forming proteins. Curr. Opin. Cell Biol. 2009, 21, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Schulte, A.; Ruamchan, S.; Khunkaewla, P.; Suginta, W. The outer membrane protein VhOmp of Vibrio harveyi: Pore-forming properties in black lipid membranes. J. Membr. Biol. 2009, 230, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Manrao, E.A.; Derrington, I.M.; Laszlo, A.H.; Langford, K.W.; Hopper, M.K.; Gillgren, N.; Pavlenok, M.; Niederweis, M.; Gundlach, J.H. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 2012, 30, 349-U174. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.A.; Jayasinghe, L.; Daltrop, O.; Mason, A.; Bayley, H. Direct transfer of membrane proteins from bacteria to planar bilayers for rapid screening by single-channel recording. Nat. Chem. Biol. 2006, 2, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Nurani, G.; Radford, M.; Charalambous, K.; O'Reilly, A.O.; Cronin, N.B.; Haque, S.; Wallace, B.A. Tetrameric bacterial sodium channels: characterization of structure, stability, and drug binding. Biochemistry 2008, 47, 8114–8121. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Aoki, S.; Hibi, T.; Kano, K.; Shirai, O. Reconstitution of the voltage-gated K+ channel KAT1 in planar lipid bilayers. Electrochem. Commun. 2008, 10, 1509–1512. [Google Scholar] [CrossRef]
- Karatekin, E.; Rothman, J.E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat. Protoc. 2012, 7, 903–920. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Svedhem, S.; Kasemo, B. Lipid transfer between charged supported lipid bilayers and oppositely charged vesicles. Langmuir 2009, 25, 5146–5158. [Google Scholar] [CrossRef] [PubMed]
- Rawle, R.J.; van Lengerich, B.; Chung, M.; Bendix, P.M.; Boxer, S.G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys. J. 2011, 101, L37–L39. [Google Scholar] [CrossRef] [PubMed]
- de Planque, M.R.R.; Mendes, G.P.; Zagnoni, M.; Sandison, M.E.; Fisher, K.H.; Berry, R.M.; Watts, A.; Morgan, H. Controlled delivery of membrane proteins to artificial lipid bilayers by nystatin-ergosterol modulated vesicle fusion. IED Proc. Nanobiotechnol. 2006, 153, 21–30. [Google Scholar] [CrossRef]
- Woodbury, D.J. Nystatin/ergosterol method for reconstituting ion channels into planar lipid bilayers. Methods Enzymol. 1999, 294, 319–339. [Google Scholar] [PubMed]
- Ataka, K.; Giess, F.; Knoll, W.; Naumann, R.; Haber-Pohlmeier, S.; Richter, B.; Heberle, J. Oriented attachment and membrane reconstitution of His-tagged cytochrome c oxidase to a gold electrode: In situ monitoring by surface-enhanced infrared absorption spectroscopy. J. Am. Chem. Soc. 2004, 126, 16199–16206. [Google Scholar] [CrossRef] [PubMed]
- Engelman, D.M. Membranes are more mosaic than fluid. Nature 2005, 438, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Trepot, S.; Mornet, S.; Benabdelhak, H.; Ducruix, A.; Brisson, A.; Lambert, O. Membrane protein selectivity oriented on solid support and reconstituted into lipid membrane. Langmuir 2007, 23, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- White, G.F.; Racher, K.I.; Lipski, A.; Hallett, F.R.; Wood, J.M. Physical properties of liposomes and proteoliposomes prepared from Escherichia coli polar lipids. Biochim. Biophys. Acta 2000, 1468, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wenk, R.M. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Lundbaek, J.A.; Birn, P.; Hansen, A.J.; Sogaard, R.; Nielsen, C.; Girshman, J.; Bruno, M.J.; Tape, S.E.; Egebjerg, J.; Greathouse, D.V.; Mattice, G.L.; Koeppe, R.E.; Andersen, O.S. Regulation of sodium channel function by bilayer elasticity: The importance of hydrophobic coupling. Effects of micelle-forming amphiphiles and cholesterol. J. Gen. Physiol. 2004, 123, 599–621. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, D.K.; Barger, W.R.; Singh, A.; Panchal, R.G.; Misakian, M.; Stanford, V.M.; Kasianowicz, J.J. Functional reconstitution of protein ion channels into planar polymerizable phospholipid membranes. Nano Lett. 2005, 5, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.E.; Rozanski, L.J.; Spratt, T.; Liu, S.C.; O'Brien, D.F.; Saavedra, S.S. Planar supported lipid bilayer polymers formed by vesicle fusion: 1. Influence of diene monomer structure and polymerization method on film properties. Langmuir 2003, 19, 1752–1765. [Google Scholar] [CrossRef]
- Subramaniam, V.; Alves, I.D.; Salgado, G.F.J.; Lau, P.W.; Wysocki, R.J.; Salamon, Z.; Tollin, G.; Hruby, V.J.; Brown, M.F.; Saavedra, S.S. Rhodopsin reconstituted into a planar-supported lipid bilayer retains photoactivity after cross-linking polymerization of lipid monomers. J. Am. Chem. Soc. 2005, 127, 5320–5321. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.W.; Sharom, F.J. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: Effects on ATPase activity, drug binding and transport. Biochemistry 2008, 47, 13686–13698. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Belli, S.; Elsener, P.M.; Wunderli-Allenspach, H.; Kramer, S.D. Cholesterol-mediated activation of P-glycoprotein: Distinct effects on basal and drug-induced ATPase activities. J. Pharm. Sci. 2009, 98, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Model membrane to investigate lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.M.; Tamm, L.K. Role of cholesterol in the formation and nature of lipid rafts in planar and spherical model membranes. Biophys. J. 2004, 86, 2965–2979. [Google Scholar] [CrossRef] [PubMed]
- Fabre, R.M.; Okeyo, G.O.; Talham, D.R. Supported lipid bilayers at skeletonized surfaces for the study of transmembrane proteins. Langmuir 2012, 28, 2835–2841. [Google Scholar] [CrossRef] [PubMed]
- Morigaki, K.; Kiyosue, K.; Taguchi, T. Micropatterned composite membranes of polymerized and fluid lipid bilayers. Langmuir 2004, 20, 7729–7735. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.M.; Bolinger, P.Y.; Hatzakis, N.S.; Mortensen, M.W.; Stamou, D. Mixing subattolitre volumes in a quantitative and highly parallel manner with soft matter nanofluidics. Nat. Nanotechnol. 2012, 7, 51–55. [Google Scholar] [CrossRef]
- Lei, G.; MacDonald, R.C. Effects on interactions of oppositely charged phospholipid vesicles of covalent attachment of polyethylene glycol oligomers to their surfaces: Adhesion, hemifusion, full fusion and “Endocytosis”. J. Membr. Biol. 2008, 221, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Clejan, S.; Krulwich, T.A.; Mondrus, K.R.; Setoyoung, D. Membrane lipid-composition of obligately and facultatively alkalophilic strains of bacillus SPP. J. Bacteriol. 1986, 168, 334–340. [Google Scholar] [PubMed]
- Baenziger, J.E.; Ryan, S.E.; Goodreid, M.M.; Vuong, N.Q.; Sturgeon, R.M.; DaCosta, C.J.B. Lipid composition alters drug action at the nicotinic acetylcholine receptor. Mol. Pharmacol. 2008, 73, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Akesson, A.; Lind, T.; Ehrlich, N.; Stamou, D.; Wacklin, H.; Cardenas, M. Composition and structure of mixed phospholipid supported bilayers formed by POPC and DPPC. Soft Matter 2012, 8, 5658–5665. [Google Scholar] [CrossRef]
- Braun, P.; LaBaer, J. High throughput protein production for functional proteomics. Trends Biotechnol. 2003, 21, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Böhm, H.J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Warne, T.; Serrano-Vega, M.J.; Baker, J.G.; Moukhametzianov, R.; Edwards, P.C.; Henderson, R.; Leslie, A.G.W.; Tate, C.G.; Schertler, G.F.X. Structure of a beta(1)-adrenergic G-protein-coupled receptor. Nature 2008, 454, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Marshall, F. The impact of GPCR structures on pharmacology and structure-based drug design. Bri. J. Pharmacol. 2010, 159, 986–996. [Google Scholar] [CrossRef]
- Fang, Y.; Lahiri, J.; Picard, L. G protein-coupled receptor microarrays for drug discovery. Drug Discov. Today 2003, 8, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J. Sensing voltage across lipid membranes. Nature 2008, 456, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, S.; Cuello, L.G.; Cortes, D.M.; Perozo, E. Structural dynamics of an isolated voltage-sensor domain in a lipid bilayer. Structure 2008, 16, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, I.; Dutzler, R. Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLoS Biol. 2011, 9, e1001101. [Google Scholar] [CrossRef] [PubMed]
- Debret, G.; Valadie, H.; Städler, A.M.; Etchebest, C. New insights of membrane environment effects on MscL channel mechanics from theoretical approaches. Proteins Struct. Funct. Bioinforma. 2008, 71, 1183–1196. [Google Scholar] [CrossRef]
- Sands, Z.A.; Sansom, M.S.P. How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure 2007, 15, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Chanda, B.; Asamoah, O.K.; Blunck, R.; Roux, B.; Bezanilla, F. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature 2005, 436, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Decoursey, T.E. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003, 83, 475–579. [Google Scholar] [PubMed]
- Gunthorpe, N.J.; Smith, G.D.; Davis, J.B.; Randall, A.D. Characterisation of a human acid-sensing ion channel (hASIC1a) endogenously expressed in HEK293 cells. Pflügers Arch. 2001, 442, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Koishi, R.; Xu, H.; Ren, D.; Navarro, B.; Spiller, B.W.; Shi, Q.; Clapham, D.E. A superfamily of voltage-gated sodium channels in bacteria. J. Biol.Chem. 2004, 279, 9532–9538. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Iwamoto, M.; Konno, T.; Nihei, A.; Sasaki, Y.C.; Oiki, S. Global twisting motion of single molecular KcsA potassium channel upon Gating. Cell 2008, 132, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Morais, C.J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Ye, S.; Alam, A.; Chen, L.; Jiang, Y. Atomic structure of a Na− and K+ conducting channel. Nature 2006, 440, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Cousin, M.A.; Ashley, R.H. Functional reconstitution of mammalian “chloride intracellular channels” CLIC1, CLIC4 and CLIC5 reveals differential regulation by cytoskeletal actin. FEBS J. 2007, 274, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Gwan, J.F.; Baumgaertner, A. Cooperative transport in a potassium ion channel. J. Chem. Phys. 2007, 127, 6306–6316. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Cuello, L.G.; Perozo, E. Voltage-dependent gating at the KcsA selectivity filter. Nat. Struct. Mol. Biol. 2006, 13, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Guo, W.L. Ion binding properties and structure stability of the NaK channel. Biochim. Biophys. Acta 2009, 1788, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Nimigean, C.M.; Miller, C. Na+ block and permeation in a K+ channel of known structure. J. Gen. Physiol 2002, 120, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Yellen, G. The voltage-gated potassium channels and their relatives. Nature 2002, 419, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Bowlby, M.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Leptihn, S.; Thompson, J.R.; Ellory, J.C.; Tucker, S.J.; Wallace, M.I. In vitro reconstitution of eukaryotic ion channels using droplet interface bilayers. J. Am. Chem. Soc. 2011, 133, 9370–9375. [Google Scholar] [CrossRef] [PubMed]
- Howitt, S.M.; Udvardi, M.K. Structure, function and regulation of ammonium transporters in plants. Biochem. Biophys. Acta 2000, 1465, 152–170. [Google Scholar] [CrossRef] [PubMed]
- Törnroth-Horsefield, S.; Wang, Y.; Hedfalk, K.; Johanson, U.; Karlsson, M.; Tajkhorshid, E.; Neutze, R.; Kjellbom, P. Structural mechanism of plant aquaporin gating. Nature 2006, 439, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.Y.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; Hoffmaster, K.A.; Ishikawa, T.; Keppler, D.; Kim, R.B.; Lee, C.A.; Niemi, M.; Polli, J.W.; Sugiyama, Y.; Swaan, P.W.; Ware, J.A.; Wright, S.H.; Yee, S.W.; Zamek-Gliszczynski, M.J.; Zhang, L.; International, T. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Demel, M.A.; Krämer, O.; Ettmayer, P.; Haaksma, E.J.; Ecker, G.F. Predicting ligand interaction with ABC transporters in ADME. Chem. Biodivers. 2009, 6, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic-Talisman, T.; Tetenbaum-Novatt, J.; McKenney, A.S.; Zilman, A.; Peters, R.; Rout, M.P.; Chait, B.T. Artificial nanopores that mimic the transport selectivity of the nuclear pore complex. Nature 2009, 457, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.Y.H.; Aebi, U.; Fahrenkrog, B. Towards reconciling structure and function in the nuclear pore complex. Histochem. Cell. Biol. 2008, 129, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Navarro, B.; Xu, H.; Yue, L.; Shi, Q.; Clapham, D.E. A prokaryotic voltage-gated sodium channel. Science 2001, 294, 2372–2375. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Xu, H.; Guffanti, A.A.; Wei, Y.; Zvi, L.; Clapham, D.E.; Krulwich, T.A. The voltage-gated Na+ channel NavBP has a role in motility, chemotaxis and pH homeostasis of an alkaliphilic Bacillus. Proc. Nat. Acad. Sci. USA 2004, 101, 10566–10571. [Google Scholar] [CrossRef] [PubMed]
- Shafrir, Y.; Durell, S.R.; Guy, H.R. Models of the structure and gating mechanisms of the pore domain of the NaChBac ion channel. Biophys. J. 2008, 95, 3650–3662. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; Furuichi, T.; Hensch, T.K.; Yamakawa, K. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A. Life at the top: The transition state of AChR gating. Sci. STKE 2003, 188. [Google Scholar] [CrossRef]
- Corringer, P.J.; Poitevin, F.; Prevost, M.S.; Sauguet, L.; Delarue, M.; Changeux, J.P. Structure and pharmacology of pentameric receptor channels: From bacteria to brain. Structure 2012, 20, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Xu, Y.C.; Li, H.L.; Wang, X.C.; Jiang, H.L.; Barrantes, F.J. Mechanics of channel gating of the nicotinic acetylcholine receptor. PLoS Comput. Biol. 2008, 4, 100–110. [Google Scholar]
- Hilf, R.J.C.; Dutzler, R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 2009, 457, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.; Gaub, H.E. Structure and mechanism of membrane proteins. Ann. Rev. Biochem. 2008, 77, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.G.; Robertson, J.W.F.; Walz, D.; Knoll, W.; Naumann, R.L.C. Electronic wiring of a multi-redox site membrane protein in a biomimetic surface architecture. Biophys. J. 2008, 94, 3698–3705. [Google Scholar] [CrossRef] [PubMed]
- Sperotto, M.M.; May, S.; Baumgaertner, A. Modelling of proteins in membranes. Chem. Phys. Lipids 2006, 141, 2–29. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Ann. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef]
- Picas, L.; Rico, F.; Scheuring, S. Direct measurement of the mechanical properties of lipid phases in supported bilayers. Biophys. J. 2012, 102, L1–L3. [Google Scholar] [CrossRef]
- Ovalle-Garcia, E.; Torres-Heredia, J.J.; Antillon, A.; Ortega-Blake, I. Simultaneous determination of the elastic properties of the lipid bilayer by atomic force microscopy: Bending, tension, and adhesion. J. Physical Chem. B 2011, 115, 4826–4833. [Google Scholar] [CrossRef]
- Steltenkamp, S.; Müller, M.M.; Desemo, M.; Hennesthal, C.; Steinem, C.; Janshoff, A. Mechanical properties of pore-spanning lipid bilayers probed by atomic force microscopy. Biophys. J. 2006, 91, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Sullan, R.M.A.; Zou, S. Atomic force microscopy force mapping in the study of supported lipid bilayers. Langmuir 2011, 27, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, K.; Booth, P.J.; Woscholski, R.; Seddon, J.M.; Templer, R.H.; Law, R.V.; Barter, L.M.C.; Ces, O. Engineering de novo membrane-mediated protein-protein communication networks. J. Am. Chem. Soc. 2012, 134, 5746–5749. [Google Scholar] [CrossRef] [PubMed]
- Veglia, G.; Ramamoorthy, A. Special issue on “membrane protein dynamics: Correlating structure to function”. Biochim. Biophys. Acta 2010, 1798, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.; Williams, C.; Waldron, G.J. Patch clamping by numbers. Drug Discov. Today 2004, 9, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Cavalli, A.; Recanatini, M. Modeling the hERG potassium channel in a phospholipid bilayer: Molecular dynamics and drug docking studies. J. Comput. Chem. 2008, 29, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Zou, B.Y.; Yu, H.B.; Babcock, J.J.; Chanda, P.; Bader, J.S.; McManus, O.B.; Li, M. Profiling diverse compounds by flux- and electrophysiology-based primary screens for inhibition of human ether-a-go-go related gene potassium channels. Assay Drug Dev. Technol. 2010, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, W.A.; Swensen, A.M.; Thomas, B.S.; Felix, J.P.; Haedo, R.J.; Solly, K.; Kiss, L.; Kaczorowski, G.J.; Garcia, M.L. A pharmacologically validated, high-capacity, functional thallium flux assay for the human ether-à-go-go related gene potassium channel. Assay Drug Dev. Technol. 2010, 8, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Poulos, J.L.; Jeon, T.J.; Damoiseaux, R.; Gillespie, E.J.; Bradley, K.A.; Schmidt, J.J. Ion channel and toxin measurement using a high throughput lipid membrane platform. Biosens. Bioelectron. 2009, 24, 1806–1810. [Google Scholar] [CrossRef] [PubMed]
- Siontorou, C.G.; Batzias, F.A. Innovation in biotechnology: Moving from academic research to product development-the case of biosensors. Crit. Rev. Biotechnol. 2010, 30, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Lodowski, D.T.; Angel, T.E.; Palczewski, K. Comparative analysis of GPCR crystal structures. Photochem. Photobiol. 2009, 85, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; George, A.M. The ABC transporter structure and mechanism: Perspectives on recent research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Landau, E.M.; Rosenbusch, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Nat. Acad. Sci. USA 1996, 93, 14532–14535. [Google Scholar] [CrossRef] [PubMed]
- Haupts, U.; Tittor, J.; Oesterhelt, D. Closing in on bacteriorhodopsin: Progress in understanding the molecule. Ann. Rev. Biophysics Biomol. Struct. 1999, 28, 367–399. [Google Scholar] [CrossRef]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; Yamamoto, M.; Miyano, M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Standfuss, J.; Edwards, P.C.; D'Antona, A.; Fransen, M.; Xie, G.F.; Oprian, D.D.; Schertler, G.F.X. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 2011, 471, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.G.; Schertler, G.F.X. Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 2009, 19, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, T.; Damian, M.; Mary, S.; Popot, J.L.; Baneres, J.L. Amphipol-assisted in vitro folding of G protein-coupled receptors. Biochemistry 2009, 48, 6516–6521. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Sligar, S.G. Membrane protein assembly into nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Gawrisch, K.; Soubias, O.; Mihailescu, M. Insights from biophysical studies on the role of polyunsaturated fatty acids for function of G-protein coupled membrane receptors. Prostagland. Leuk. Essent. Fat. Acids 2008, 79, 131–134. [Google Scholar] [CrossRef]
- Jonsson, M.P.; Jönsson, P.; Dahlin, A.B.; Höök, F. Supported lipid bilayer formation and lipid-membrane-mediated biorecognition reactions studied with a new nanoplasmonic sensor template. Nano Lett. 2007, 7, 3462–3468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tamm, L. FTIR and fluorescence studies of interaction of synaptic fusion proteins in polymer-supported bilayers. Langmuir 2003, 19, 1838–1846. [Google Scholar] [CrossRef]
- Kundu, J.; Levin, C.S.; Halas, N.J. Real-time monitoring of lipid transfer between vesicles and hybrid bilayers on Au nanoshells using surface enhanced Raman scattering (SERS). Nanoscale 2009, 1, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Frederix, P.L.T.M.; Bosshart, P.D.; Engel, A. Atomic force microscopy of biological membranes. Biophys. J. 2009, 96, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sackmann, E. Polymer-supported membranes as models for the cell surface. Nature 2005, 437, 656–663. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tiefenauer, L.; Demarche, S. Challenges in the Development of Functional Assays of Membrane Proteins. Materials 2012, 5, 2205-2242. https://doi.org/10.3390/ma5112205
Tiefenauer L, Demarche S. Challenges in the Development of Functional Assays of Membrane Proteins. Materials. 2012; 5(11):2205-2242. https://doi.org/10.3390/ma5112205
Chicago/Turabian StyleTiefenauer, Louis, and Sophie Demarche. 2012. "Challenges in the Development of Functional Assays of Membrane Proteins" Materials 5, no. 11: 2205-2242. https://doi.org/10.3390/ma5112205
APA StyleTiefenauer, L., & Demarche, S. (2012). Challenges in the Development of Functional Assays of Membrane Proteins. Materials, 5(11), 2205-2242. https://doi.org/10.3390/ma5112205