A Series of Supramolecular Complexes for Solar Energy Conversion via Water Reduction to Produce Hydrogen: An Excited State Kinetic Analysis of Ru(II),Rh(III),Ru(II) Photoinitiated Electron Collectors

Abstract
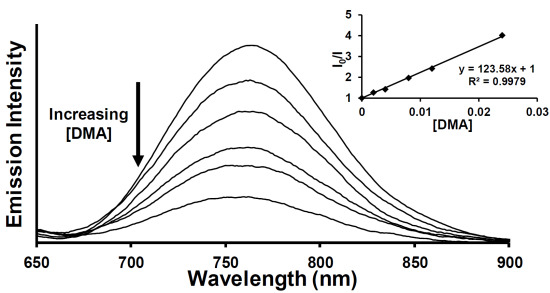
:
1. Introduction
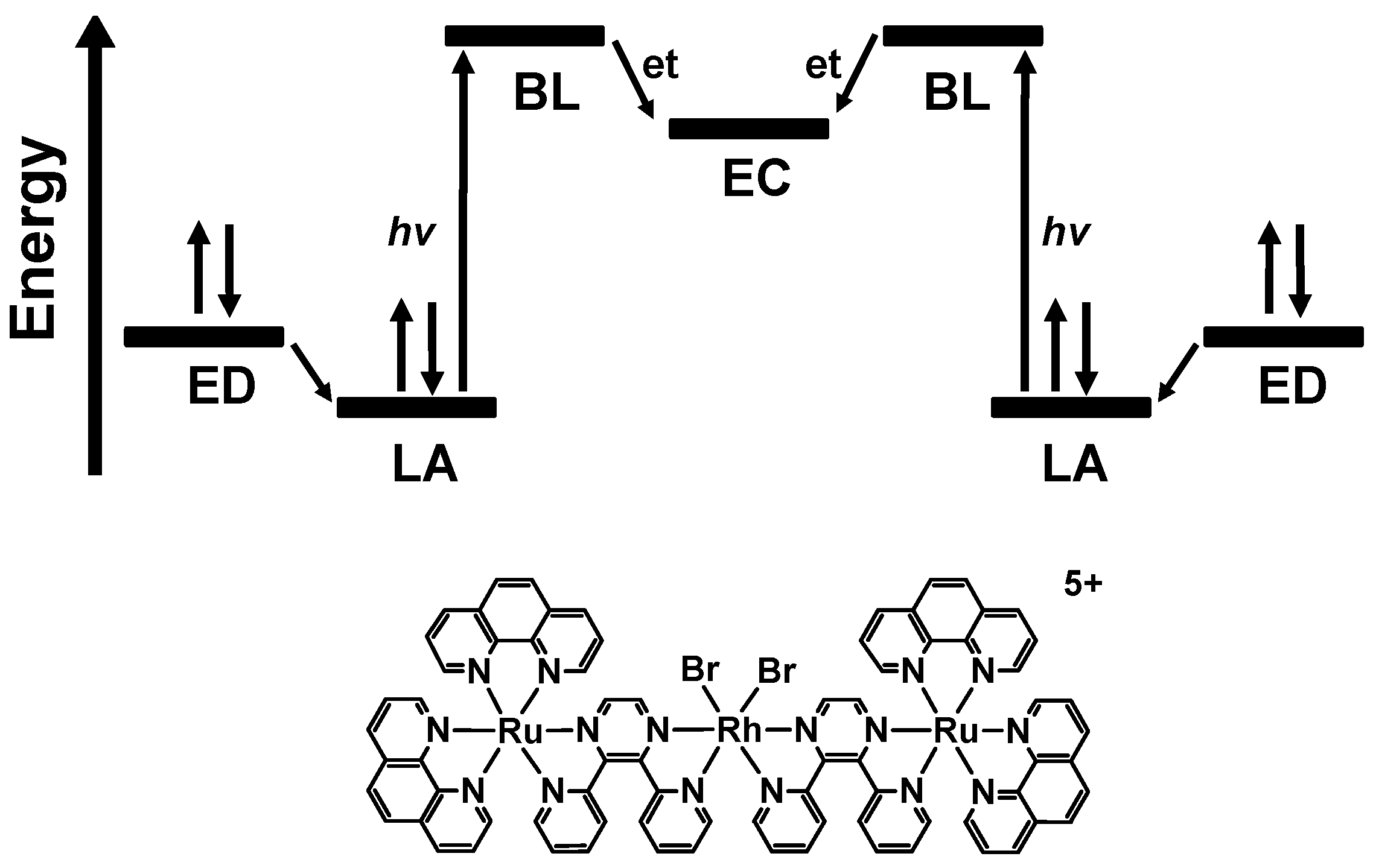
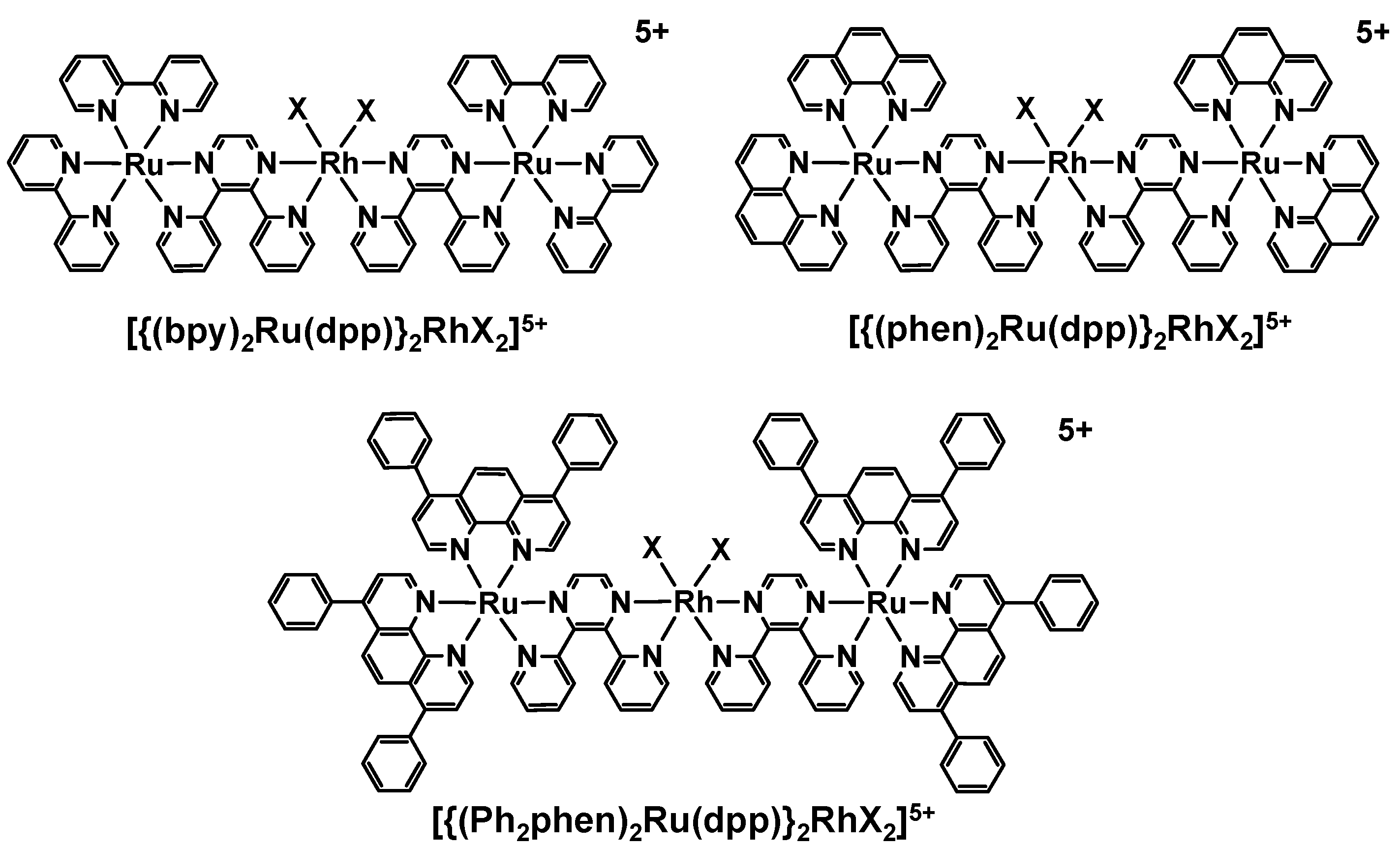
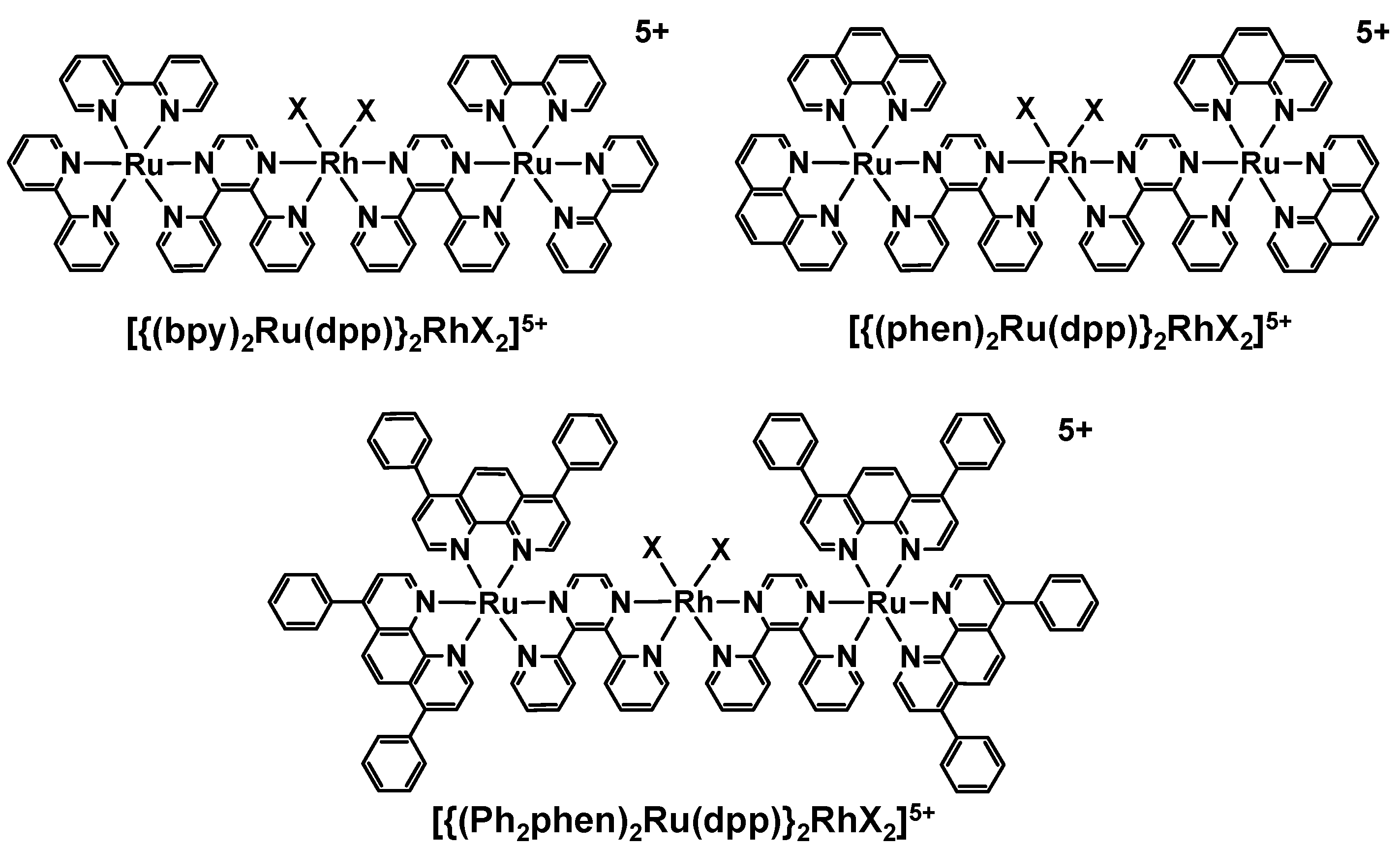
2. Results and Discussion
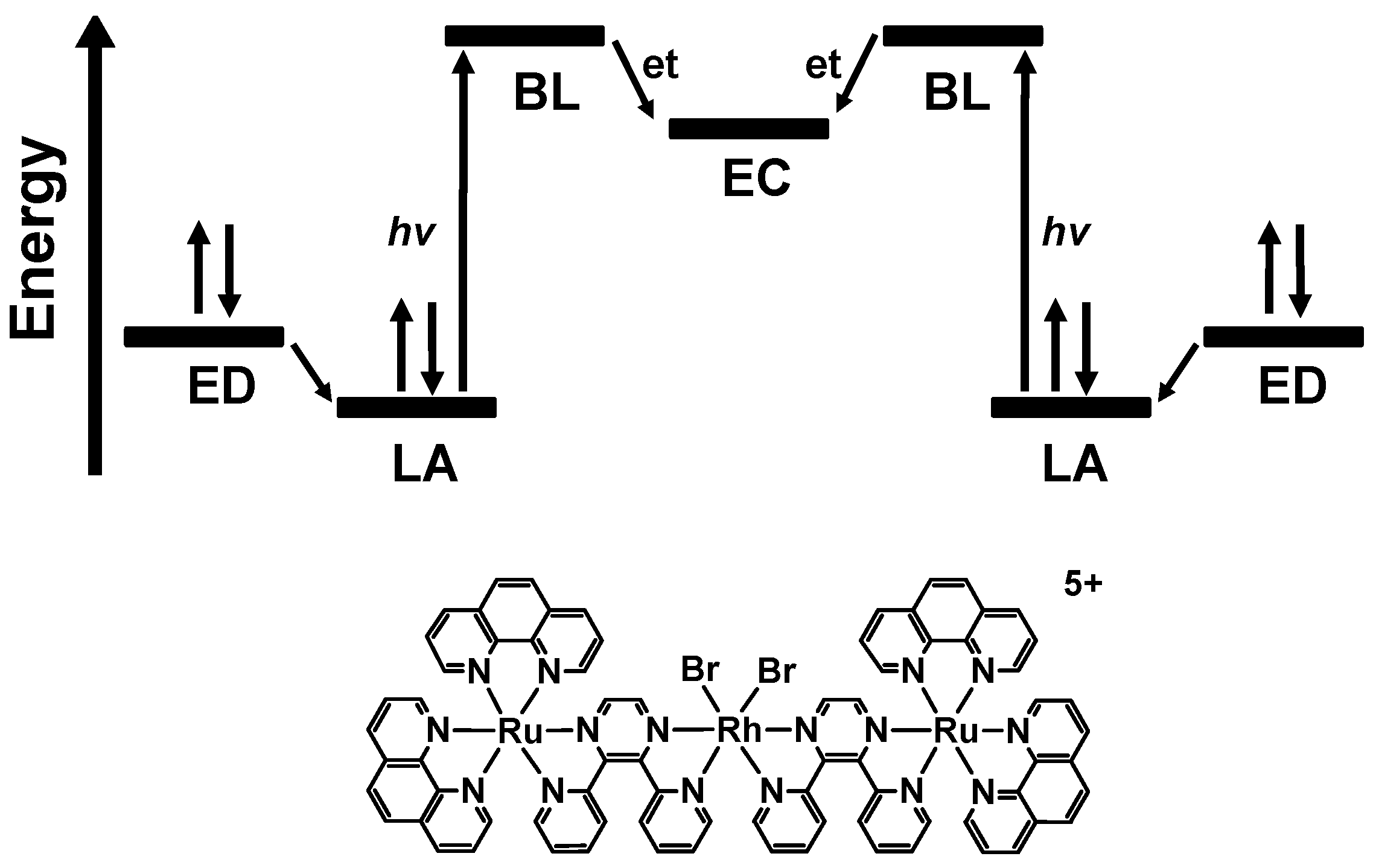
2.1. Photophysical Properties
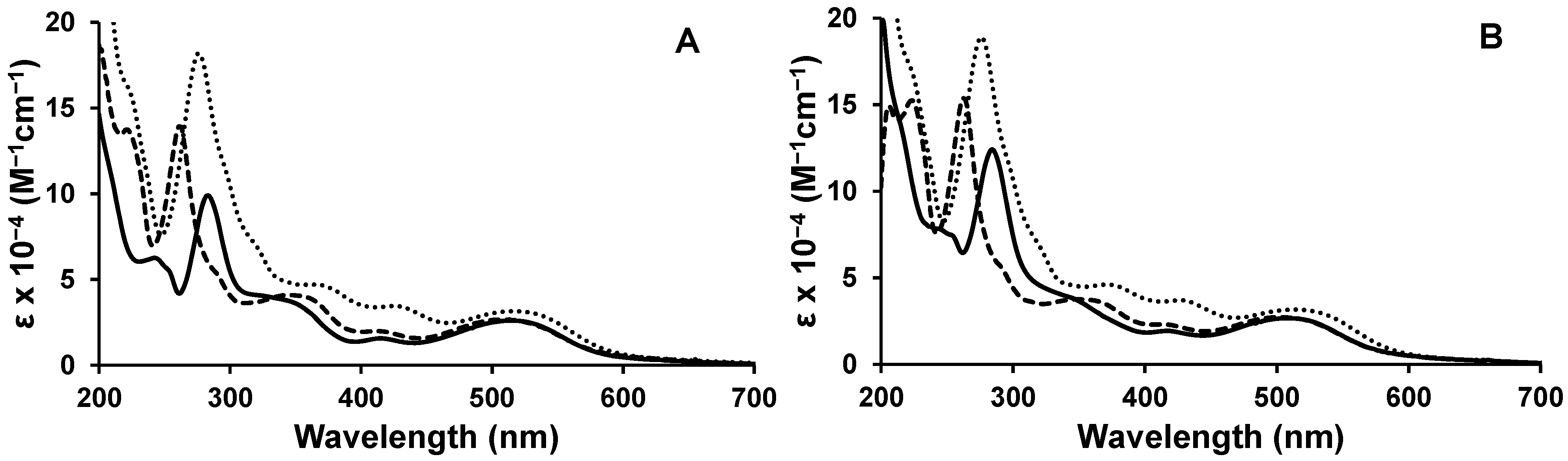

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
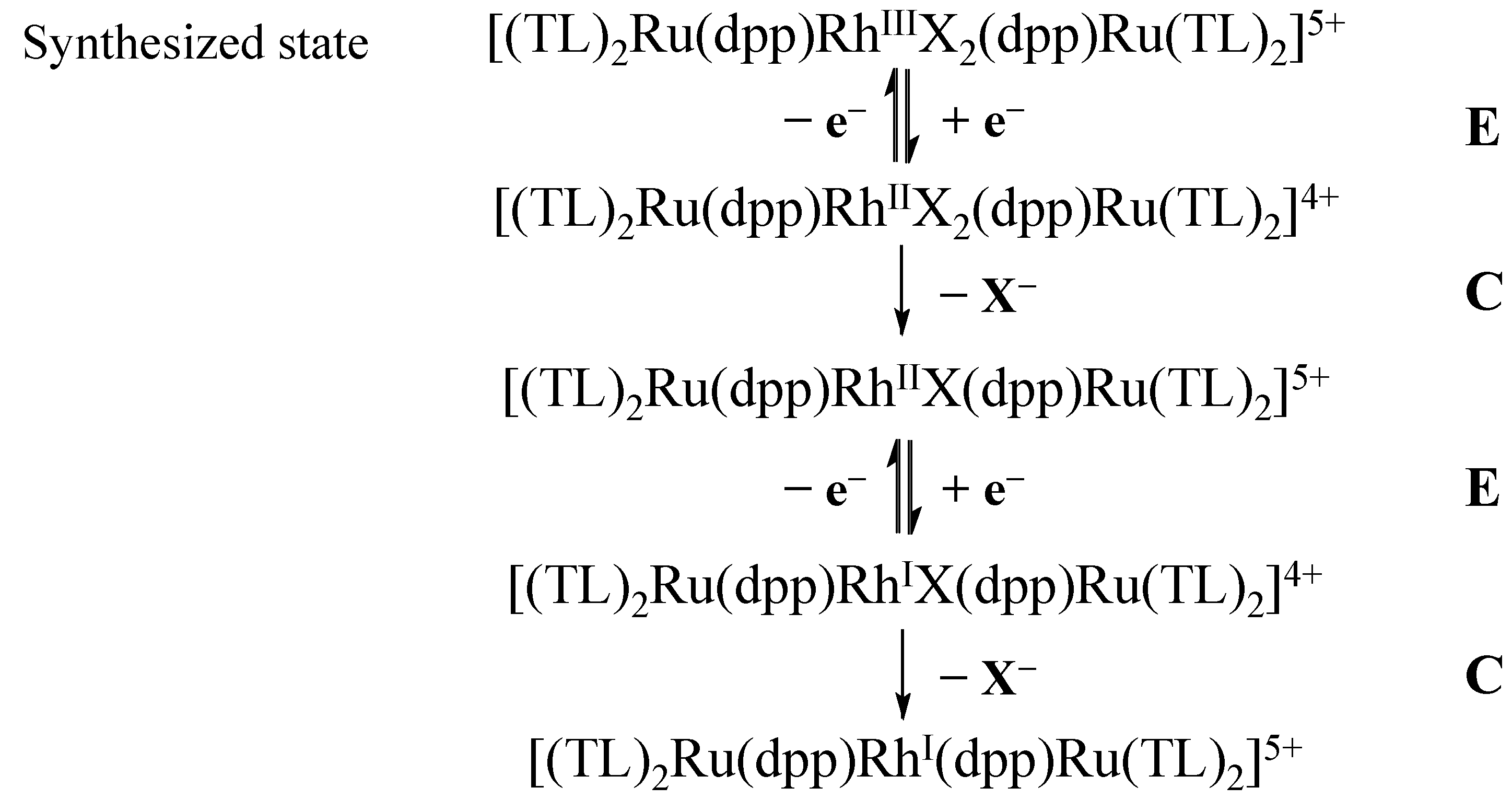{kind=link}
{kind=link}
{kind=link}
| Complex | RT a | 77 K b | ||||||
|---|---|---|---|---|---|---|---|---|
| λmaxem (nm) | ΦMLCTem (10−3) c | τ (ns) d | kr (103 s−1) | knr (106 s−1) | ket (107 s−1) | λmaxem (nm) | τ (μs) d | |
| [(bpy)2Ru(dpp)Ru(bpy)2]4+ | 752 | 0.97 | 145 | 6.7 | 6.9 | 730 | 2.4 | |
| [(phen)2Ru(dpp)Ru(phen)2]4+ | 750 | 1.6 | 170 | 9.4 | 5.9 | 695 | 2.0 | |
| [(Ph2phen)2Ru(dpp)Ru(Ph2phen)2]4+ | 754 | 1.7 | 192 | 9.0 | 5.2 | 698 | 2.0 | |
| [{(bpy)2Ru(dpp)}2RhCl2]5+ | 776 | 0.26 | 38 | 6.7 | 6.9 | 1.9 | 716 | 1.9 |
| [{(bpy)2Ru(dpp)}2RhBr2]5+ | 776 | 0.14 | 34 | 6.7 | 6.9 | 2.3 | 716 | 1.9 |
| [{(phen)2Ru(dpp)}2RhCl2]5+ | 760 | 0.22 | 35 | 9.4 | 5.9 | 2.3 | 706 | 1.8 |
| [{(phen)2Ru(dpp)}2RhBr2]5+ | 760 | 0.17 | 30 | 9.4 | 5.9 | 2.8 | 706 | 1.9 |
| [{(Ph2phen)2Ru(dpp)}2RhCl2]5+ | 770 | 0.24 | 52 | 9.0 | 5.2 | 1.4 | 696 | 1.8 |
| [{(Ph2phen)2Ru(dpp)}2RhBr2]5+ | 770 | 0.20 | 40 | 9.0 | 5.2 | 2.0 | 696 | 1.9 |
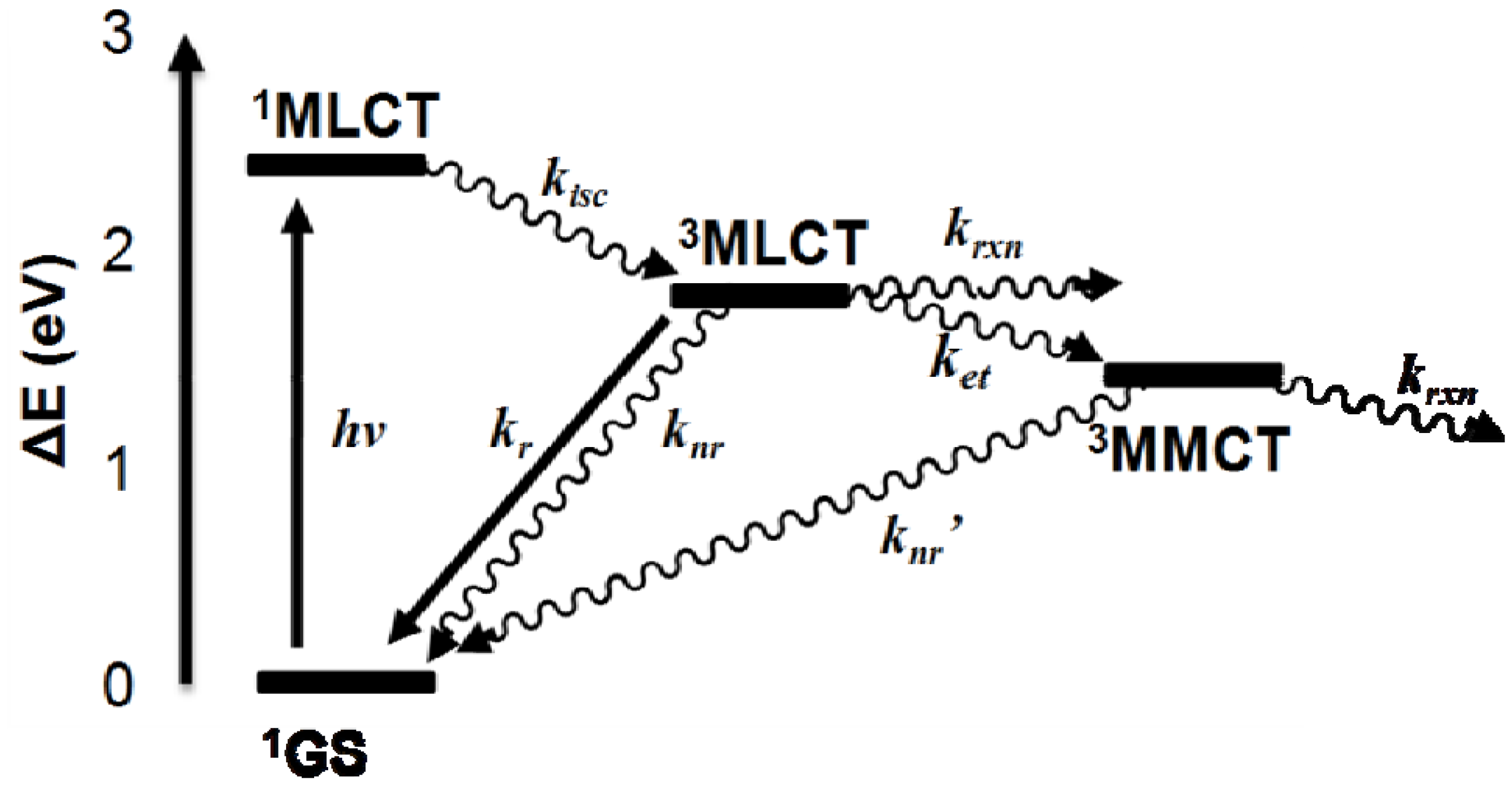
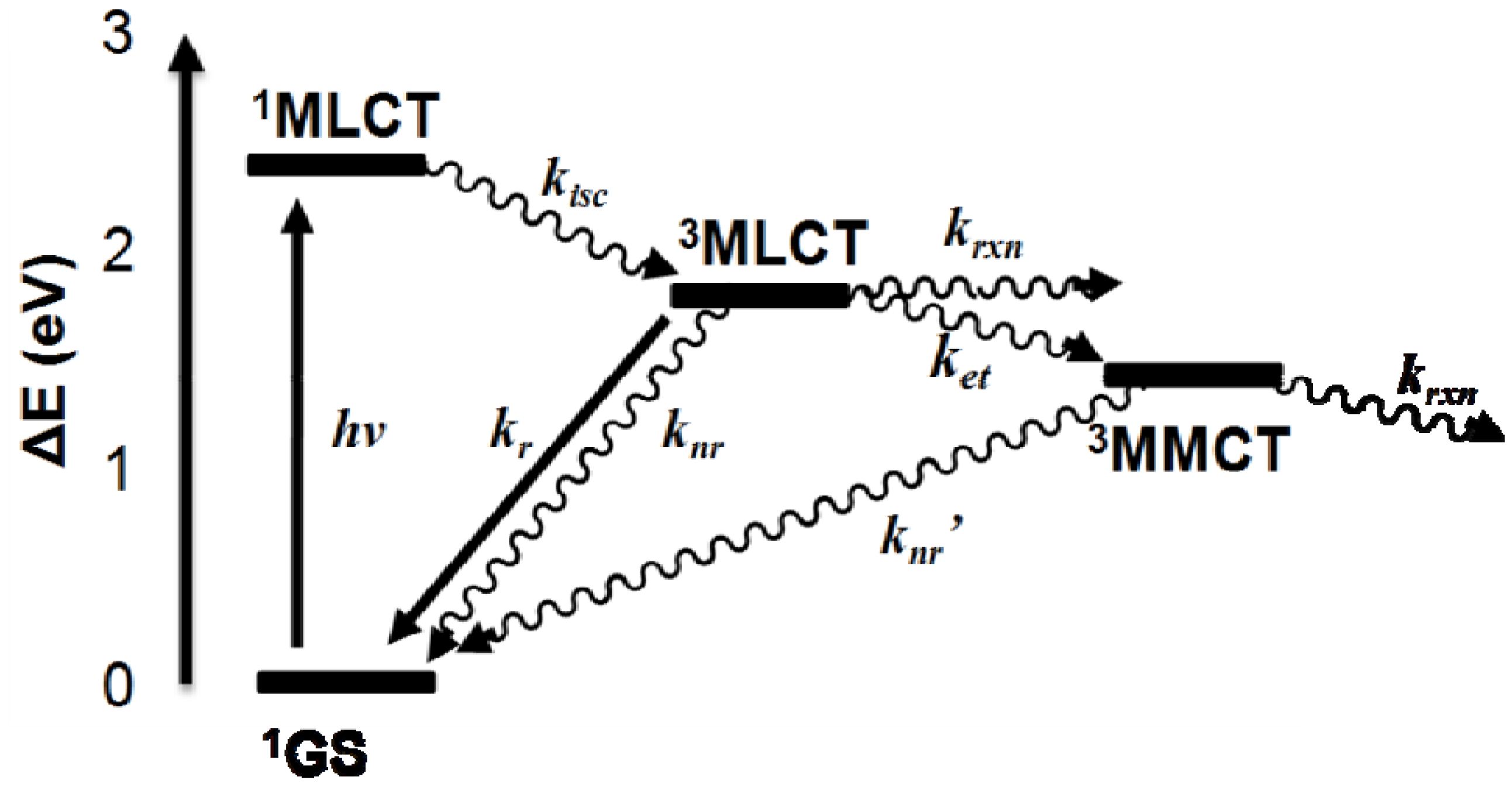
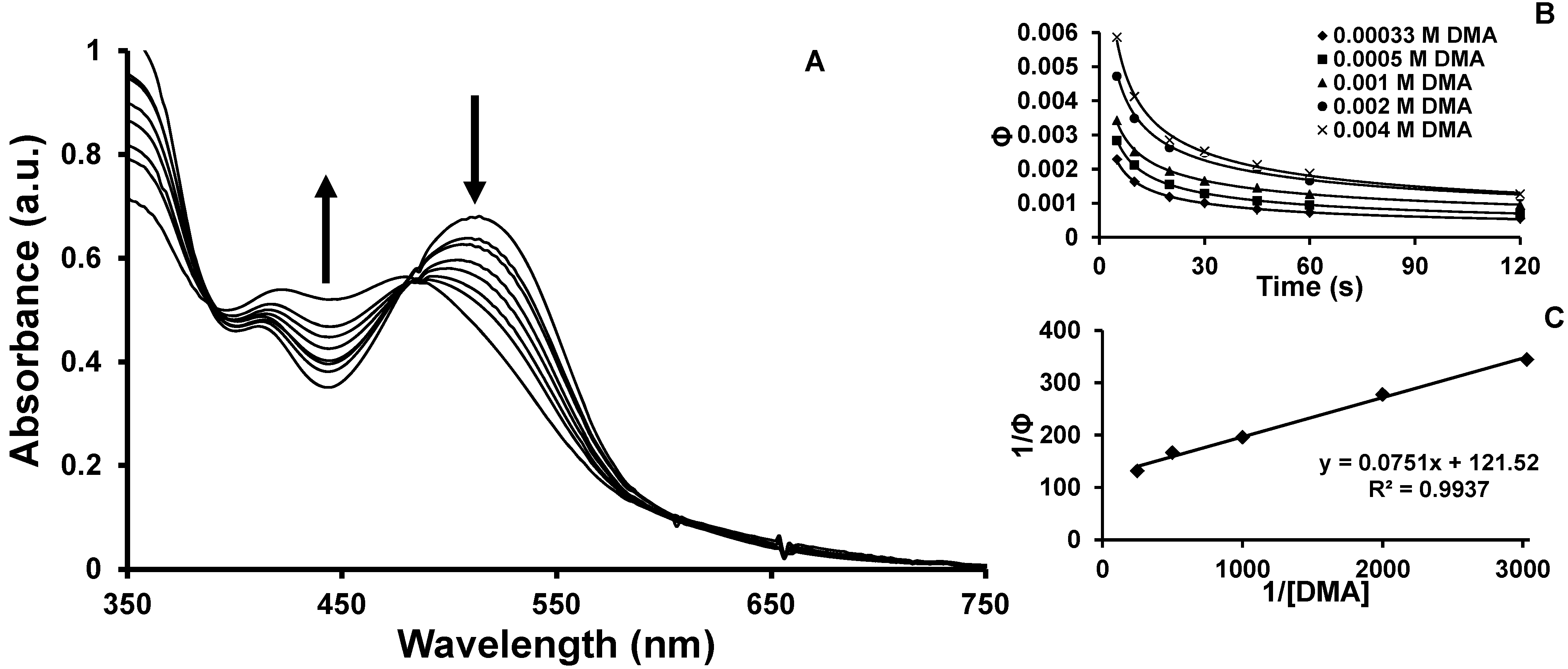
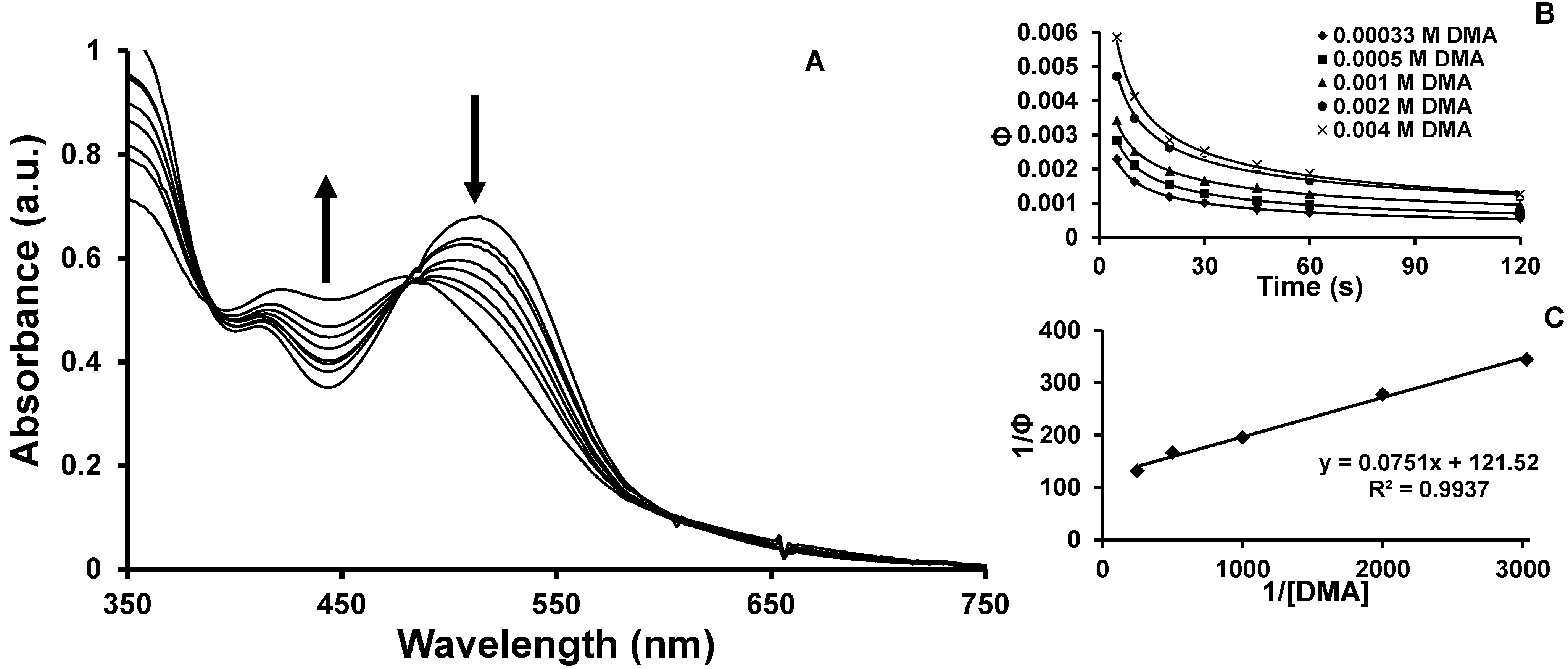
2.2. Photochemical Properties

- MLCT Excitation:
- Unimolecular Deactivation of 3MLCT State:
- Bimolecular Deactivation of 3MLCT State:
- Reductive Quenching of 3MLCT State:
- Intramolecular Electron Transfer:
- Unimolecular Deactivation of 3MMCT State:
- Bimolecular Deactivation of 3MMCT State:
- Reductive Quenching of 3MLCT State:
2.3. Emission Quenching
| Complex | E(*CATn+/CAT(n − 1)+) 3MLCT (V)a | E(*CATn+/CAT(n − 1)+) 3MMCT (V)a | Eredox 3MLCT (V)b | Eredox 3MMCT (V)b | kq + k2 (M−1s−1)c |
|---|---|---|---|---|---|
| [Ru(bpy)3]2+ e | +0.82 | -- | −0.04 | -- | 7.1 × 107 d |
| [Ru(bpz)3]2+ f | +1.50 | -- | +0.64 | -- | 8.4 × 109 d |
| [{(bpy)2Ru(dpp)}2RhCl2]5+ | +1.35 | +0.94 | +0.49 | +0.08 | 2.5 × 109 |
| [{(bpy)2Ru(dpp)}2RhBr2]5+ | +1.38 | +0.99 | +0.52 | +0.13 | 3.2 × 109 |
| [{(phen)2Ru(dpp)}2RhCl2]5+ | +1.41 | +1.01 | +0.55 | +0.15 | 3.9 × 109 |
| [{(phen)2Ru(dpp)}2RhBr2]5+ | +1.44 | +1.05 | +0.58 | +0.19 | 5.9 × 109 |
| [{(Ph2phen)2Ru(dpp)}2RhCl2]5+ | +1.43 | +1.04 | +0.57 | +0.18 | 1.5 × 109 |
| [{(Ph2phen)2Ru(dpp)}2RhBr2]5+ | +1.46 | +1.09 | +0.60 | +0.23 | 2.9 × 109 |
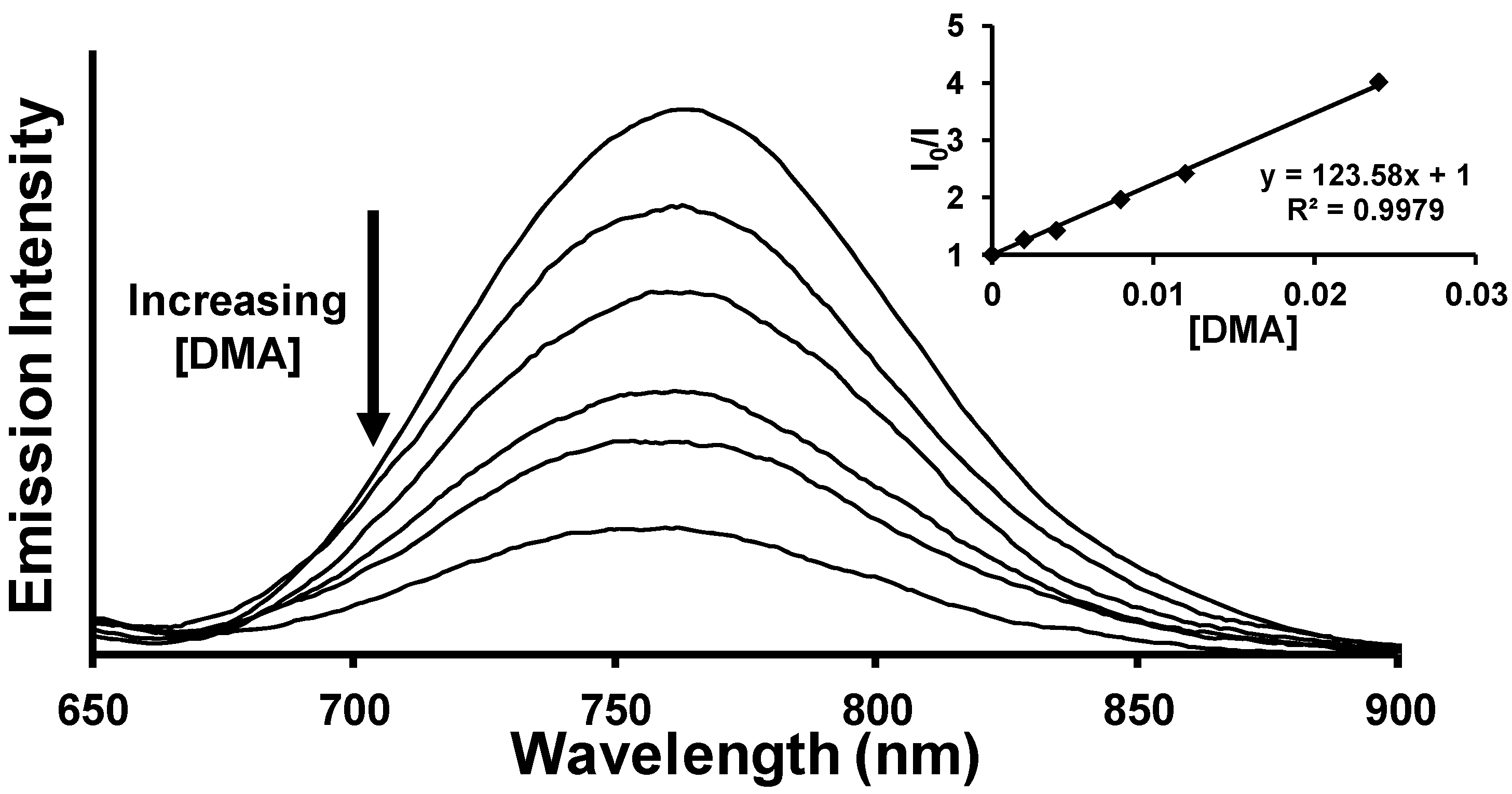
2.4. Product Formation
| Complex | kq (108 M−1s−1) a | k2 (109 M−1s−1) b | Φ3MMCT c | k4/kq2 d |
|---|---|---|---|---|
| [{(bpy)2Ru(dpp)}2RhCl2]5+ | 2.1 | 2.3 | 0.74 | 0.094 |
| [{(bpy)2Ru(dpp)}2RhBr2]5+ | 3.5 | 2.9 | 0.77 | 0.066 |
| [{(phen)2Ru(dpp)}2RhCl2]5+ | 3.8 | 3.5 | 0.79 | 0.060 |
| [{(phen)2Ru(dpp)}2RhBr2]5+ | 4.3 | 5.5 | 0.82 | 0.063 |
| [{(Ph2phen)2Ru(dpp)}2RhCl2]5+ | 2.5 | 1.3 | 0.73 | 0.056 |
| [{(Ph2phen)2Ru(dpp)}2RhBr2]5+ | 4.2 | 2.4 | 0.79 | 0.047 |
| Complex a | H2 (μmol) | TON b | ΦH2 c |
|---|---|---|---|
| [{(bpy)2Ru(dpp)}2RhCl2]5+ | 7.2 ± 0.7 | 25 ± 2 | 0.0025 |
| [{(bpy)2Ru(dpp)}2RhBr2]5+ | 8.9 ± 0.4 | 31 ± 1 | 0.0055 |
| [{(phen)2Ru(dpp)}2RhCl2]5+ | 4.1 ± 0.2 | 14 ± 1 | 0.0017 |
| [{(phen)2Ru(dpp)}2RhBr2]5+ | 5.9 ± 0.7 | 20 ± 3 | 0.0026 |
| [{(Ph2phen)2Ru(dpp)}2RhCl2]5+ | 33 ± 3 | 110 ± 10 | 0.012 |
| [{(Ph2phen)2Ru(dpp)}2RhBr2]5+ | 40 ± 4 | 140 ± 10 | 0.019 |
3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Electronic Absorption Spectroscopy
3.2.2. Steady State Luminescence Spectroscopy
3.2.3. Excited State Emission Quenching
3.2.4. Photochemical Product Formation
3.2.5. Photocatalytic Hydrogen Production
4. Conclusions
Acknowledgments
References
- McDaniel, N.D.; Bernhard, S. Solar fuels: Thermodynamics, candidates, tactics, and figures of merit. Dalton Trans. 2010, 39, 10021–10030. [Google Scholar] [PubMed]
- Teets, T.S.; Nocera, D.G. Photocatalytic hydrogen production. Chem. Commun. 2011, 47, 9268–9274. [Google Scholar] [CrossRef]
- Bard, A.J.; Fox, M.A. Artificial photosynthesis: Solar splitting of water to hydrogen and oxygen. Acc. Chem. Res. 1995, 28, 141–145. [Google Scholar] [CrossRef]
- Balzani, V.; Juris, A.; Venturi, M.; Campagna, S.; Serroni, S. Luminescent and redox-active polynuclear transition metal complexes. Chem. Rev. 1996, 96, 759–834. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Moggi, L.; Scandola, F. Supramolecular Photochemistry; Balzani, V., Ed.; Reidel: Dordrecht, The Netherlands, 1987; pp. 1–28. [Google Scholar]
- Molnar, S.M.; Nallas, G.; Bridgewater, J.S.; Brewer, K.J. Photoinitiated electron collection in a mixed-metal trimetallic complex of the form [{(bpy)2Ru(dpb)]2IrCl2}(PF6)5 (bpy = 2,2′-bipyridine and dpb = 2,3-bis(2-pyridyl)benzoquinoxaline). J. Am. Chem. Soc. 1994, 116, 5206–5210. [Google Scholar] [CrossRef]
- Konduri, R.; Ye, H.; MacDonnell, F.M.; Serroni, S.; Campagna, S.; Rajeshwar, K. Ruthenium photocatalysts capable of reversibly storing up to four electrons in a single acceptor ligand: A step closer to artificial photosynthesis. Angew. Chem. Int. Ed. 2002, 41, 3185–3187. [Google Scholar] [CrossRef]
- Kim, M.-J.; Konduri, R.; Ye, H.; MacDonnell, F.M.; Puntoriero, F.; Serroni, S.; Campagna, S.; Holder, T.; Kinsel, G.; Rajeshwar, K. Dinuclear Ruthenium(II) polypyridyl complexes containing large, redox-active, aromatic bridging ligands: Synthesis, characterization, and intramolecular quenching of MLCT excited states. Inorg. Chem. 2002, 41, 2471–2476. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, D.; Cabelli, D.; Muckerman, J.T.; Fujita, E.; Koizumi, T.; Fukushima, T.; Wada, T.; Tanaka, K. Photochemical and radiolytic production of an organic hydride donor with a Ru-II complex containing an NAD(+) model ligand. Angew. Chem. Int. Ed. 2007, 46, 4169–4172. [Google Scholar] [CrossRef]
- Polyansky, D.E.; Cabelli, D.; Muckerman, J.T.; Fukushima, T.; Tanaka, K.; Fujita, E. Mechanism of hydride donor generation using a Ru(II) complex containing an NAD(+) model ligand: Pulse and steady-state radiolysis studies. Inorg. Chem. 2008, 47, 3958–3968. [Google Scholar] [CrossRef] [PubMed]
- Elvington, M.; Brewer, K.J. Photoinitiated electron collection at a metal in a rhodium-centered mixed-metal supramolecular complex. Inorg. Chem. 2006, 45, 5242–5244. [Google Scholar] [CrossRef] [PubMed]
- Arachchige, S.M.; Brown, J.; Brewer, K.J. Photochemical hydrogen production from water using the new photocatalyst [{(bpy)2Ru(dpp)}2RhBr2](PF6)5. J. Photochem. Photobiol. A Chem. 2008, 197, 13–17. [Google Scholar] [CrossRef]
- Arachchige, S.M.; Brown, J.R.; Chang, E.; Jain, A.; Zigler, D.F.; Rangan, K.; Brewer, K.J. Design considerations for a system for photocatalytic hydrogen production from water employing mixed-metal photochemical molecular devices for photoinitiated electron collection. Inorg. Chem. 2009, 48, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Elvington, M.; Brown, J.; Arachchige, S.M.; Brewer, K.J. Photocatalytic hydrogen production from water employing a Ru, Rh, Ru molecular device for photoinitiated electron collection. J. Am. Chem. Soc. 2007, 129, 10644–10645. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Higgins, S.L.H.; Arachchige, S.M.; Brewer, K.J. Efficient photocatalytic hydrogen production in a single-component system using Ru,Rh,Ru supramolecules containing 4,7-diphenyl-1,10-phenanthroline. Angew. Chem. Int. Ed. 2011, 50, 12209–12213. [Google Scholar] [CrossRef]
- White, T.A.; Rangan, K.; Brewer, K.J. Synthesis, characterization, and study of the photophysics and photocatalytic properties of the photoinitiated electron collector [{(phen)2Ru(dpp)}2RhBr2](PF6)5. J. Photochem. Photobiol. A Chem. 2010, 209, 203–209. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Nelson, J.; Kirkpatrick, J.; Ravirajan, P. Factors limiting the efficiency of molecular photovoltaic devices. Phys. Rev. B 2004, 69, 035337:1–035337:11. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Kochi, J.K. Fresh look at electron-transfer mechanisms via the donor/acceptor bindings in the critical encounter complex. Acc. Chem. Res. 2008, 41, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Navon, G.; Sutin, N. Mechanism of the quenching of the phosphorescence of tris(2,2′-bipyridine) ruthenium(II) by some cobalt(III) and ruthenium(III) complexes. Inorg. Chem. 1974, 13, 2159–2164. [Google Scholar] [CrossRef]
- Sutin, N.; Creutz, C. Electron-transfer reactions of excited states. J. Chem. Educ. 1983, 60, 809–814. [Google Scholar] [CrossRef]
- Bock, C.R.; Connor, J.A.; Gutierrez, A.R.; Meyer, T.J.; Whitten, D.G.; Sullivan, B.P.; Nagle, J.K. Estimation of excited-state redox potentials by electron-transfer quenching. Application of electron-transfer theory to excited-state redox processes. J. Am. Chem. Soc. 1979, 101, 4815–4824. [Google Scholar] [CrossRef]
- Creutz, C.; Sutin, N. Electron-transfer reactions of excited states: Direct evidence for reduction of the charge-transfer excited state of tris(2,2′-bipyridine)ruthenium(II). J. Am. Chem. Soc. 1976, 98, 6384–6385. [Google Scholar] [CrossRef]
- Indelli, M.T.; Bignozzi, C.A.; Harriman, A.; Schoonover, J.R.; Scandola, F. Four intercomponent processes in a Ru(II)-Rh(III) polypyridine dyad: Electron transfer from excited donor, electron transfer to excited acceptor, charge recombination, and electronic energy transfer. J. Am. Chem. Soc. 1994, 116, 3768–3779. [Google Scholar] [CrossRef]
- Indelli, M.T.; Chiorboli, C.; Flamigni, L.; de Cola, L.; Scandola, F. Photoinduced electron transfer across oligo-p-phenylene bridges. Distance and conformational effects in Ru(II)/Rh(III) dyads. Inorg. Chem. 2007, 46, 5630–5641. [Google Scholar] [CrossRef] [PubMed]
- Indelli, M.T.; Scandola, F.; Collin, J.-P.; Sauvage, J.-P.; Sour, A. Photoinduced electron and energy transfer in rigidly bridged Ru(II)/Rh(III) binuclear complexes. Inorg. Chem. 1996, 35, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Indelli, M.T.; Scandola, F.; Flamigni, L.; Collin, J.P.; Sauvage, J.P.; Sour, A. Photoinduced electron transfer in ruthenium(II)/rhodium(III) terpyridine dyads. Inorg. Chem. 1997, 36, 4247–4250. [Google Scholar] [CrossRef]
- Furue, M.; Hirata, M.; Kinoshita, S.; Kushida, T.; Kamachi, M. Intramolecular electron-transfer of covalently-linked polypyridine ruthenium(II)/rhodium(III) binuclear complexes in the excited state. Observation of the marcus inverted region. Chem. Lett. 1990, 19, 2065–2068. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Graetzel, M.; Nazeeruddin, M.K. Excited-state interactions in ligand-bridged chromophore-quencher complexes containing rhodium(III) and ruthenium(II) polypyridyl units. J. Phys. Chem. 1992, 96, 5865–5872. [Google Scholar] [CrossRef]
- Lee, J.-D.; Vrana, L.M.; Bullock, E.R.; Brewer, K.J. A tridentate-bridged ruthenium/rhodium complex as a stereochemically defined light-absorber/electron-acceptor Dyad. Inorg. Chem. 1998, 37, 3575–3580. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Arachchige, S.; Sedai, B.; Brewer, K. Emission spectroscopy as a probe into photoinduced intramolecular electron transfer in polyazine bridged Ru(II),Rh(III) supramolecular complexes. Materials 2010, 3, 4328–4354. [Google Scholar] [CrossRef]
- Zigler, D.F.; Wang, J.; Brewer, K.J. Ruthenium(II)-polyazine light absorbers bridged to reactive cis-dichlororhodium(III) centers in a bimetallic molecular architecture. Inorg. Chem. 2008, 47, 11342–11350. [Google Scholar] [CrossRef] [PubMed]
- Kew, G.; DeArmond, K.; Hanck, K. Electrochemistry of rhodium-dipyridyl complexes. J. Phys. Chem. 1974, 78, 727–734. [Google Scholar] [CrossRef]
- Rasmussen, S.C.; Richter, M.M.; Yi, E.; Place, H.; Brewer, K.J. Synthesis and characterization of a series of novel rhodium and iridium complexes containing polypyridyl bridging ligands: Potential uses in the development of multimetal catalysts for carbon dioxide reduction. Inorg. Chem. 1990, 29, 3926–3932. [Google Scholar] [CrossRef]
- Anderson, C.P.; Salmon, D.J.; Meyer, T.J.; Young, R.C. Photochemical generation of Ru(bpy)3+ and O2−. J. Am. Chem. Soc. 1977, 99, 1980–1982. [Google Scholar] [CrossRef]
- Haga, M.; Dodsworth, E.S.; Eryavec, G.; Seymour, P.; Lever, A.B.P. Luminescence quenching of the tris(2,2′-bipyrazine)ruthenium(II) cation and its monoprotonated complex. Inorg. Chem. 1985, 24, 1901–1906. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Berlin, Heidelberg, Germany, 2006. [Google Scholar]
- Ballardini, R.; Varani, G.; Indelli, M.T.; Scandola, F.; Balzani, V. Free energy correlation of rate constants for electron transfer quenching of excited transition metal complexes. J. Am. Chem. Soc. 1978, 100, 7219–7223. [Google Scholar] [CrossRef]
- Brown, J.R.; Elvington, M.; Mongelli, M.T.; Zigler, D.F.; Brewer, K.J. Analytical methods development for supramolecular design in solar hydrogen production. Proc. SPIE 2006, 6340. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
White, T.A.; Knoll, J.D.; Arachchige, S.M.; Brewer, K.J. A Series of Supramolecular Complexes for Solar Energy Conversion via Water Reduction to Produce Hydrogen: An Excited State Kinetic Analysis of Ru(II),Rh(III),Ru(II) Photoinitiated Electron Collectors. Materials 2012, 5, 27-46. https://doi.org/10.3390/ma5010027
White TA, Knoll JD, Arachchige SM, Brewer KJ. A Series of Supramolecular Complexes for Solar Energy Conversion via Water Reduction to Produce Hydrogen: An Excited State Kinetic Analysis of Ru(II),Rh(III),Ru(II) Photoinitiated Electron Collectors. Materials. 2012; 5(1):27-46. https://doi.org/10.3390/ma5010027
Chicago/Turabian StyleWhite, Travis A., Jessica D. Knoll, Shamindri M. Arachchige, and Karen J. Brewer. 2012. "A Series of Supramolecular Complexes for Solar Energy Conversion via Water Reduction to Produce Hydrogen: An Excited State Kinetic Analysis of Ru(II),Rh(III),Ru(II) Photoinitiated Electron Collectors" Materials 5, no. 1: 27-46. https://doi.org/10.3390/ma5010027
APA StyleWhite, T. A., Knoll, J. D., Arachchige, S. M., & Brewer, K. J. (2012). A Series of Supramolecular Complexes for Solar Energy Conversion via Water Reduction to Produce Hydrogen: An Excited State Kinetic Analysis of Ru(II),Rh(III),Ru(II) Photoinitiated Electron Collectors. Materials, 5(1), 27-46. https://doi.org/10.3390/ma5010027