Calcium Orthophosphates in Nature, Biology and Medicine

Abstract
:1. Introduction
2. Geological and Biological Ooccurrence of Calcium Orthophosphates

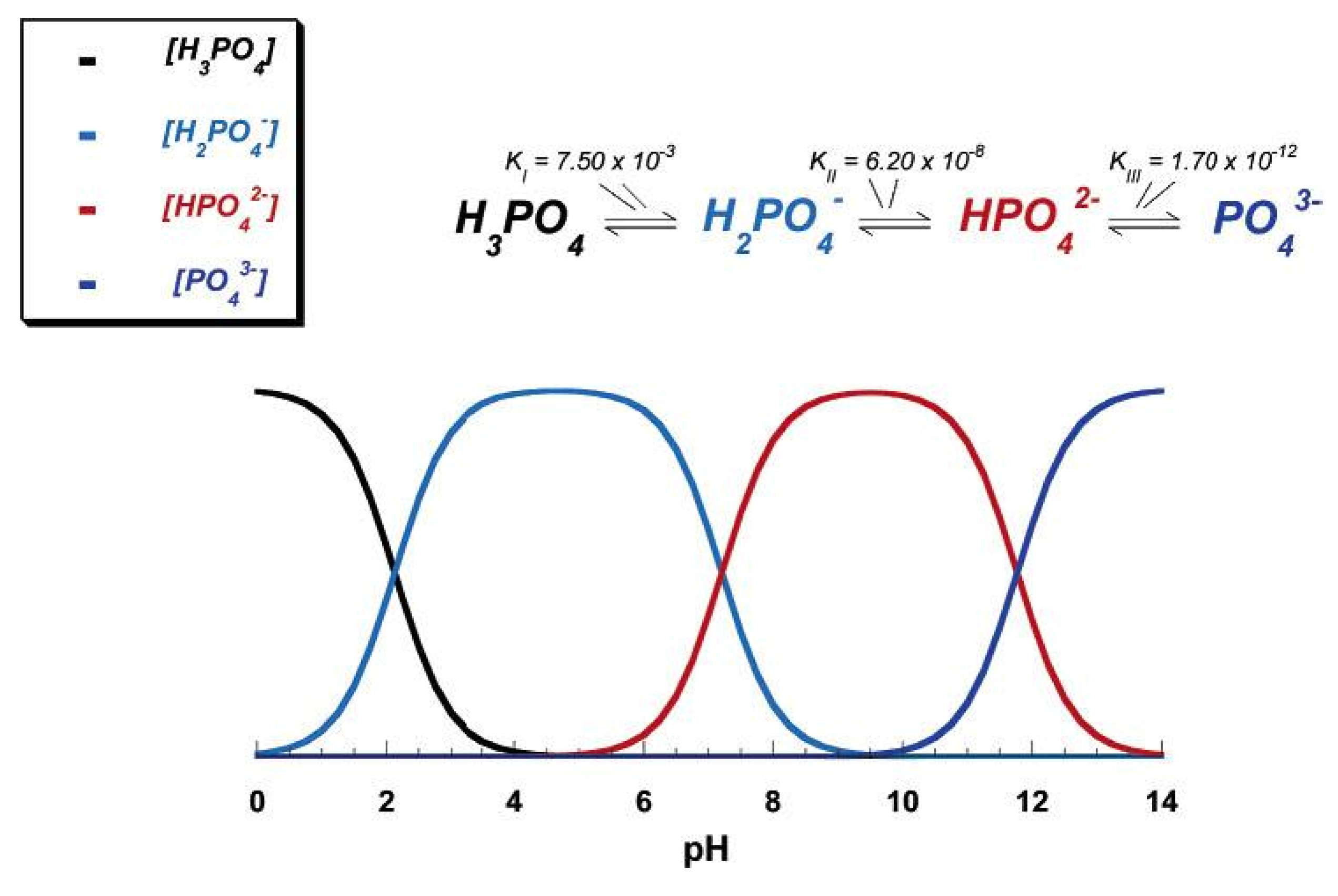
3. The Members of the Calcium Orthophosphate Family

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition, wt.% | Enamel | Dentin | Cementum | Bone | HA |
|---|---|---|---|---|---|
| Calcium[a] | 36.5 | 35.1 | [c] | 34.8 | 39.6 |
| Phosphorus (as P)[a] | 17.7 | 16.9 | [c] | 15.2 | 18.5 |
| Ca/P (molar ratio)[a] | 1.63 | 1.61 | [c] | 1.71 | 1.67 |
| Sodium[a] | 0.5 | 0.6 | [c] | 0.9 | - |
| Magnesium[a] | 0.44 | 1.23 | [c] | 0.72 | - |
| Potassium[a] | 0.08 | 0.05 | [c] | 0.03 | - |
| Carbonate (as CO32-)[b] | 3.5 | 5.6 | [c] | 7.4 | - |
| Fluoride[a] | 0.01 | 0.06 | [c] | 0.03 | - |
| Chloride[a] | 0.30 | 0.01 | [c] | 0.13 | - |
| Pyrophosphate (as P2O74-)[b] | 0.022 | 0.10 | [c] | 0.07 | - |
| Total inorganic[b] | 97 | 70 | 60 | 65 | 100 |
| Total organic[b] | 1.5 | 20 | 25 | 25 | - |
| Water[b] | 1.5 | 10 | 15 | 10 | - |
| Crystallographic properties: Lattice parameters ( ± 0.003 Å) | |||||
| a-axis, Å | 9.441 | 9.421 | [c] | 9.41 | 9.430 |
| c-axis, Å | 6.880 | 6.887 | [c] | 6.89 | 6.891 |
| Crystallinity index, (HA = 100) | 70 – 75 | 33 – 37 | [c] | 33 – 37 | 100 |
| Typical crystal sizes (nm) [311, 362, 364] | 105×50×50 | 35×25×4 | [c] | 50×25×4 | 200 – 600 |
| Ignition products (800 ºC) | β-TCP + HA | β-TCP+ HA | β-TCP+ HA | HA + CaO | HA |
| Elastic modulus (GPa) | 80 | 15 | [c] | 0.34 – 13.8 | 10 |
| Tensile strength (MPa) | 10 | 100 | [c] | 150 | 100 |
| Ca/P ionic ratio | Compound | Chemical formula | Solubility at 25 ºC, –log(Ks) | Solubility at 25 ºC, g/L | pH stability range in aqueous solutions at 25°C |
|---|---|---|---|---|---|
| 0.5 | Monocalcium phosphate monohydrate (MCPM) | Ca(H2PO4)2·H2O | 1.14 | ~ 18 | 0.0 – 2.0 |
| 0.5 | Monocalcium phosphate anhydrous (MCPA) | Ca(H2PO4)2 | 1.14 | ~ 17 | [c] |
| 1.0 | Dicalcium phosphate dihydrate (DCPD), mineral brushite | CaHPO4·2H2O | 6.59 | ~ 0.088 | 2.0 – 6.0 |
| 1.0 | Dicalcium phosphate anhydrous (DCPA), mineral monetite | CaHPO4 | 6.90 | ~ 0.048 | [c] |
| 1.33 | Octacalcium phosphate (OCP) | Ca8(HPO4)2(PO4)4·5H2O | 96.6 | ~ 0.0081 | 5.5 – 7.0 |
| 1.5 | α-Tricalcium phosphate (α-TCP) | α-Ca3(PO4)2 | 25.5 | ~ 0.0025 | [a] |
| 1.5 | β-Tricalcium phosphate (β-TCP) | β-Ca3(PO4)2 | 28.9 | ~ 0.0005 | [a] |
| 1.2 – 2.2 | Amorphous calcium phosphate (ACP) | CaxHy(PO4)z·nH2O, n = 3 – 4.5; 15 – 20% H2O | [b] | [b] | ~ 5 – 12 [d] |
| 1.5 – 1.67 | Calcium-deficient hydroxyapatite (CDHA)[e] | Ca10- x(HPO4)x(PO4)6-x(OH)2-x[f] (0<x<1) | ~ 85.1 | ~ 0.0094 | 6.5 – 9.5 |
| 1.67 | Hydroxyapatite (HA) | Ca10(PO4)6(OH)2 | 116.8 | ~ 0.0003 | 9.5 – 12 |
| 1.67 | Fluorapatite (FA) | Ca10(PO4)6F2 | 120.0 | ~ 0.0002 | 7 – 12 |
| 2.0 | Tetracalcium phosphate (TTCP), mineral hilgenstockite | Ca4(PO4)2O | 38 – 44 | ~ 0.0007 | [a] |
| Compound | Space group | Unit cell parameters | Z[a] | Density, g cm-3 |
|---|---|---|---|---|
| MCPM | triclinic P triclinic P | a = 5.6261(5), b = 11.889(2), c = 6.4731(8) Å, | 2 | 2.23 |
| α = 98.633(6)º, β = 118.262(6)º, γ = 83.344(6)º | ||||
| MCPA | triclinic P triclinic P | a = 7.5577(5), b = 8.2531(6), c = 5.5504(3) Å, | 2 | 2.58 |
| α = 109.87(1)º, β = 93.68(1)º, γ = 109.15(1)º | ||||
| DCPD | monoclinic Ia | a = 5.812(2), b = 15.180(3), c = 6.239(2) Å, β = 116.42(3)º | 4 | 2.32 |
| DCPA | triclinic P triclinic P | a = 6.910(1), b = 6.627(2), c = 6.998(2) Å, | 4 | 2.89 |
| α = 96.34(2)º, β = 103.82(2)º, γ = 88.33(2)º | ||||
| OCP | triclinic P triclinic P | a = 19.692(4), b = 9.523(2), c = 6.835(2) Å, α = 90.15(2)º, β = 92.54(2)º, γ = 108.65(1)º | 1 | 2.61 |
| α-TCP | monoclinic P21/a | a = 12.887(2), b = 27.280(4), c = 15.219(2) Å, β = 126.20(1)º | 24 | 2.86 |
| β-TCP | rhombohedral R3cH | a = b = 10.4183(5), c = 37.3464(23) Å, γ = 120° | 21[b] | 3.08 |
| HA | monoclinic P21/b | a = 9.84214(8), b = 2a, c = 6.8814(7) Å, γ = 120° (monoclinic); | 4 | 3.16 |
| or hexagonal P63/m | a = b = 9.4302(5), c = 6.8911(2) Å, γ = 120º (hexagonal) | 2 | ||
| FA | hexagonal P63/m | a = b = 9.367, c = 6.884 Å, γ = 120º | 2 | 3.20 |
| TTCP | monoclinic P21 | a = 7.023(1), b = 11.986(4), c = 9.473(2) Å, β = 90.90(1)º | 4 | 3.05 |
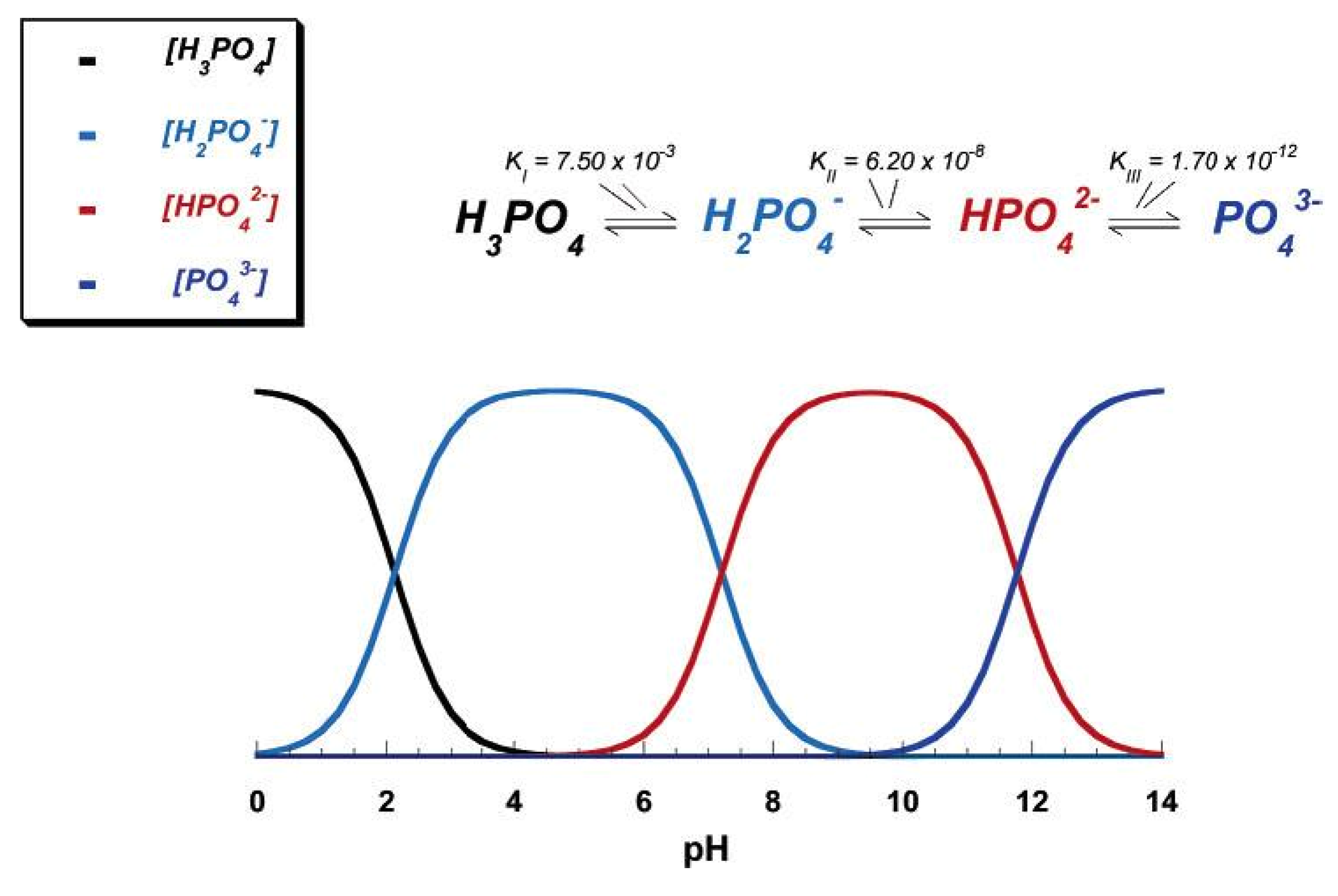
3.1. MCPM
3.2. MCPA
3.3. DCPD
3.4. DCPA
3.5. OCP
3.6. β-TCP
3.7. α-TCP
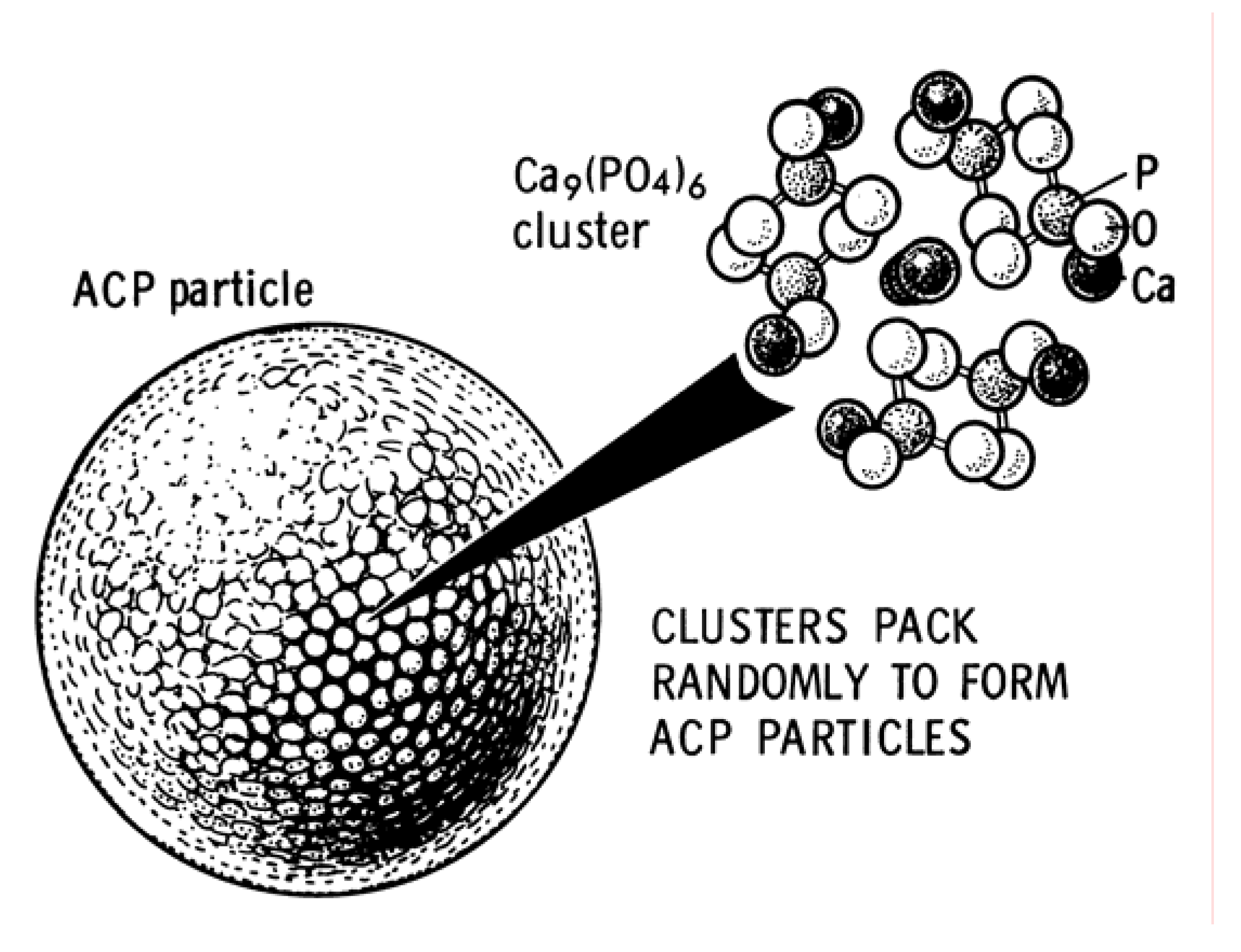
3.8. ACP
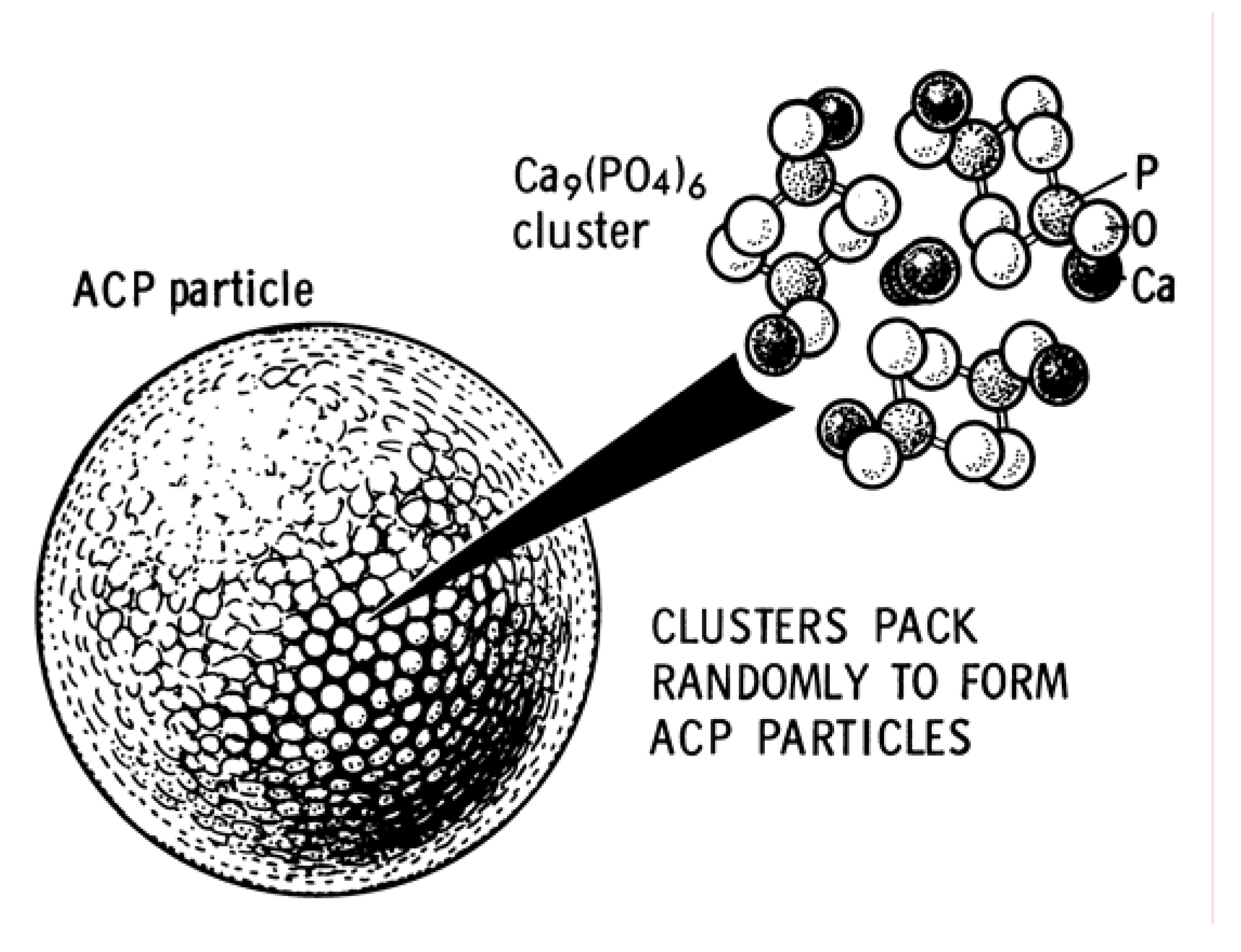
3.9. CDHA
3.10. HA
3.11. FA
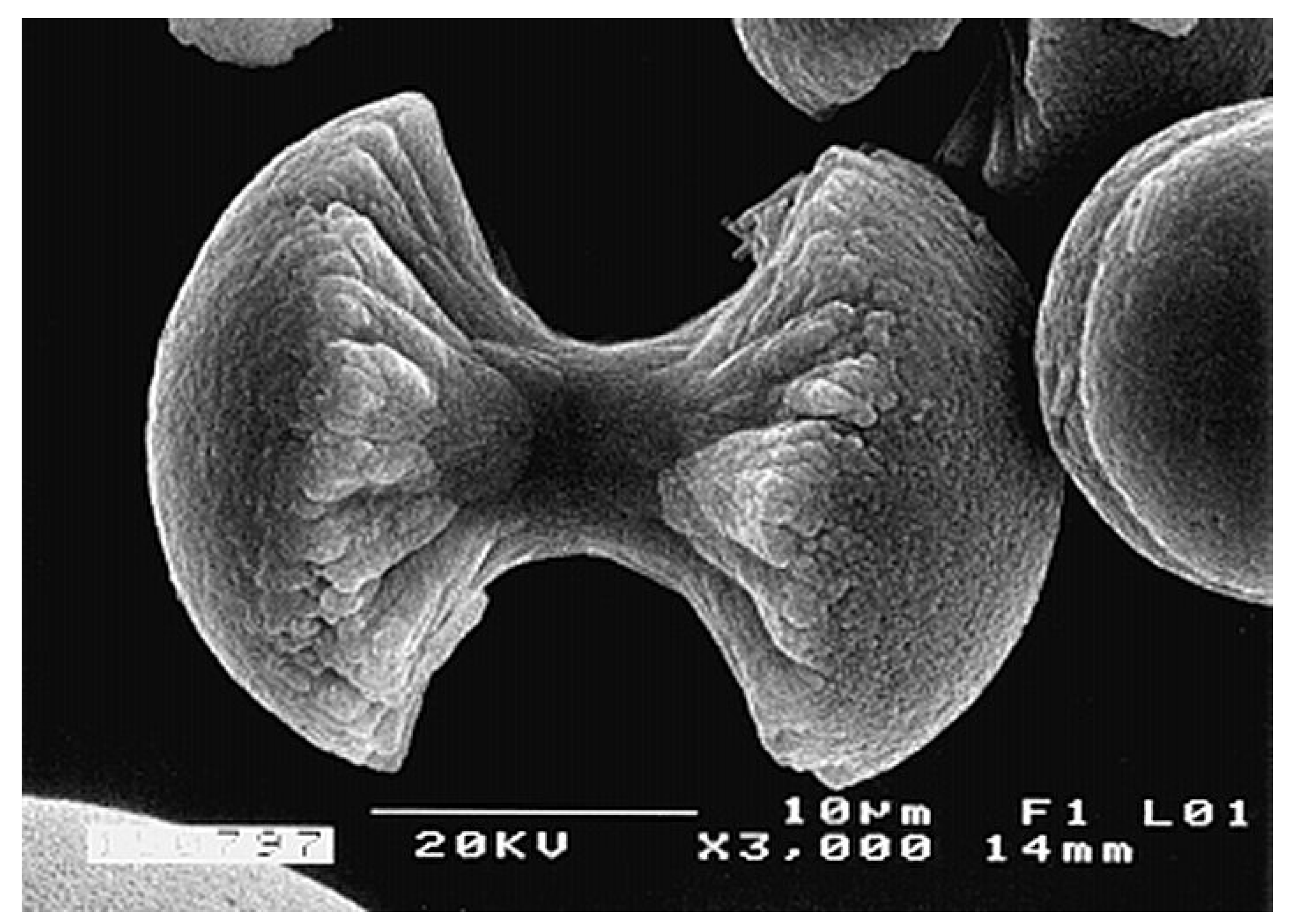
3.12. TTCP
3.13. Substituted Calcium Orthophosphates
4. Biological Calcium Orthophosphate Hard Tissues
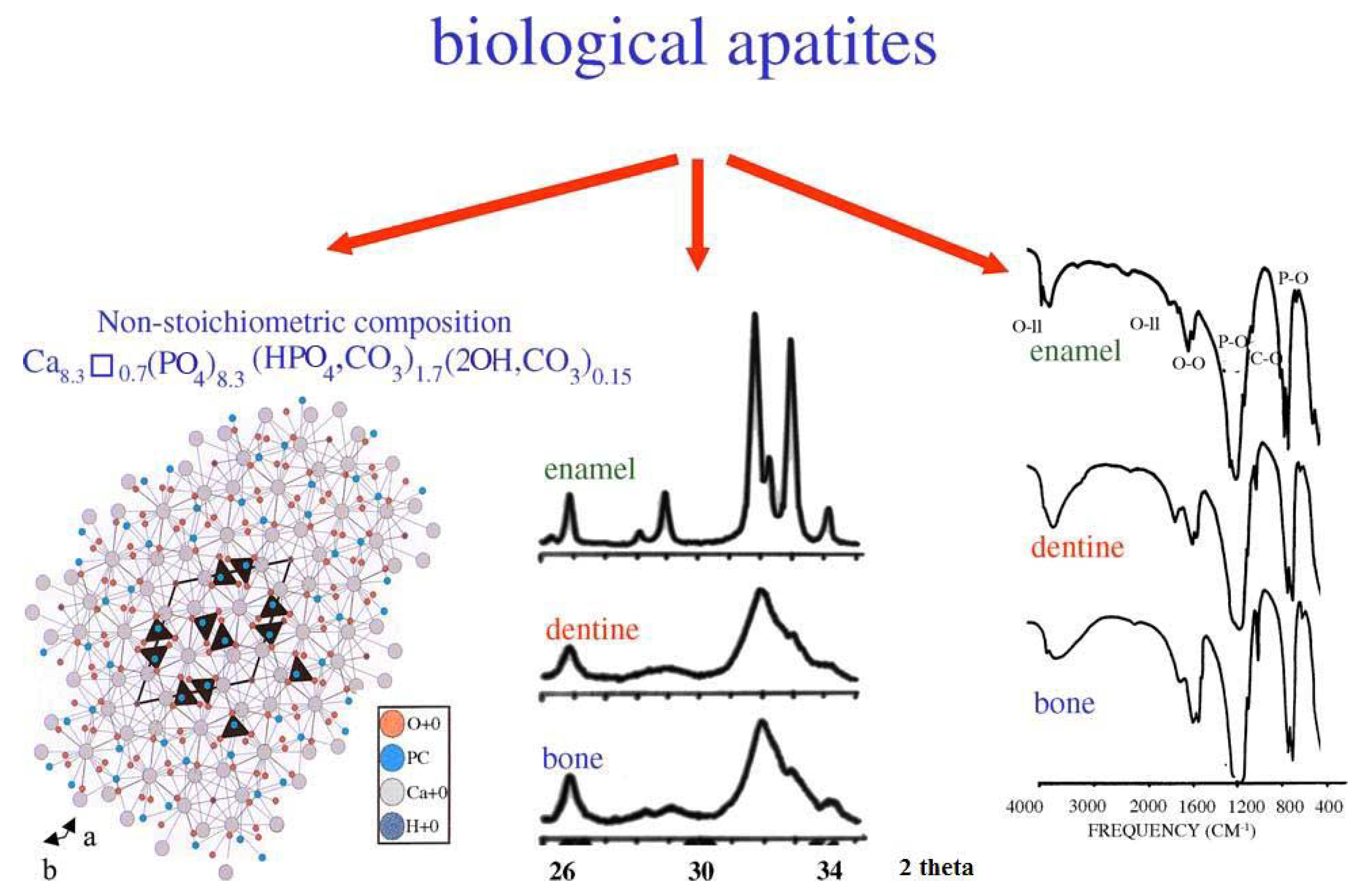
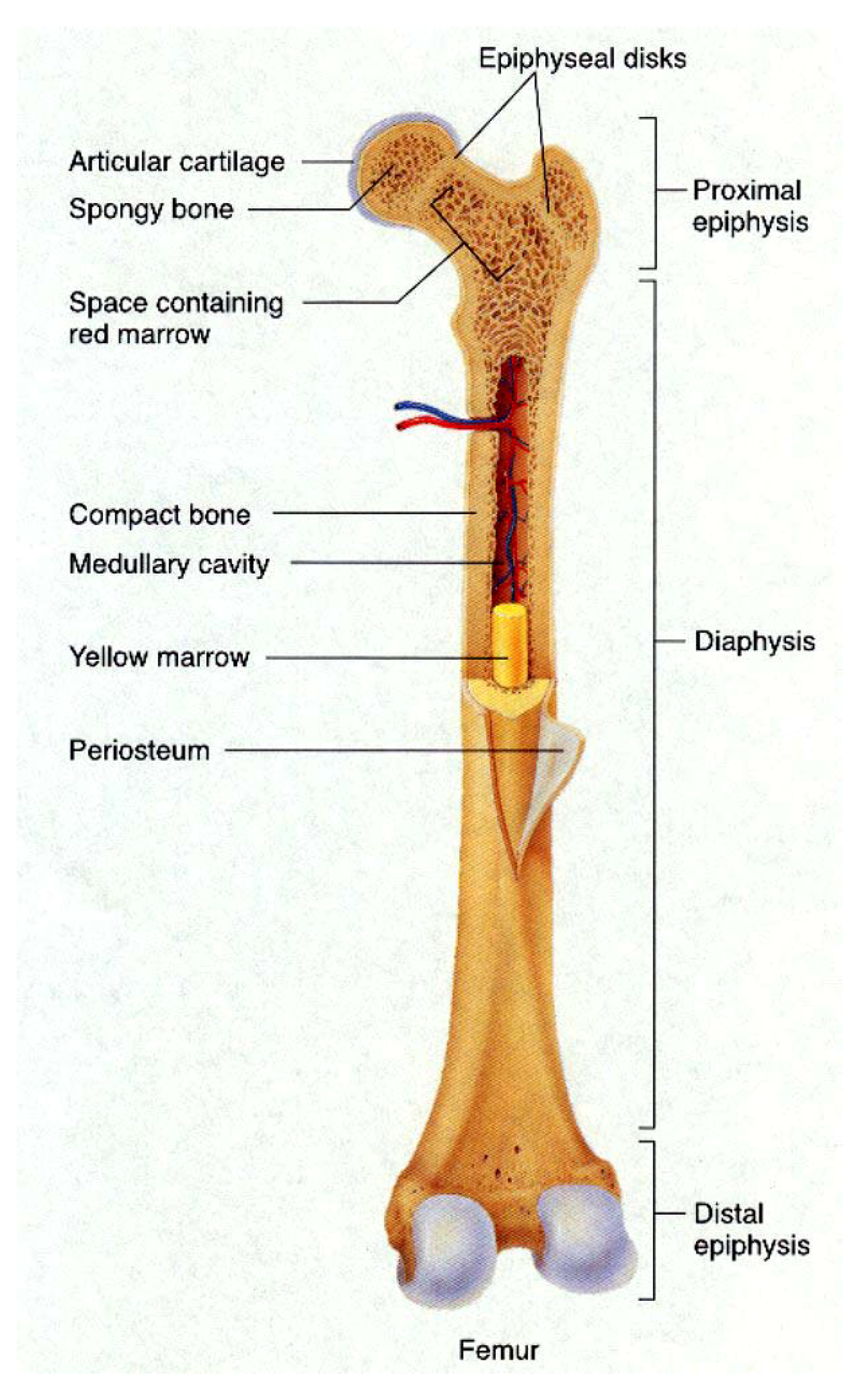
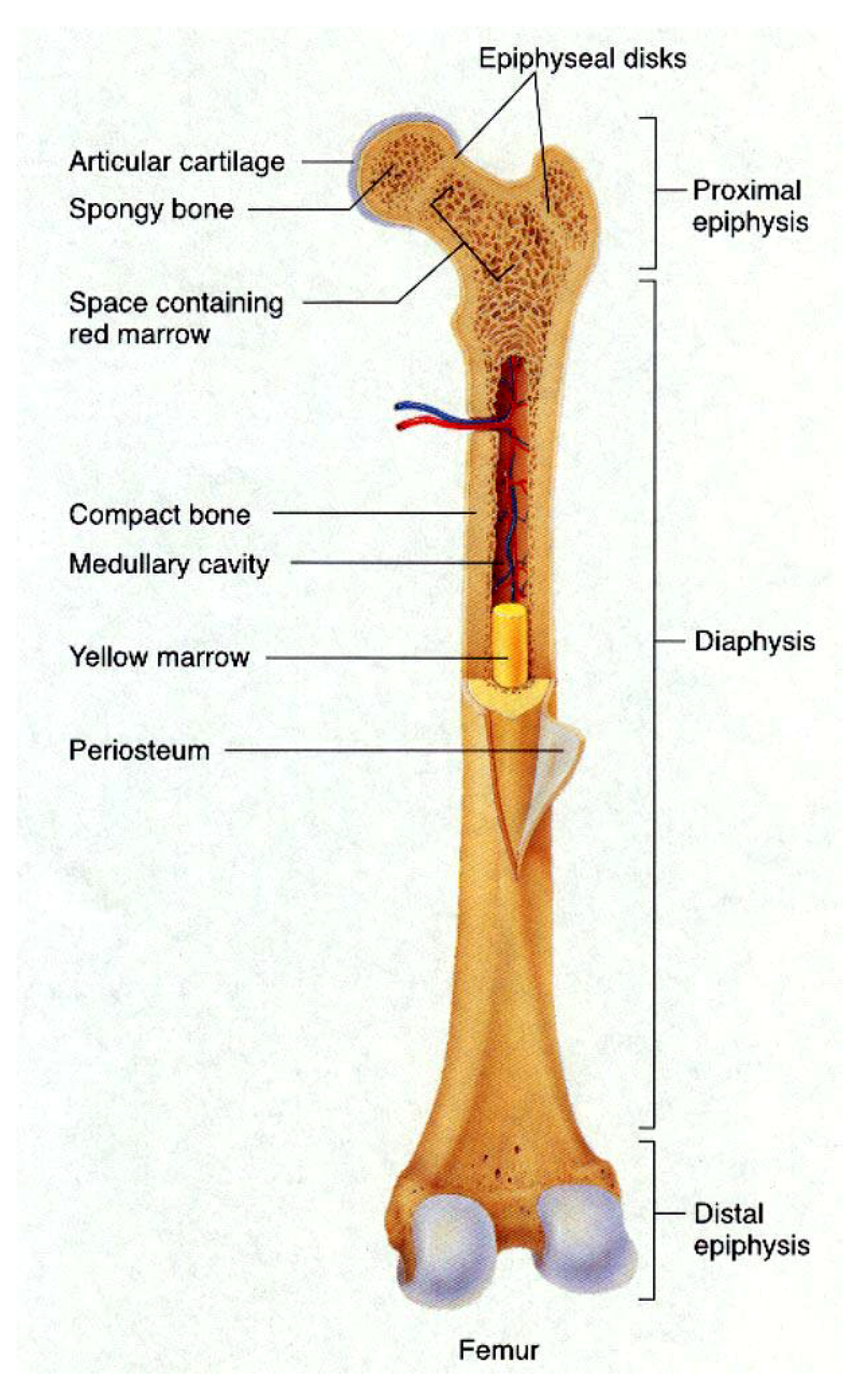
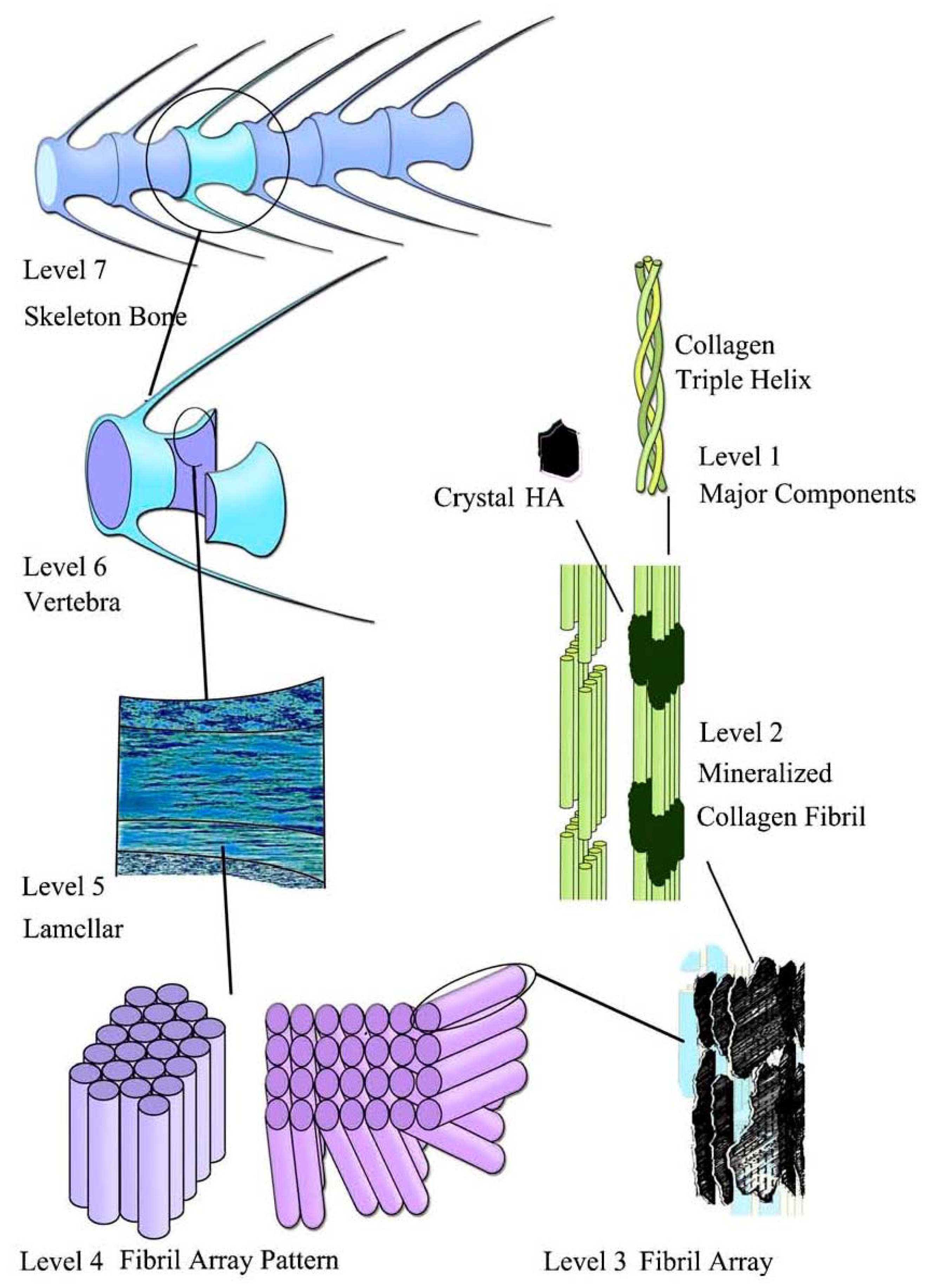
4.1. Bone
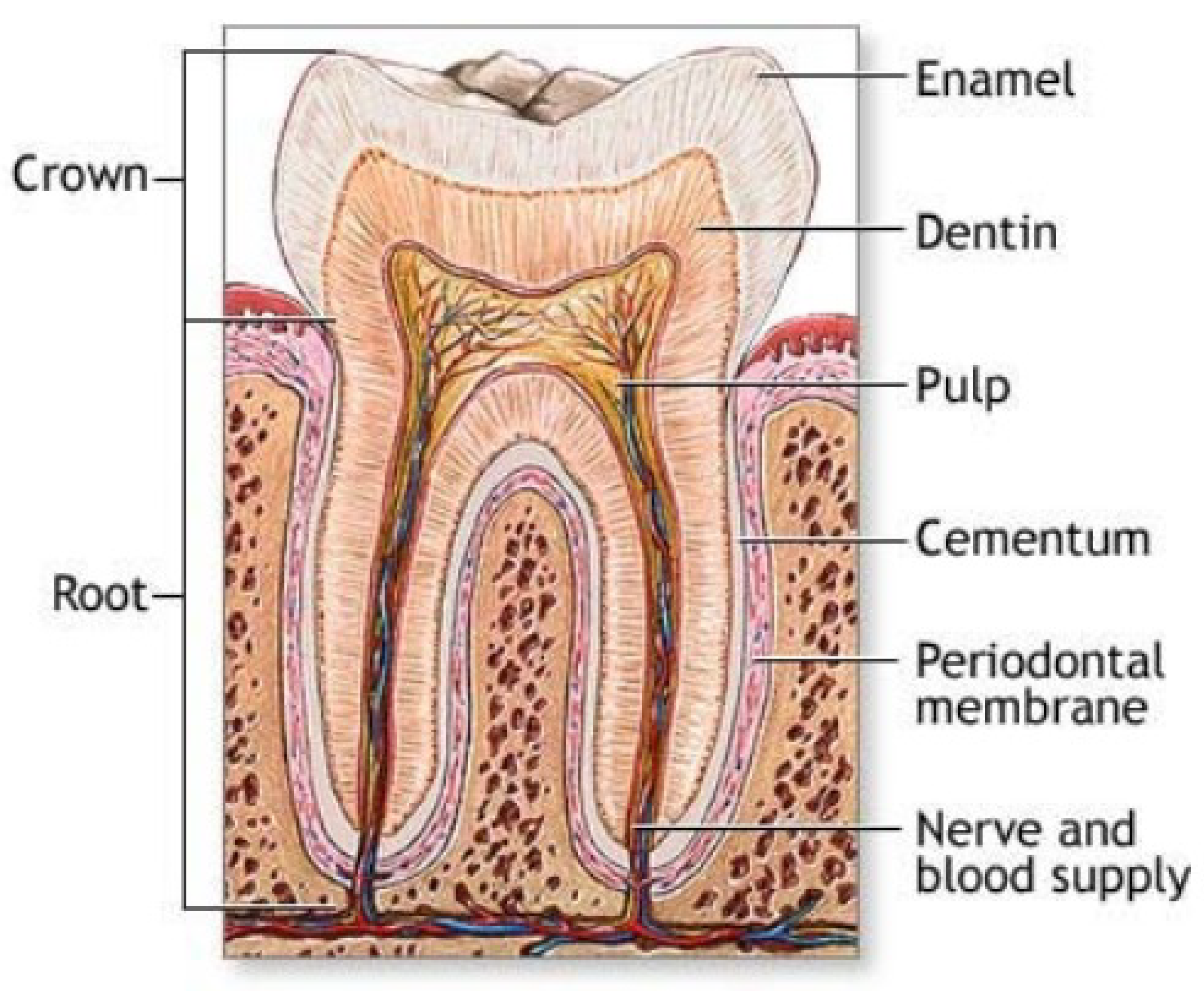
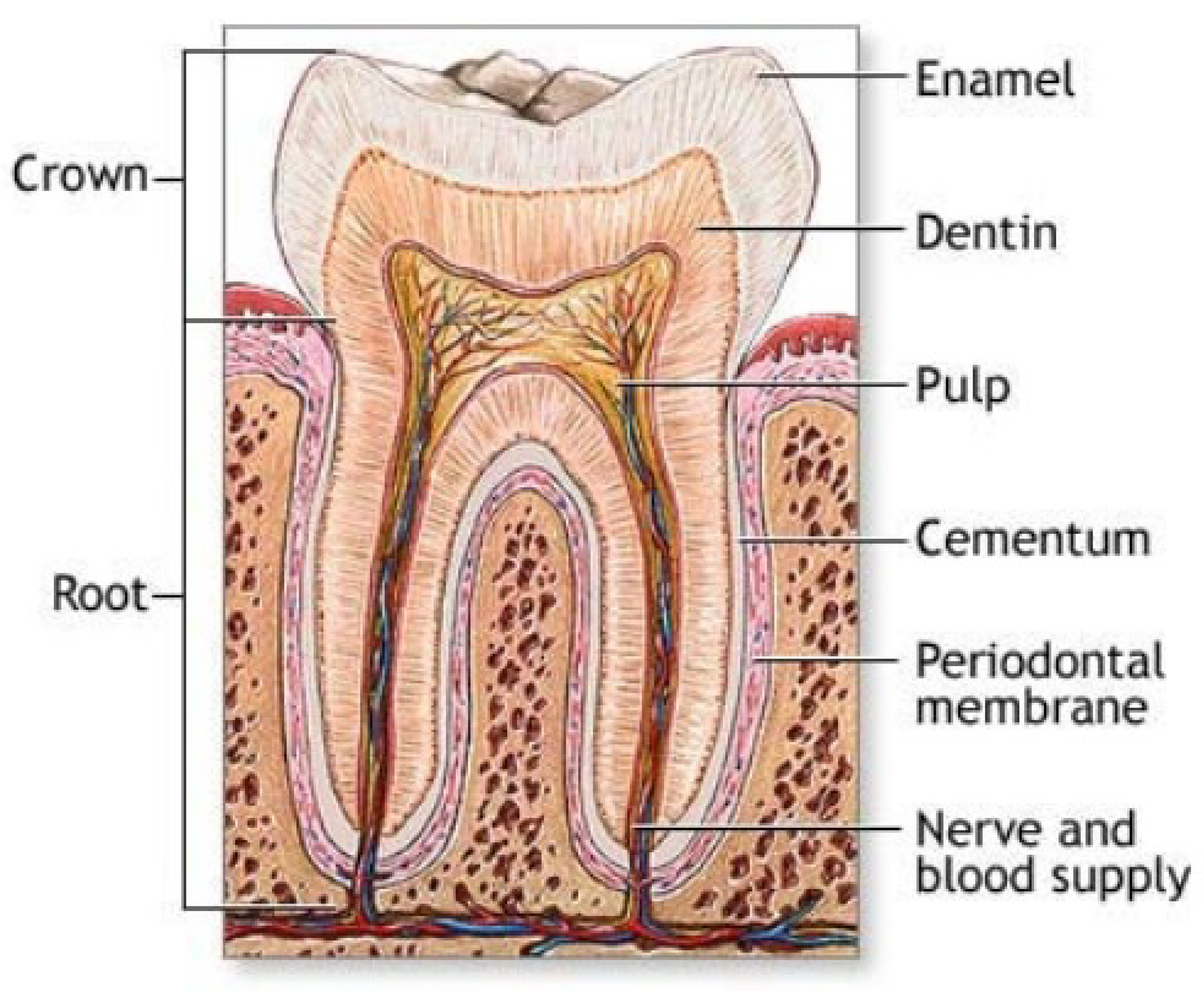
4.2. Teeth

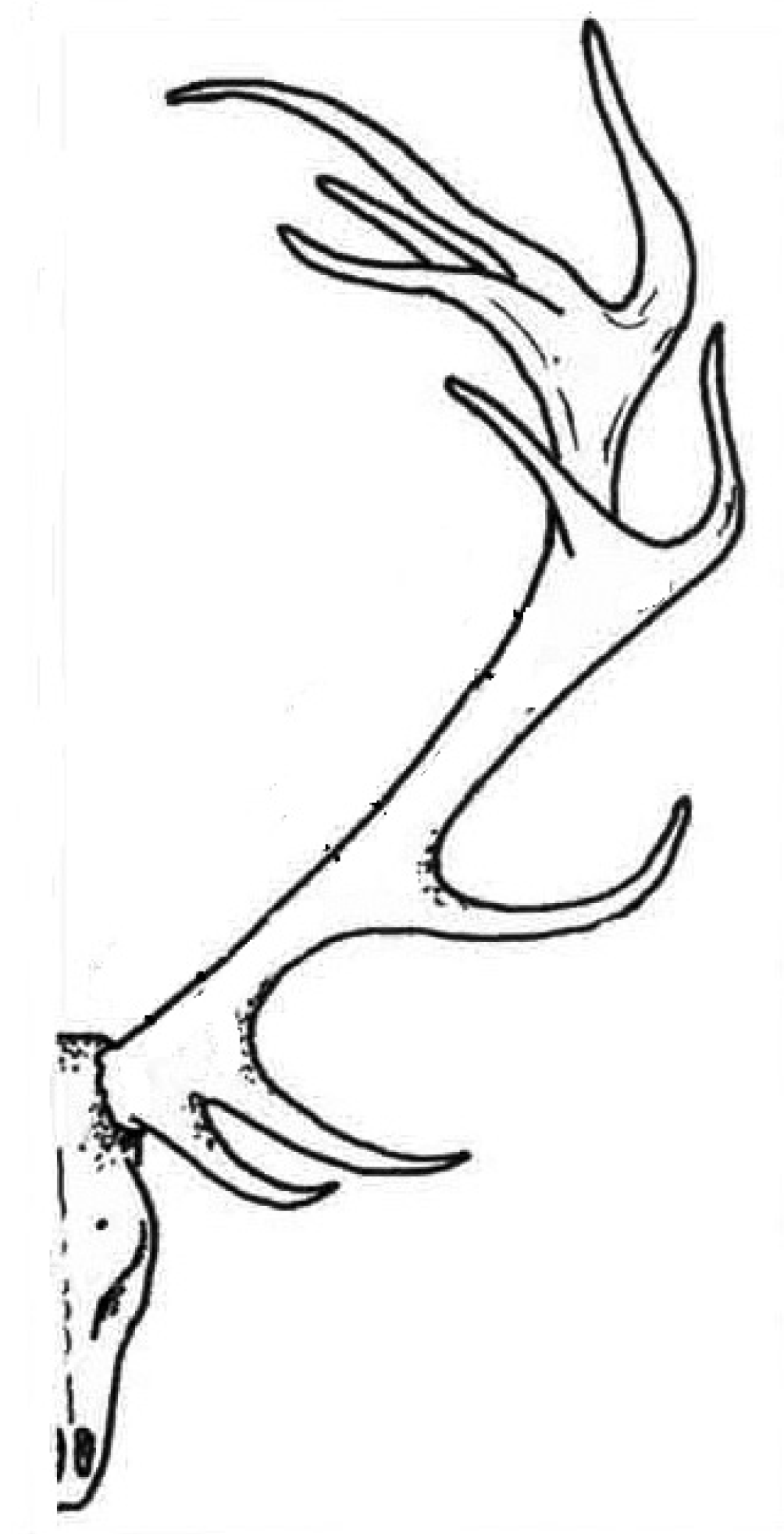
4.3. Antlers
5. Pathological Calcification of Calcium Orthophosphates
| Calcium phosphate | Occurrence |
|---|---|
| biological apatite | enamel, dentin, bone, dental calculi, stones, urinary stones, soft-tissue deposits |
| OCP | dental calculi and urinary stones |
| DCPD | dental calculi, crystalluria, chrondrocalcinosis, in some carious lesions |
| β-(Ca, Mg)3(PO4)2 | dental calculi, salivary stones, arthritic cartilage, soft-tissue deposits |
| Ca2P2O7·2H2O | pseudo-gout deposits in synovium fluids |
| ACP | heart calcifications in uremic patients, kidney stones |
6. Calcium Orthophosphates as Biomaterials and Bioceramics
| Technique | Thickness | Advantages | Disadvantages |
|---|---|---|---|
| Thermal spraying | 30 – 200 μm | High deposition rates; low cost | Line of sight technique; high temperatures induce decomposition; rapid cooling produces amorphous coatings |
| Sputter coating | 0.5 – 3 μm | Uniform coating thickness on flat substrates; dense coating | Line of sight technique; expensive; time consuming; produces amorphous coatings |
| Pulsed laser deposition | 0.05-5 μm | Coating by crystalline and amorphous phases; dense and porous coating | Line of sight technique |
| Dynamic mixing method | 0.05-1.3 μm | High adhesive strength | Line of sight technique; expensive; produces amorphous coatings |
| Dip coating | 0.05 – 0.5 mm | Inexpensive; coatings applied quickly; can coat complex substrates | Requires high sintering temperatures; thermal expansion mismatch |
| Sol-gel technique | < 1 μm | Can coat complex shapes; low processing temperatures; relatively cheap as coatings are very thin | Some processes require controlled atmosphere processing; expensive raw materials |
| Electrophoretic deposition | 0.1 – 2.0 mm | Uniform coating thickness; rapid deposition rates; can coat complex substrates | Difficult to produce crack-free coatings; requires high sintering temperatures |
| Biomimetic coating | < 30 μm | Low processing temperatures; can form bonelike apatite; can coat complex shapes; can incorporate bone growth stimulating factors | Time consuming; requires replenishment and a pH constancy of simulated body fluid |
| Hot isostatic pressing | 0.2 – 2.0 μm | Produces dense coatings | Cannot coat complex substrates; high temperature required; thermal expansion mismatch; elastic property differences; expensive; removal/interaction of encapsulation material |
| Electrochemical deposition | 0.05 – 0.5 mm | Uniform coating thickness; rapid deposition rates; can coat complex substrates; moderate temperature, low cost | The coating/substrate bonding is not strong enough |

7. Biomimetic Crystallization of Calcium Orthophosphates
- (i)
- In vitro crystallization normally occurs at permanently depleting concentrations of calcium and orthophosphate, while the concentrations of all ions and molecules are kept strictly constant during biological mineralization (the same is valid for the solution pH);
- (ii)
- Chemical crystallization is a fast process (time scale of minutes to days), while the biological process is a slow one (time scale of weeks to years);
- (iii)
- Many inorganic, bioorganic, biological and polymeric compounds are present in biological liquids (blood plasma, serum, saliva). Each of these compounds might act as an inhibitor, promoter, nucleator or even as a template for the growth of biological apatite [355]. In addition, each of them somehow influences the crystallization kinetics and might be either incorporated into the solid structure or co-precipitated with calcium orthophosphates.
- (iv)
8. Calcium Orthophosphates in Tissue Engineering
9. Conclusions and Outlook
References and Notes
- Berzelius, J. Ueber basische phosphorsaure kalkerde. Ann. Chem. Pharma. 1845, 53, 286–288. [Google Scholar] [CrossRef]
- Cameron, F.K.; Seidell, A. The phosphates of calcium. I. J. Am. Chem. Soc. 1905, 27, 1503–1512. [Google Scholar] [CrossRef]
- Cameron, F.K.; Bell, J.M. The phosphates of calcium. II. J. Am. Chem. Soc. 1905, 27, 1512–1514. [Google Scholar] [CrossRef]
- Cameron, F.K.; Bell, J.M. The phosphates of calcium, III; Superphosphate. J. Am. Chem. Soc. 1906, 28, 1222–1229. [Google Scholar] [CrossRef]
- Cameron, F.K.; Bell, J.M. The phosphates of calcium. IV. J. Am. Chem. Soc. 1910, 32, 869–873. [Google Scholar] [CrossRef]
- Bassett, H., Jr. Beiträge zum Studium der Calciumphosphate. I. Die Hydrate der Calcium-Hydroorthophosphate. Z. Anorg. Chem. 1907, 53, 34–48. [Google Scholar] [CrossRef]
- Bassett, H., Jr. Beiträge zum Studium der Calciumphosphate. II. Die Einwirkung von Ammoniakgas auf Calcium-Hydroorthophosphate. Z. Anorg. Chem. 1907, 53, 49–62. [Google Scholar] [CrossRef]
- Bassett, H., Jr. Beiträge zum Studium der Calciumphosphate. III. Das System CaO – P2O5 – H2O. Z. Anorg. Chem. 1908, 59, 1–55. [Google Scholar] [CrossRef]
- Bassett, H., Jr. The phosphates of calcium. Part IV. The basic phosphates. J. Chem. Soc. 1917, 111, 620–642. [Google Scholar] [CrossRef]
- As a mineral species, apatite was first recognized by the father of German geology Abraham Gottlob Werner (1750 – 1817) in 1786 and named by him from the ancient Greek απατάω (apatao) – “to mislead” or “to deceive”, because it had previously been mistaken for other minerals, such as beryl, tourmaline, chrysolite, amethyst, fluorite, etc.
- Hausen, H. Die Apatite, deren chemische zusammensetzung, und ihrer Verhältnis zu physikalischen und morphologischen Eigenschaften. Acta Acad. Abo. Ser. B: Mat. Phys. Mat. Natur. Teknik. 1929, 5, 62–65. [Google Scholar]
- Lide, D.R. The CRC handbook of chemistry and physics, 86th Ed. ed; CRC Press: Boca Raton, Florida, USA, 2005; p. 2544. [Google Scholar]
- LeGeros, R.Z. Calcium phosphates in oral biology and medicine; Karger: Basel, Switzerland, 1991; p. 201. [Google Scholar]
- Elliot, J.C. Structure and chemistry of the apatites and other calcium orthophosphates. Stud. Inorg. Chem. 1994, 18, 389. [Google Scholar]
- Amjad, Z. (Ed.) Calcium phosphates in biological and industrial systems; Kluwer Academic Publishers: Boston, MA, USA, 1997; p. 529.
- Cantelar, E.; Lifante, G.; Calderón, T.; Meléndrez, R.; Millán, A.; Alvarez, M.A.; Barboza-Flores, M. Optical characterisation of rare earths in natural fluorapatite. J. Alloys Compd. 2001, 323, 851–854. [Google Scholar] [CrossRef]
- Ribeiro, H.B.; Guedes, K.J.; Pinheiro, M.V.B.; Greulich-Weber, S.; Krambrock, K. About the blue and green colours in natural fluorapatite. Phys. Status Solidi C 2005, 2, 720–723. [Google Scholar] [CrossRef]
- Dorozhkin, S.V.; Epple, M. Biological and medical significance of calcium phosphates. Angew. Chem. Int. Ed. Engl. 2002, 41, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- McConnell, D. Apatite: its crystal chemistry, mineralogy, utilization, and geologic and biologic occurrences; Springer-Verlag: Vienna and New York, USA, 1973; p. 111. [Google Scholar]
- Becker, P. Phosphates and phosphoric acid: raw materials technology and economics of the wet process, 2nd Ed. ed; In Fertilizer science and technology seriesMarcel Dekker: New York, USA, 1989; p. 760. [Google Scholar]
- Smith, D.K. Calcium phosphate apatites in nature. In Hydroxyapatite and related materials; Brown, P.W., Constantz, B., Eds.; CRC Press Inc.: Boca Raton, FL, USA, 1994; pp. 29–44. [Google Scholar]
- Angelov, A.I.; Levin, B.V.; Chernenko, Yu.D. Phosphate ore, A reference book; (in Russian). Nedra Busyness Centre: Moscow, Russia, 2000; p. 120. [Google Scholar]
- Phosphate deposits of the world: phosphate rock resources; Cook, P.J.; Shergold, J.H.; Davidson, D.F. (Eds.) Cambridge University Press: Cambridge, MA, USA, 2005; Volume 2, p. 600.
- Ford, A.K. A remarkable crystal of apatite from Mt. Apatite, Auburn, Maine. Am. J. Sci. 1917, 44, 245–246. [Google Scholar] [CrossRef]
- Hogarth, D.D. The discovery of apatite on the Lievre River, Quebec. Mineral. Rec. 1974, 5, 178–182. [Google Scholar]
- van Velthuizen, J. Giant fluorapatite crystals: a question of locality. Mineral. Rec. 1992, 23, 459–463. [Google Scholar]
- Pan, Y.; Fleet, M.E. Compositions of the apatite-group minerals: substitution mechanisms and controlling factors. In Phosphates: geochemical, geobiological and materials importance; Series: Reviews in Mineralogy and Geochemistry; Vol. 48, Hughes, J.M., Kohn, M., Rakovan, J., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2002; pp. 13–49. [Google Scholar]
- Zanin, Yu.N. The classification of calcium phosphates of phosphorites. Lithol. Miner. Resour. 2004, 39, 281–282. [Google Scholar] [CrossRef]
- Rogers, A.F. Collophane, a much neglected mineral. Am. J. Sci. 1922, 3, 269–276. [Google Scholar] [CrossRef]
- http://www.mindat.org/min-10072.html accessed April 2009.
- Elorza, J.; Astibia, H.; Murelaga, X.; Pereda-Suberbiola, X. Francolite as a diagenetic mineral in dinosaur and other upper cretaceous reptile bones (Lano, Iberian peninsula): microstructural, petrological and geochemical features. Cretaceous Res. 1999, 20, 169–187. [Google Scholar] [CrossRef]
- Hubert, B.; Álvaro, J.J.; Chen, J.Y. Microbially mediated phosphatization in the Neoproterozoic Doushantuo Lagerstätte, South China. Bull. Soc. Géol. Fr. 2005, 176, 355–361. [Google Scholar] [CrossRef]
- Chakhmouradian, A.R.; Medici, L. Clinohydroxylapatite: a new apatite-group mineral from northwestern Ontario (Canada), and new data on the extent of Na-S substitution in natural apatites. Eur. J. Mineral. 2006, 18, 105–112. [Google Scholar] [CrossRef]
- http://www.mindat.org/gallery.php?min=9293 accessed April 2009.
- Klein, C. Brushite from the island of Mona (between Haiti and Puerto Rico). Sitzber. K. Preuss. Aka. 1901, 720–725. [Google Scholar]
- Merrill, G.P. On the calcium phosphate in meteoric stones. Am. J. Sci. 1917, 43, 322–324. [Google Scholar] [CrossRef]
- Tyrrell, G.W. Apatite, nepheline, and rare-earth mining in the Kola Peninsula. Nature 1938, 141, 354–355. [Google Scholar] [CrossRef]
- Trueman, N.A. Substitutions for phosphate ions in apatite. Nature 1966, 210, 937–938. [Google Scholar] [CrossRef]
- White, T.; Ferraris, C.; Kim, J.; Madhavi, S. Apatite – an adaptive framework structure. In Micro- and mesoporous mineral phases; Series: Reviews in Mineralogy and Geochemistry; Vol. 57, Ferraris, G., Merlino, S., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2005; pp. 307–401. [Google Scholar]
- Jacob, K.D.; Reynolds, D.S. Reduction of tricalcium phosphate by carbon. Ind. Eng. Chem. 1928, 20, 1204–1210. [Google Scholar] [CrossRef]
- Copson, R.L.; Newton, R.H.; Lindsay, J.D. Superphosphate manufacture – mixing phosphate rook with concentrated phosphoric acid. Ind. Eng. Chem. 1936, 28, 923–927. [Google Scholar] [CrossRef]
- Newton, R.H.; Copson, R.L. Superphosphate manufacture – composition of superphosphate made from phosphate rock and concentrated phosphoric acid. Ind. Eng. Chem. 1936, 28, 1182–1186. [Google Scholar] [CrossRef]
- Magda, A.; Pode, V.; Niculescu, M.; Muntean, C.; Bandur, G.; Iovi, A. Studies on process of obtaining the fertilizers based on ammonium phosphates with addition of boric acid. Rev. Chim. (Bucharest) 2008, 59, 1340–1344. [Google Scholar]
- Omelon, S.J.; Grynpas, M.D. Relationships between polyphosphate chemistry, biochemistry and apatite biomineralization. Chem. Rev. 2008, 108, 4694–4715. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C.; Drouet, C.; Sfihi, H. Chemical diversity of apatites. Adv. Sci. Technol. 2006, 49, 27–36. [Google Scholar] [CrossRef]
- O’Neill, W.C. The fallacy of the calcium – phosphorus product. Kidney Int. 2007, 72, 792–796. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Formation and transformation of calcium phosphates: relevance to vascular calcification. Z Kardiol. 2001, 90 Suppl. 3, III116–III125. [Google Scholar] [CrossRef]
- Becker, A.; Epple, M.; Müller, K.M.; Schmitz, I. A comparative study of clinically well-characterized human atherosclerotic plaques with histological, chemical, and ultrastructural methods. J. Inorg. Biochem. 2004, 98, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng., C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- Pasteris, J.D.; Wopenka, B.; Valsami-Jones, E. Bone and tooth mineralization: why apatite? Elements 2008, 4, 97–104. [Google Scholar] [CrossRef]
- Sun, Y.; Hanley, E.N., Jr. Calcium-containing crystals and osteoarthritis. Curr. Opin. Orthoped. 2007, 18, 472–478. [Google Scholar] [CrossRef]
- Occasionally “biological apatite” is called “bioapatite” [53,54,55].
- Danil’chenko, S.N.; Kulik, A.N.; Bugai, A.N.; Pavlenko, P.A.; Kalinichenko, T.G.; Ul’yanchich, N.V.; Sukhodub, L.F. Thermally activated diffusion of magnesium from bioapatite crystals. J. Appl. Spectrosc. 2005, 72, 899–905. [Google Scholar] [CrossRef]
- Passey, B.H.; Robinson, T.F.; Ayliffe, L.K.; Cerling, T.E.; Sphonheimer, M.; Dearing, M.D.; Roeder, B.L.; Ehleringer, J.R. Carbon isotopic fractionation between diet, breath and bioapatite in different mammals. J. Arch. Sci. 2005, 32, 1459–1470. [Google Scholar] [CrossRef]
- Meneghini, C.; Dalconi, M.C.; Nuzzo, S.; Mobilio, S.; Wenk, R.H. Rietveld refinement on X-ray diffraction patterns of bioapatite in human fetal bones. Biophys. J. 2003, 84, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- There are reports that dahllite belongs to the francolite group. Natural dahllite might be a rock forming mineral [57]. For example, it was found in some phosphorite concretions of Podolia [58, 59]. In addition, it was found in both massive and accretionary crustal phosphorites [28].
- Rogers, A.F. Dahllite from Tonopah, Nevada; Voelckerite, a new basic calcium phosphate; comments on the chemical composition of apatite and phosphorite. Z. Krystallogr. Mineral. 1912, 52, 209–217. [Google Scholar]
- Schaller, W.T. The probable identity of podolite with dahllite. Am. J. Sci. 1910, 30, 309–310. [Google Scholar] [CrossRef]
- Schaller, W.T. On the likely identity of podolite and dahllite. Z. Krystallogr. Mineral. 1910, 48, 559–561. [Google Scholar]
- Collagens are fibrous, insoluble proteins found in the connective tissues, including skin, bone, ligaments and cartilage.
- Skinner, H.C.W. Biominerals. Mineral. Mag. 2005, 69, 621–641. [Google Scholar] [CrossRef]
- Daculsi, G. Physicochemical and ultrastructural analysis of bone bioactive interface. Biomater. Tissue Int. 1992, 10, 296–304. [Google Scholar]
- Daculsi, G.; Bouler, J.M.; LeGeros, R.Z. Adaptive crystal formation in normal and pathological calcifications in synthetic calcium phosphate and related biomaterials. Int. Rev. Cytology 1997, 172, 129–191. [Google Scholar]
- Driessens, F.C.M.; Verbeeck, R.M.H. Biominerals; CRC Press: Boca Raton, FL, USA, 1990; p. 440. [Google Scholar]
- Clark, N.A. The system P2O5-CaO- H2O and the recrystallization of monocalcium phosphate. J. Phys. Chem. 1931, 35, 1232–1238. [Google Scholar] [CrossRef]
- Brown, P.W. Phase relationships in the ternary system CaO - P2O5 - H2O at 25°C. J. Am. Ceram. Soc. 1992, 75, 17–22. [Google Scholar] [CrossRef]
- Martin, R.I.; Brown, P.W. Phase equilibria among acid calcium phosphates. J. Am. Ceram. Soc. 1997, 80, 1263–1266. [Google Scholar] [CrossRef]
- In literature occasionally one might find notes on the 12th calcium orthophosphate, namely oxyapatite (Ca10(PO4)6O). A mixture of oxyapatite and HA might be prepared by dehydration of HA at temperatures exceeding ~ 900°C (e.g., during plasma-spray of HA) only in the absence of water vapor [14,15,69,70]. It also might be crystallized in glass-ceramics [71]. Computer modeling techniques have been employed to qualitatively and quantitatively investigate the dehydration of HA to oxyapatite [72]. Oxyapatite has the hexagonal space group symmetry Р6 (174) of cesanite type [73], while the space group symmetry for partially dehydrated HA was found to change from hexagonal P63/m to triclinic Р6 when more than ca. 35% of the structurally bound water had been removed [70]. Oxyapatite is very reactive and transforms to HA in contact with water vapor [69]. Oxyapatite is still very poorly known; however, the following data on the solubility constant (Ks ~ 10-69 at 25ºC) are available [14].
- Gross, K.A.; Berndt, C.C.; Dinnebier, R.; Stephens, P. Oxyapatite in hydroxyapatite coatings. J. Maert. Sci. Mater. Med. 1998, 33, 3985–3991. [Google Scholar] [CrossRef]
- Alberius-Henning, P.; Adolfsson, E.; Grins, J.; Fitch, A. Triclinic oxy-hydroxyapatite. J. Mater. Sci. 2001, 36, 663–668. [Google Scholar] [CrossRef]
- van’t Hoen, C.; Rheinberger, V.; Höland, W.; Apel, E. Crystallization of oxyapatite in glass-ceramics. J. Eur. Ceram. Soc. 2007, 27, 1579–1584. [Google Scholar] [CrossRef]
- de Leeuw, N.H.; Bowe, J.R.; Rabone, J.A.L. A computational investigation of stoichiometric and calcium-deficient oxy- and hydroxy-apatites. Faraday Disc. 2007, 134, 195–214. [Google Scholar] [CrossRef]
- White, T.J.; Dong, Z.L. Structural derivation and crystal chemistry of apatites. Acta Crystallogr., Sect. B: Struc. Sci. 2003, B59, 1–16. [Google Scholar] [CrossRef]
- Mathew, M.; Takagi, S. Structures of biological minerals in dental research. J. Res. Natl. Inst. Stand. Technol. 2001, 106, 1035–1044. [Google Scholar] [CrossRef]
- Hughes, J.M.; Rakovan, J. The crystal structure of apatite, Ca5(PO4)3(F,OH,Cl). In Phosphates: geochemical, geobiological and materials importance; Series: Reviews in Mineralogy and Geochemistry; Vol. 48, Hughes, J.M., Kohn, M., Rakovan, J., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2002; pp. 1–12. [Google Scholar]
- Wang, L.; Nancollas, G.H. Calcium orthophosphates: crystallization and dissolution. Chem. Rev. 2008, 108, 4628–4669. [Google Scholar] [CrossRef] [PubMed]
- Lynn, A.K.; Bonfield, W. A novel method for the simultaneous, titrant-free control of pH and calcium phosphate mass yield. Acc. Chem. Res. 2005, 38, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Gil, F.J.; Ginebra, M.P.; Driessens, F.C.M.; Planell, J.A.; Best, S.M. Calcium phosphate bone cements for clinical applications. Part I: solution chemistry. J. Mater. Sci. Mater. Med. 1999, 10, 169–176. [Google Scholar] [CrossRef] [PubMed]
- McDowell, H.; Gregory, T.M.; Brown, W.E. Solubility of Ca5(PO4)3OH in the system Ca(OH)2 -H3PO4 - H2O at 5, 15, 25, and 37 degree. C. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1977, 81A, 273–281. [Google Scholar] [CrossRef]
- Pan, H.B.; Darvell, B.W. Calcium phosphate solubility: the need for re-evaluation. Cryst. Growth Des. 2009, 9, 639–645. [Google Scholar] [CrossRef]
- Biocompatibility is the ability of a material to perform with an appropriate host response in a specific application [82].
- Williams, D.F. The Williams dictionary of biomaterials; Liverpool University Press: Liverpool, UK, 1999; p. 368. [Google Scholar]
- Köster, K.; Heide, H.; König, R. Resorbierbare Calciumphosphatkeramik im Tierexperiment unter Belastung. Langenbecks Arch. Chir. 1977, 343, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, O.; Boltong, M.G.; Driessens, F.C.M.; Planell, J.A. Optimization of a calcium orthophosphate cement formulation occurring in the combination of monocalcium phosphate monohydrate with calcium oxide. J. Mater. Sci. Mater. Med. 1994, 5, 67–71. [Google Scholar] [CrossRef]
- Bermúdez, O.; Boltong, M.G.; Driessens, F.C.M.; Planell, J.A. Development of some calcium phosphate cements from combinations of α-TCP, MCPM and CaO. J. Mater. Sci. Mater. Med. 1994, 5, 160–163. [Google Scholar] [CrossRef]
- Driessens, F.C.M.; Boltong, M.G.; Bermúdez, O.; Planell, J.A.; Ginebra, M.P.; Fernández, E. Effective formulations for the preparation of calcium phosphate bone cements. J. Mater. Sci. Mater. Med. 1994, 5, 164–170. [Google Scholar] [CrossRef]
- Huan, Z.; Chang, J. Novel bioactive composite bone cements based on the β-tricalcium phosphate – monocalcium phosphate monohydrate composite cement system. Acta. Biomater. 2009, 5, 1253–1264. [Google Scholar]
- The Merck Index: An encyclopedia of chemicals, drugs, and biologicals, 12th Ed.; Budavari, S.; O’Neil, M.J.; Smith, A.; Heckelman, P.E.; Kinneary, J.F. (Eds.) Chapman & Hall: USA, 1996; p. 1741.
- Stein, H.H.; Kadzere, C.T.; Kim, S.W.; Miller, P.S. Influence of dietary phosphorus concentration on the digestibility of phosphorus in monocalcium phosphate by growing pigs. J. Animal Sci. 2008, 86, 1861–1867. [Google Scholar] [CrossRef]
- To honor Prof. George Jarvis Brush (1831-1912), an American mineralogist, Yale University, New Haven, Connecticut, USA.
- Arsic, J.; Kaminski, D.; Poodt, P.; Vlieg, E. Liquid ordering at the brushite-{010}-water interface. Phys. Rev. B 2004, 69, 245406, (4 pages). [Google Scholar] [CrossRef]
- Qiu, S.R.; Orme, C.A. Dynamics of biomineral formation at the near-molecular level. Chem. Rev. 2008, 108, 4784–4822. [Google Scholar] [CrossRef] [PubMed]
- Kurashina, K.; Kurita, H.; Hirano, M.; Kotani, A.; Klein, C.P.; de Groot, K. In vivo study of calcium phosphate cements: implantation of an α-tricalcium phosphate / dicalcium phosphate dibasic / tetracalcium phosphate monoxide cement paste. Biomaterials 1997, 18, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Driessens, F.C.M.; Planell, J.A.; Boltong, M.G.; Khairoun, I.; Ginebra, M.P. Osteotransductive bone cements. Proc. Inst. Mech. Eng. H: J. Eng. Med. 1998, 212, 427–435. [Google Scholar] [CrossRef]
- Takagi, S.; Chow, L.C.; Ishikawa, K. Formation of hydroxyapatite in new calcium phosphate cements. Biomaterials 1998, 19, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Niwa, S.; Hori, M.; Hattori, T.; Sawai, K.; Aoki, S.; Hirano, M.; Takeuchi, H. Mechanical strength of calcium phosphate cement in vivo and in vitro. Biomaterials 1998, 19, 1587–1591. [Google Scholar] [CrossRef] [PubMed]
- Crall, J.J.; Bjerga, J.M. Effects of DCPD / APF application and prolonged exposure to fluoride on caries-like lesion formation in vitro. J. Oral Pathol. Med. 1987, 16, 488–491. [Google Scholar] [CrossRef]
- Wefel, J.S.; Harless, J.D. The use of saturated DCPD in remineralization of artificial caries lesions in vitro. J. Dent. Res. 1987, 66, 1640–1643. [Google Scholar] [CrossRef] [PubMed]
- Hoppenbrouwers, P.M.; Groenendijk, E.; Tewarie, N.R.; Driessens, F.C.M. Improvement of the Caries Resistance of human dental roots by a two-step conversion of the root mineral into fluoridated hydroxylapatite. J. Dent. Res. 1988, 67, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Gaffar, A.; Blake-Haskins, J.; Mellberg, J. In vivo studies with a dicalcium phosphate / MFP system for caries prevention. Int. Dent. J. 1993, 43 (Suppl. 1), 81–88. [Google Scholar] [PubMed]
- Sullivan, R.J.; Charig, A.; Blake-Haskins, J.; Zhang, Y.P.; Miller, S.M.; Strannick, M.; Gaffar, A.; Margolis, H.C. In vivo detection of calcium from dicalcium phosphate dihydrate dentifrices in demineralized human enamel and plaque. Adv. Dent. Res. 1997, 11, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Mostashari, S.M.; Haddadi, H.; Hashempoor, Z. Effect of deposited calcium hydrogen phosphate dihydrate on the flame retardancy imparted to cotton fabric. Asian J. Chem. 2006, 18, 2388–2390. [Google Scholar]
- Powditch, H.I.; Bosworth, A.W. Studies on infant feeding IX. The availability of dicalcium phosphate when present as a constituent of infant’s food. Boston Med. Surg. J. 1917, 177, 864–867. [Google Scholar] [CrossRef]
- For Moneta (now Monito) Island (archipelago of Puerto Rico), which contains a notable occurrence.
- Eshtiagh-Hosseini, H.; Houssaindokht, M.R.; Chahkandhi, M.; Youssefi, A. Preparation of anhydrous dicalcium phosphate, DCPA, through sol-gel process, identification and phase transformation evaluation. J. Non-Cryst. Solids 2008, 354, 3854–3857. [Google Scholar] [CrossRef]
- Fukase, Y.; Eanes, E.D.; Takagi, S.; Chow, L.C.; Brown, W.E. Setting reactions and compressive strengths of calcium phosphate cements. J. Dent. Res. 1990, 69, 1852–1856. [Google Scholar] [CrossRef] [PubMed]
- TenHuisen, K.S.; Brown, P.W. The formation of hydroxyapatite-ionomer cements at 38 degrees C. J. Dent. Res. 1994, 73, 598–606. [Google Scholar] [PubMed]
- Fernández, E.; Ginebra, M.P.; Boltong, M.G.; Driessens, F.C.M.; J. Ginebra, de Maeyer, E.A.; Verbeeck, R.M.H.; Planell, J.A. Kinetic study of the setting reaction of a calcium phosphate bone cement. J. Biomed. Mater. Res. 1996, 32, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Gil, F.J.; Best, S.M.; Ginebra, M.P.; Driessens, F.C.M.; Planell, J.A. The cement setting reaction in the CaHPO4-α-Ca3(PO4)2 system: an X-ray diffraction study. J. Biomed. Mater. Res. 1998, 42, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Gil, F.J.; Ginebra, M.P.; Driessens, F.C.M.; Planell, J.A.; Best, S.M. Production and characterization of new calcium phosphate bone cements in the CaHPO4 – α-Ca3(PO4)2 system: pH, workability and setting times. J. Mater. Sci. Mater. Med. 1999, 10, 223–230. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Preparation of octacalcium phosphate (OCP): a direct fast method. Calcif. Tissue Int. 1985, 37, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Boanini, E.; Borghi, M.; Cojazzi, G.; Panzavolta, S.; Roveri, N. Synthesis and hydrolysis of octacalcium phosphate: effect of sodium polyacrylate. J. Inorg. Biochem. 1999, 75, 145–151. [Google Scholar] [CrossRef]
- Nakahira, A.; Aoki, S.; Sakamoto, K.; Yamaguchi, S. Synthesis and evaluation of various layered octacalcium phosphates by wet-chemical processing. J. Mater. Sci. Mater. Med. 2001, 12, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Shelton, R.M.; Liu, Y.; Cooper, P.R.; Gbureck, U.; German, M.J.; Barralet, J.E. Bone marrow cell gene expression and tissue construct assembly using octacalcium phosphate microscaffolds. Biomaterials 2006, 27, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, N.; Kishimoto, K.N.; Anada, T.; Imaizumi, H.; Itoi, E.; Suzuki, O. Effect of partial hydrolysis of octacalcium phosphate on its osteoconductive characteristics. Biomaterials 2009, 30, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Jiménez, M.J.; García-García, R.; Reyes-Gasga, J. Synthesis and hydrolysis of octacalcium phosphate and its characterization by electron microscopy and X-ray diffraction. J. Phys. Chem. Solids 2009, 70, 390–395. [Google Scholar] [CrossRef]
- Brown, W.E.; Mathew, M.; Tung, M.S. Crystal chemistry of octacalcium phosphate. Prog. Cryst. Growth Charact. 1981, 4, 59–87. [Google Scholar] [CrossRef]
- LeGeros, R.Z. Variations in the crystalline components of human dental calculus: I. crystallographic and spectroscopic methods of analysis. J. Dent. Res. 1974, 53, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H. Formation and inhibition of dental calculus. J. Periodontol. 1969, 40, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Octacalcium phosphate; Chow, L.C.; Eanes, E.D. (Eds.) Karger: Basel, Switzerland, 2001; p. 167.
- Brown, W.E. Octacalcium phosphate and hydroxyapatite: crystal structure of octacalcium phosphate. Nature 1962, 196, 1048–1050. [Google Scholar] [CrossRef]
- Brown, W.E.; Smith, J.P.; Lehr, J.R.; Frazier, A.W. Octacalcium phosphate and hydroxyapatite: crystallographic and chemical relations between octacalcium phosphate and hydroxyapatite. Nature 1962, 196, 1050–1055. [Google Scholar] [CrossRef]
- Brown, W.E. Crystal growth of bone mineral. Clin. Orthop. Relat. Res. 1966, 44, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.G.A.; Wood, G.J.; Barry, J.C.; Featherstone, J.D.B. The structure of (100) defects in carbonated apatite crystallites: a high-resolution electron microscope study. Ultramicroscopy 1986, 19, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Nelson, D.G.A.; Pan, Y.; Kreinbrink, A.T.; Adachi, M.; Goto, T.; Moriwaki, Y. Fluoride analysis of apatite crystals with a central planar OCP inclusion: concerning the role of F- ions on apatite / OCP / apatite structure formation. Calcif. Tissue Int. 1996, 59, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Bodier-Houllé, P.; Steuer, P.; Voegel, J.C.; Cuisinier, F.J.G. First experimental evidence for human dentine crystal formation involving conversion of octacalcium phosphate to hydroxyapatite. Acta Crystallogr. Sect D: Biol. Crystallogr. 1998, 54, 1377–1381. [Google Scholar] [CrossRef]
- Aoba, T.; Komatsu, H.; Shimazu, Y.; Yagishita, H.; Taya, Y. Enamel mineralization and an initial crystalline phase. Connect. Tissue Res. 1998, 38, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Tomazic, B.B.; Brown, W.E.; Shoen, F.J. Physicochemical properties of calcific deposits isolated from porcine bioprosthetic heart valves removed from patients following 2-13 years function. J. Biomed. Mater. Res. 1994, 28, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Nancollas, G.H.; Wu, W. Biomineralization mechanisms: a kinetics and interfacial energy approach. J. Cryst. Growth 2000, 211, 137–142. [Google Scholar] [CrossRef]
- Kamakura, S.; Sasano, Y.; Homma, H.; Suzuki, O.; Kagayama, M.; Motegi, K. Implantation of octacalcium phosphate (OCP) in rat skull defects enhances bone repair. J. Dent. Res. 1999, 78, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, S.; Sasano, Y.; Homma, H.; Suzuki, O.; Kagayama, M.; Motegi, K. Implantation of octacalcium phosphate nucleates isolated bone formation in rat skull defects. Oral Dis. 2001, 7, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei-Aval, F.; Sobhani, A.; Arab, M.R.; Sarani, S.A.; Heydari, M.H. The efficacy of implant of octacalcium phosphate in combination with bone matrix gelatin (BMG) on bone regeneration in skull defects in rat. Iran. J. Med. Sci. 2004, 29, 124–129. [Google Scholar]
- Suzuki, O.; Kamakura, S.; Katagiri, T.; Nakamura, M.; Zhao, B.; Honda, Y.; Kamijo, R. Bone formation enhanced by implanted octacalcium phosphate involving conversion into Ca-deficient hydroxyapatite. Biomaterials 2006, 27, 2671–2681. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, O.; Imaizumi, H.; Kamakura, S.; Katagiri, T. Bone regeneration by synthetic octacalcium phosphate and its role in biological mineralization. Cur. Med. Chem. 2008, 15, 305–313. [Google Scholar] [CrossRef]
- Kikawa, T.; Kashimoto, O.; Imaizumi, H.; Kokubun, S.; Suzuki, O. Intramembranous bone tissue response to biodegradable octacalcium phosphate implant. Acta Biomater 2009. (early view). [Google Scholar]
- In 1941, to honor Mr. Herbert Percy Whitlock (1868 – 1948), an American mineralogist, the curator of the American Museum of Natural History, New York City, New York, USA, the term whitlockite was coined as a synonym for β-TCP identified by its X-ray diffraction pattern in phosphate rocks [137,138,139]. Therefore, strictly speaking, β-TCMP should be called as a “magnesium whitlokite”. Its solubility is less than that of β-TCP [140]. An iron-containing whitlockite with chemical formula Ca9(Mg,Fe2+)(PO4)6(PO3, OH) exists in nature: is a relatively rare natural mineral but is found in granitic pegmatite and has also been found in meteorites. It can form small, but distinct and well-formed crystals [141, 142].
- Jensen, A.T.; Rowles, S.L. Magnesium whitlockite, a major constituent of dental calculus. Acta Odont. Scand. 1957, 16, 121–139. [Google Scholar] [CrossRef]
- Frondel, C. Whitlockite: a new calcium phosphate, Ca3(PO4)2. Am. Mineral. 1941, 26, 145–152. [Google Scholar]
- Frondel, C. Mineralogy of the calcium phosphates in insular phosphate rock. Am. Mineral. 1943, 28, 215–232. [Google Scholar]
- Li, X.; Ito, A.; Sogo, Y.; Wang, X.; LeGeros, R.Z. Solubility of Mg-containing β-tricalcium phosphate at 25 °C. Acta Biomater. 2009, 5, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Calvo, C.; Gopal, R. The crystal structure of whitlockite from the Palermo Quarry. Am. Mineral. 1975, 60, 120–133. [Google Scholar]
- http://rruff.geo.arizona.edu/doclib/hom/whitlockite.pdf accessed April 2009.
- Kodaka, T.; Debari, K.; Higashi, S. Magnesium-containing crystals in human dental calculus. J. Electron Microsc. (Tokyo) 1988, 37, 73–80. [Google Scholar]
- Mirtchi, A.A.; Lemaître, J.; Munting, E. Calcium phosphate cements: study of the β-tricalcium phosphate – dicalcium phosphate – calcite cements. Biomaterials 1990, 11, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Mirtchi, A.A.; Lemaître, J.; Munting, E. Calcium phosphate cements: effect of fluorides on the setting and hardening of β-tricalcium phosphate – dicalcium phosphate – calcite cements. Biomaterials 1991, 12, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, J.; Munting, E.; Mirtchi, A.A. Setting, hardening and resorption of calcium phosphate ionic cements. Rev. Stomatol. Chir. Maxillofac. 1992, 93, 163–165. [Google Scholar] [PubMed]
- Ohura, K.; Bohner, M.; Hardouin, P.; Lemaître, J.; Pasquier, G.; Flautre, B. Resorption of, and bone formation from, new β-tricalcium phosphate-monocalcium phosphate cements: an in vivo study. J. Biomed. Mater. Res. 1996, 30, 193–200. [Google Scholar] [CrossRef] [PubMed]
- The term biphasic calcium phosphate (BCP) was first used by Nery et. al. [149] to describe the bioceramic, that consisted of a mixture of HA and β-TCP, based on the X-ray diffraction analysis which showed that the ‘tricalcium phosphate” preparation material used in their early publication [150] was in fact a mixture of ~ 20% HA and ~ 80% β-TCP.
- Ellinger, R.F.; Nery, E.B.; Lynch, K.L. Histological assessment of periodontal osseous defects following implantation of hydroxyapatite and biphasic calcium phosphate ceramics: a case report. Int. J. Periodont. Restor. Dent. 1986, 3, 22–33. [Google Scholar]
- Nery, E.B.; Lynch, K.L.; Hirthe, W.M.; Mueller, K.H. Bioceramic implants in surgically produced infrabony defects. J. Periodontol. 1975, 46, 328–347. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, A.; Gautier, H.; Bouler, J.M.; Gouyette, A.; Pegon, Y.; Daculsi, G.; Merle, C. Biphasic calcium phosphate: a comparative study of interconnected porosity in two ceramics. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 84B, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tancret, F.; Bouler, J.M.; Chamousset, J.; Minois, L.M. Modelling the mechanical properties of microporous and macroporous biphasic calcium phosphate bioceramics. J. Eur. Ceram. Soc. 2006, 26, 3647–3656. [Google Scholar] [CrossRef]
- Bouler, J.M.; Trecant, M.; Delecrin, J.; Royer, J.; Passuti, N.; Daculsi, G. Macroporous biphasic calcium phosphate ceramics: Influence of five synthesis parameters on compressive strength. J. Biomed. Mater. Res. 1996, 32, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, W.; Li, Y.; Fan, S.; Weng, J.; Zhang, X. Biological evaluation of biphasic calcium phosphate ceramic vertebral laminae. Biomaterials 1998, 19, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G. Biphasic calcium phosphate concept applied to artificial bone, implant coating and injectable bone substitute. Biomaterials 1998, 19, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G.; Weiss, P.; Bouler, J.M.; Gauthier, O.; Millot, F.; Aguado, E. Biphasic calcium phosphate / hydrosoluble polymer composites: a new concept for bone and dental substitution biomaterials. Bone 1999, 25 (Suppl. 2), 59S–61S. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; Lin, S.; Rohanizadeh, R.; Mijares, D.; LeGeros, J.P. Biphasic calcium phosphate bioceramics: preparation, properties and applications. J. Mater. Sci. Mater. Med. 2003, 14, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G.; Laboux, O.; Malard, O.; Weiss, P. Current state of the art of biphasic calcium phosphate bioceramics. J. Mater. Sci. Mater. Med. 2003, 14, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Alam, I.; Asahina, I.; Ohmamiuda, K.; Enomoto, S. Comparative study of biphasic calcium phosphate ceramics impregnated with rhBMP-2 as bone substitutes. J. Biomed. Mater. Res. 2001, 54, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G. Biphasic calcium phosphate granules concept for injectable and mouldable bone substitute. Adv. Sci. Technol. 2006, 49, 9–13. [Google Scholar] [CrossRef]
- Metsger, D.S.; Driskell, T.D.; Paulsrud, J.R. Tricalcium phosphate ceramic – a resorbable bone implant: review and current status. J. Am. Dent. Assoc. 1982, 105, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Nurse, R.W.; Welch, J.B.; Gun, W. High-temperature phase equilibria in the system dicalcium silicate – tricalcium phosphate. J. Chem. Soc. 1959, 1077–1083. [Google Scholar] [CrossRef]
- Langstaff, S.D.; Sayer, M.; Smith, T.J.N.; Pugh, S.M.; Hesp, S.A.M.; Thompson, W.T. Resorbable bioceramics based on stabilized calcium phosphates. Part I: Rational design, sample preparation and material characterization. Biomaterials 1999, 20, 1727–1741. [Google Scholar] [CrossRef]
- Langstaff, S.D.; Sayer, M.; Smith, T.J.N.; Pugh, S.M. Resorbable bioceramics based on stabilized calcium phosphates. Part II: Evaluation of biological response. Biomaterials 2001, 22, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Sayer, M.; Stratilatov, A.D.; Reid, J.W.; Calderin, L.; Stott, M.J.; Yin, X.; MacKenzie, M.; Smith, T.J.N.; Hendry, J.A.; Langstaff, S.D. Structure and composition of silicon-stabilized tricalcium phosphate. Biomaterials 2003, 24, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.W.; Pietak, A.M.; Sayer, M.; Dunfield, D.; Smith, T.J.N. Phase formation and evolution in the silicon substituted tricalcium phosphate / apatite system. Biomaterials 2005, 26, 2887–2897. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.W.; Tuck, L.; Sayer, M.; Fargo, K.; Hendry, J.A. Synthesis and characterization of single-phase silicon substituted α-tricalcium phosphate. Biomaterials 2006, 27, 2916–2925. [Google Scholar] [CrossRef] [PubMed]
- Astala, R.; Calderin, L.; Yin, X.; Stott, M.J. Ab initio simulation of Si-doped hydroxyapatite. Chem. Mater. 2006, 18, 413–422. [Google Scholar] [CrossRef]
- Yin, X.; Stott, M.J.; Rubio, A. α- and β-tricalcium phosphate: a density functional study. Phys. Rev. B 2003, 68, 205205, (7 pages). [Google Scholar] [CrossRef]
- Constantz, B.R.; Ison, I.C.; Fulmer, M.T.; Poser, R.D.; Smith, S.T.; Vanwagoner, M.; Ross, J.; Goldstein, S.A.; Jupiter, J.B.; Rosenthal, D.I. Skeletal repair by in situ formation of the mineral phase of bone. Science 1995, 267, 1796–1799. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Takeuchi, A.; Lin, X.; Matsuya, S.; Ishikawa, K. Effects of liquid phase on basic properties of α-tricalcium phosphate-based apatite cement. Dent. Mater. J. 2008, 27, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Stott, M.J. Theoretical insights into bone grafting Si-stabilized α-tricalcium phosphate. J. Chem. Phys. 2005, 122, 024709, (9 pages). [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Stott, M.J. Surface and adsorption properties of α-tricalcium phosphate. J. Chem. Phys. 2006, 124, 124701, (9 pages). [Google Scholar] [CrossRef] [PubMed]
- Termine, J.D.; Eanes, E.D. Comparative chemistry of amorphous and apatitic calcium phosphate preparations. Calcif. Tissue Res. 1972, 10, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Eanes, E.D.; Termine, J.D.; Nylen, M.U. An electron microscopic study of the formation of amorphous calcium phosphate and its transformation to crystalline apatite. Calcif. Tissue Res. 1973, 12, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.L.; Eanes, E.D. A thermodynamic analysis of the amorphous to crystalline calcium phosphate transformation. Calcif. Tissue Res. 1978, 28, 59–68. [Google Scholar] [CrossRef]
- Meyer, J.L.; Eanes, E.D. A thermodynamic analysis of the secondary transition in the spontaneous precipitation of calcium phosphate. Calcif. Tissue Res. 1978, 28, 209–216. [Google Scholar] [CrossRef]
- Wuthier, R.E.; Rice, G.S.; Wallace, J.E.; Weaver, R.L.; LeGeros, R.Z. In vitro precipitation of calcium phosphate under intracellular conditions: formation of brushite from an amorphous precursor in the absence of ATP. Calcif. Tissue Int. 1985, 37, 401–410. [Google Scholar]
- Sinyaev, V.A.; Shustikova, E.S.; Levchenko, L.V.; Sedunov, A.A. Synthesis and dehydration of amorphous calcium phosphate. Inorg. Mater. 2001, 37, 619–622. [Google Scholar] [CrossRef]
- Termine, J.D.; Peckauskas, R.A.; Posner, A.S. Calcium phosphate formation in vitro. II. Effects of environment on amorphous-crystalline transformation. Arch. Biochem. Biophys. 1970, 140, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Elliot, J.C. Recent studies of apatites and other calcium orthophosphates. In Les matériaux en phosphate de calcium. Aspects fondamentaux. / Calcium phosphate materials. Fundamentals; Brès, E., Hardouin, P., Eds.; Sauramps Medical: Montpellier, France, 1998; pp. 25–66. [Google Scholar]
- Li, Y.; Weng, W. In vitro synthesis and characterization of amorphous calcium phosphates with various Ca/P atomic ratios. J. Mater. Sci. Mater. Med. 2007, 18, 2303–2308. [Google Scholar] [CrossRef] [PubMed]
- Tadic, D.; Peters, F.; Epple, M. Continuous synthesis of amorphous carbonated apatites. Biomaterials 2002, 23, 2553–2559. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L.; Posner, A.S. Conversion of amorphous calcium phosphate to microcrystalline hydroxyapatite. A pH-dependent, solution-mediated, solid-solid conversion. J. Phys. Chem. 1973, 77, 2313–2317. [Google Scholar] [CrossRef]
- Posner, A.S.; Betts, F. Synthetic amorphous calcium phosphate and its relation to bone mineral structure. Acc. Chem. Res. 1975, 8, 273–281. [Google Scholar] [CrossRef]
- Harries, J.E.; Hukins, D.W.L.; Hasnain, S.S. Analysis of the EXAFS spectrum of hydroxyapatite. J. Phys. C: Solid State Phys. 1986, 19, 6859–6872. [Google Scholar] [CrossRef]
- Harries, J.E.; Hukins, D.W.L.; Holt, C.; Hasnain, S.S. Conversion of amorphous calcium phosphate into hydroxyapatite investigated by EXAFS spectroscopy. J. Cryst. Growth 1987, 84, 563–570. [Google Scholar] [CrossRef]
- Taylor, M.G.; Simkiss, K.; Simmons, J.; Wu, L.N.Y.; Wuthier, R.E. Structural studies of a phosphatidyl serine-amorphous calcium phosphate complex. Cell. Mol. Life Sci. 1998, 54, 192–202. [Google Scholar] [CrossRef]
- Peters, F.; Schwarz, K.; Epple, M. The structure of bone studied with synchrotron X-ray diffraction, X-ray absorption spectroscopy and thermal analysis. Thermochim. Acta 2000, 361, 131–138. [Google Scholar] [CrossRef]
- Posner, A.S.; Betts, F.; Blumenthal, N.C. Formation and structure of synthetic and bone hydroxyapatite. Progr. Cryst. Growth Char. 1980, 3, 49–64. [Google Scholar] [CrossRef]
- Boskey, A.L. Amorphous calcium phosphate: the contention of bone. J. Dent. Res. 1997, 76, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Onuma, K.; Ito, A. Cluster growth model for hydroxyapatite. Chem. Mater. 1998, 10, 3346–3351. [Google Scholar] [CrossRef]
- Skrtic, D.; Hailer, A.W.; Takagi, S.; Antonucci, J.M.; Eanes, E.D. Quantitative assessment of the efficacy of amorphous calcium phosphate / methacrylate composites in remineralizing caries-like lesions artificially produced in bovine enamel. J. Dent. Res. 1996, 75, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Improved properties of amorphous calcium phosphate fillers in remineralizing resin composites. Dent. Mater. 1996, 12, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Amorphous calcium phosphate-based bioactive polymeric composites for mineralized tissue regeneration. J. Res. Natl. Inst. Stand. Technol. 2003, 108, 167–182. [Google Scholar] [CrossRef]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D.; Eichmiller, F.C.; Schumacher, G.E. Physicochemical evaluation of bioactive polymeric composites based on hybrid amorphous calcium phosphates. J. Biomed. Mater. Res. B Appl. Biomater. 2000, 53, 381–391. [Google Scholar] [CrossRef]
- Schiller, C.; Siedler, M.; Peters, F.; Epple, M. Functionally graded materials of biodegradable polyesters and bone-like calcium phosphates for bone replacement. Ceram. Transact. 2001, 114, 97–108. [Google Scholar]
- Linhart, W.; Peters, F.; Lehmann, W.; Schilling, A.F.; Schwarz, K.; Amling, M.; Rueger, J.M.; Epple, M. Biologically and chemically optimized composites of carbonated apatite and polyglycolide as bone substitution materials. J. Biomed. Mater. Res. 2001, 54, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Tadic, D.; Beckmann, F.; Schwarz, K.; Epple, M. A novel method to produce hydroxylapatite objects with interconnecting porosity that avoids sintering. Biomaterials 2004, 25, 3335–3340. [Google Scholar] [CrossRef] [PubMed]
- Tadic, D.; Epple, M. Amorphous calcium phosphates as bone substitution materials. Eur. J. Trauma 2002, 28, 136–137. [Google Scholar]
- Eanes, E.D. Amorphous calcium phosphate. In Octacalcium phosphate; Chow, L.C., Eanes, E.D., Eds.; Karger: Basel, Switzerland, 2001; pp. 130–147. [Google Scholar]
- In some research papers, CDHA is defined as “precipitated HA” [203,204,205].
- Knowles, J.C.; Callcut, S.; Georgiou, G. Characterisation of the rheological properties and zeta potential of a range of hydroxyapatite powders. Biomaterials 2000, 21, 1387–1392. [Google Scholar]
- Kumar, R.; Prakash, K.H.; Cheang, P.; Khor, K.A. Temperature driven morphological changes of chemically precipitated hydroxyapatite nanoparticles. Langmuir 2004, 20, 5196–5200. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Lee, J.C.; Yang, J.W. Crystallization and sintering characteristics of chemically precipitated hydroxyapatite nanopowder. J. Cryst. Growth 2004, 262, 467–472. [Google Scholar] [CrossRef]
- Brès, E.F.; Duhoo, T.; Leroy, N.; Lemaitre, J. Evidence of a transient phase during the hydrolysis of calcium-deficient hydroxyapatite. Zeitschrift fuer Metallkunde / Mater. Res. Adv. Tech. 2005, 96, 503–506. [Google Scholar]
- Dorozhkina, E.I.; Dorozhkin, S.V. Mechanism of the solid-state transformation of a calcium-deficient hydroxyapatite (CDHA) into biphasic calcium phosphate (BCP) at elevated temperatures. Chem. Mater. 2002, 14, 4267–4272. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Mechanism of solid-state conversion of non-stoichiometric hydroxyapatite to diphase calcium phosphate. Russ. Chem. Bull. Int. Ed. 2003, 52, 2369–2375. [Google Scholar] [CrossRef]
- Rodríguez-Lorenzo, L. Studies on calcium deficient apatites structure by means of MAS-NMR spectroscopy. J. Mater. Sci. Mater. Med. 2005, 16, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Elliott, J.C.; Dowker, S.E.P. Formate incorporation in the structure of Ca-deficient apatite: Rietveld structure refinement. J. Solid State Chem. 2003, 174, 132–140. [Google Scholar] [CrossRef]
- Zahn, D.; Hochrein, O. On the composition and atomic arrangement of calcium-deficient hydroxyapatite: an ab-initio analysis. J. Solid State Chem. 2008, 181, 1712–1716. [Google Scholar] [CrossRef]
- Brown, P.W.; Martin, R.I. An analysis of hydroxyapatite surface layer formation. J. Phys. Chem. B 1999, 103, 1671–1675. [Google Scholar] [CrossRef]
- Honghui, Z.; Hui, L.; Linghong, G. Molecular and crystal structure characterization of calcium-deficient apatite. Key Eng. Mater. 2007, 330-332, 119–122. [Google Scholar]
- Mortier, A.; Lemaitre, J.; Rodrique, L.; Rouxhet, P.G. Synthesis and thermal behavior of well-crystallized calcium-deficient phosphate apatite. J. Solid State Chem. 1989, 78, 215–219. [Google Scholar] [CrossRef]
- Jeanjean, J.; McGrellis, S.; Rouchaud, J.C.; Fedoroff, M.; Rondeau, A.; Perocheau, S.; Dubis, A. A crystallographic study of the sorption of cadmium on calcium hydroxyapatites: incidence of cationic vacancies. J. Solid State Chem. 1996, 126, 195–201. [Google Scholar] [CrossRef]
- Wilson, R.M.; Elliott, J.C.; Dowker, S.E.P.; Rodriguez-Lorenzo, L.M. Rietveld refinements and spectroscopic studies of the structure of Ca-deficient apatite. Biomaterials 2005, 26, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T. I.; Frank-Kamenetskaya, O.V.; Kol’tsov, A.B.; Ugolkov, V.L. Crystal structure of calcium-deficient carbonated hydroxyapatite thermal decomposition. J. Solid State Chem. 2001, 160, 340–349. [Google Scholar] [CrossRef]
- Matsunaga, K. Theoretical investigation of the defect formation mechanism relevant to nonstoichiometry in hydroxyapatite. Phys. Rev. B 2008, 77, 104106, (14 pages). [Google Scholar] [CrossRef]
- Tsuchida, T.; Yoshioka, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Synthesis of biogasoline from ethanol over hydroxyapatite catalyst. Ind. Eng. Chem. Res. 2008, 47, 1443–1452. [Google Scholar] [CrossRef]
- It is worth noting that the chemically correct name would be hydroxylapatite (perhaps, hydroxidapatite would be even better because it relates to calcium hydroxide) while by the medical and material communities it is usually called as hydroxyapatite.
- Elliott, J.C.; Mackie, P.E.; Young, R.A. Monoclinic hydroxyapatite. Science 1973, 180, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Rangavittal, N.; Landa-Cánovas, A.R.; González-Calbet, J.M.; Vallet-Regi, M. Structural study and stability of hydroxyapatite and β-tricalcium phosphate: two important bioceramics. J. Biomed. Mater. Res. 2000, 51, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Fenton, R.R.; Hunter, B.A.; Kennedy, B.J. Powder diffraction studies of synthetic calcium and lead apatites. Austr. J. Chem. 2000, 53, 679–686. [Google Scholar] [CrossRef]
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal structure of hydroxyapatite. Nature 1964, 204, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Treboux, G.; Layrolle, P.; Kanzaki, N.; Onuma, K.; Ito, A. Existence of Posner’s cluster in vacuum. J. Phys. Chem. A 2000, 104, 5111–5114. [Google Scholar] [CrossRef]
- Calderin, L.; Stott, M.J.; Rubio, A. Electronic and crystallographic structure of apatites. Phys. Rev. B 2003, 67, 134106, (6 pages). [Google Scholar] [CrossRef]
- Rulis, P.; Ouyang, L.; Ching, W.Y. Electronic structure and bonding in calcium apatite crystals: hydroxyapatite, fluorapatite, chlorapatite and bromapatite. Phys. Rev. B 2004, 70, 155104, (8 pages). [Google Scholar] [CrossRef]
- Treboux, G.; Layrolle, P.; Kanzaki, N.; Onuma, K.; Ito, A. Symmetry of Posner’s cluster. J. Am. Chem. Soc. 2000, 122, 8323–8324. [Google Scholar] [CrossRef]
- Yin, X.; Stott, M.J. Biological calcium phosphates and Posner's cluster. J. Chem. Phys. 2003, 118, 3717–3723. [Google Scholar] [CrossRef]
- Kanzaki, N.; Treboux, G.; Onuma, K.; Tsutsumi, S.; Ito, A. Calcium phosphate clusters. Biomaterials 2001, 22, 2921–2929. [Google Scholar] [CrossRef] [PubMed]
- Calderin, L.; Dunfield, D.; Stott, M.J. Shell-model study of the lattice dynamics of hydroxyapatite. Phys. Rev. B 2005, 72, 224304, (12 pages). [Google Scholar] [CrossRef]
- Briak-Ben, El; Abdeslam, H.; Ginebra, M.P.; Vert, M.; Boudeville, P. Wet or dry mechanochemical synthesis of calcium phosphates? Influence of the water content on DCPD – CaO reaction kinetics. Acta Biomater. 2008, 4, 378–386. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; LeGeros, J.P. Dense hydroxyapatite. In An introduction to bioceramics; Hench, L.L., Wilson, J., Eds.; World Scientific: London, UK, 1993; pp. 139–180. [Google Scholar]
- Rodriguez-Lorenzo, L.M.; Vallet-Regi, M. Controlled crystallization of calcium phosphate apatites. Chem. Mater. 2000, 12, 2460–2465. [Google Scholar] [CrossRef]
- Cazalbou, S.; Combes, C.; Eichert, D.; Rey, C. Adaptive physico-chemistry of bio-related calcium phosphates. J. Mater. Chem. 2004, 14, 2148–2153. [Google Scholar] [CrossRef]
- Markovic, M.; Fowler, B.O.; Tung, M.S. Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 553–568. [Google Scholar] [CrossRef]
- Ioku, K.; Kawachi, G.; Sasaki, S.; Fujimori, H.; Goto, S. Hydrothermal preparation of tailored hydroxyapatite. J. Mater. Sci. 2006, 41, 1341–1344. [Google Scholar] [CrossRef]
- Layrolle, P.; Lebugle, A. Characterization and reactivity of nanosized calcium phosphates prepared in anhydrous ethanol. Chem. Mater. 1994, 6, 1996–2004. [Google Scholar] [CrossRef]
- Layrolle, P.; Lebugle, A. Synthesis in pure ethanol and characterization of nanosized calcium phosphate fluoroapatite. Chem. Mater. 1996, 8, 134–144. [Google Scholar] [CrossRef]
- Yeong, B.; Junmin, X.; Wang, J. Mechanochemical synthesis of hydroxyapatite from calcium oxide and brushite. J. Am. Ceram. Soc. 2001, 84, 465–467. [Google Scholar] [CrossRef]
- Roy, D.M.; Linnehan, S.K. Hydroxyapatite formed from coral skeletal carbonate by hydrothermal exchange. Nature 1974, 247, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, B. Natural bioceramics: from coral to bone and beyond. Curr. Opin. Solid State Mater. Sci. 2003, 7, 283–288. [Google Scholar] [CrossRef]
- Ben-Nissan, B.; Milev, A.; Vago, R. Morphology of sol-gel derived nano-coated coralline hydroxyapatite. Biomaterials 2004, 25, 4971–4975. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C.; Young, R.A. Conversion of single crystals of chlorapatite into single crystals of hydroxyapatite. Nature 1967, 214, 904–906. [Google Scholar] [CrossRef]
- Tao, J.; Jiang, W.; Pan, H.; Xu, X.; Tang, R. Preparation of large-sized hydroxyapatite single crystals using homogeneous releasing controls. J. Cryst. Growth 2007, 308, 151–158. [Google Scholar] [CrossRef]
- Vallet-Regi, M.; Gutiėrrez Rios, M.T.; Alonso, M.P.; de Frutos, M.I.; Nicolopoulos, S. Hydroxyapatite particles synthesized by pyrolysis of an aerosol. J. Solid State Chem. 1994, 112, 58–64. [Google Scholar] [CrossRef]
- Montero, M.L.; Sáenz, A.; Rodríguez, J.G.; Arenas, J.; Castaño, V.M. Electrochemical synthesis of nanosized hydroxyapatite. J. Mater. Sci. 2006, 41, 2141–2144. [Google Scholar] [CrossRef]
- Kumar, A.R.; Kalainathan, S. Growth and characterization of nano-crystalline hydroxyapatite at physiological conditions. Crystal Res. Technol. 2008, 43, 640–644. [Google Scholar] [CrossRef]
- Kalita, S.J.; Bhardwaj, A.; Bhatt, H.A. Nanocrystalline calcium phosphate ceramics in biomedical engineering. Mater. Sci. Eng., C 2007, 27, 441–449. [Google Scholar] [CrossRef]
- Melikhov, I.V.; Komarov, V.F.; Severin, A.V.; Bozhevolnov, V.E.; Rudin, V.N. Two-dimensional crystalline hydroxyapatite. Doklady Phys. Chem. 2000, 373, 125–128. [Google Scholar]
- Suvorova, E.I.; Buffat, P.A. Electron diffraction and HRTEM characterization of calcium phosphate precipitation from aqueous solutions under biomineralization conditions. Eur. Cell Mater. 2001, 1, 27–42. [Google Scholar] [PubMed]
- Suvorova, E.I.; Buffat, P.A. Model of the mechanism of Ca loss by bones under microgravity and earth conditions. J. Biomed. Mater. Res. 2002, 63, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, K.; Takagi, M.; Honda, T.; Uchida, N.; Saito, K. Transparent hydroxyapatite prepared by hot isostatic pressing of filter cake. J. Am. Ceram. Soc. 1989, 72, 1476–1478. [Google Scholar] [CrossRef]
- Takikawa, K.; Akao, M. Fabrication of transparent hydroxyapatite and application to bone marrow derived cell / hydroxyapatite interaction observation in vivo. J. Mater. Sci. Mater. Med. 1996, 7, 439–445. [Google Scholar] [CrossRef]
- Watanabe, Y.; Ikoma, T.; Monkawa, A.; Suetsugu, Y.; Yamada, H.; Tanaka, J.; Moriyoshi, Y. Fabrication of transparent hydroxyapatite sintered body with high crystal orientation by pulse electric current sintering. J. Am. Ceram. Soc. 2005, 88, 243–245. [Google Scholar] [CrossRef]
- Kotobuki, N.; Ioku, K.; Kawagoe, D.; Fujimori, H.; Goto, S.; Ohgushi, H. Observation of osteogenic differentiation cascade of living mesenchymal stem cells on transparent hydroxyapatite ceramics. Biomaterials 2005, 26, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Perloff, A.; Posner, A.S. Preparation of pure hydroxyapatite crystals. Science 1956, 124, 583–584. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; LeGeros, J.P.; Daculsi, G.; Kijkowska, R. Calcium phosphate biomaterials: preparation, properties and biodegradation. In Encyclopedic handbook of biomaterials and bioengineering; Part A: Materials; Vol. 2, Wise, D.L., Trantolo, D.J., Altobelli, D.E., Yaszemski, M.J., Gresser, J.D., Schwartz, E.R., Eds.; Marcel Dekker: New York, USA, 1995; pp. 1429–1463. [Google Scholar]
- Narasaraju, T.S.B.; Phebe, D.E. Some physico-chemical aspects of hydroxylapatite. J. Mater. Sci. 1996, 31, 1–21. [Google Scholar] [CrossRef]
- Orlovskii, V.P.; Barinov, S.M. Hydroxyapatite and hydroxyapatite-matrix materials: a survey. Russ. J. Inorg. Chem. 2001, 46 (Suppl. 2), S129–S149. [Google Scholar]
- Orlovskii, V.P.; Komlev, V.S.; Barinov, S.M. Hydroxyapatite and hydroxyapatite-based ceramics. Inorg. Mater. 2002, 38, 973–984. [Google Scholar] [CrossRef]
- Peňa, J.; Vallet-Regi, M. Hydroxyapatite, tricalcium phosphate and biphasic materials prepared by a liquid mix technique. J. Eur. Ceram. Soc. 2003, 23, 1687–1696. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: a review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Rakovan, J. Growth and surface properties of apatite. In Phosphates: geochemical, geobiological and materials importance; Series: Reviews in Mineralogy and Geochemistry; Vol. 48, Hughes, J.M., Kohn, M., Rakovan, J., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2002; pp. 51–86. [Google Scholar]
- Dorozhkin, S.V. A review on the dissolution models of calcium apatites. Prog. Cryst. Growth Charact. Mater. 2002, 44, 45–61. [Google Scholar] [CrossRef]
- Suchanek, W.; Yoshimura, M. Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J. Mater. Res. 1998, 13, 94–117. [Google Scholar] [CrossRef]
- Willmann, G. Coating of implants with hydroxyapatite – material connections between bone and metal. Adv. Eng. Mater. 1999, 1, 95–105. [Google Scholar] [CrossRef]
- Sun, L.; Berndt, C.C.; Gross, K.A.; Kucuk, A. Review: material fundamentals and clinical performance of plasma sprayed hydroxyapatite coatings. J. Biomed. Mater. Res. B Appl. Biomater. 2001, 58, 570–592. [Google Scholar] [CrossRef]
- Ong, J.L.; Chan, D.C.N. Hydroxyapatites and their use as coatings in dental implants: a review. Crit. Rev. Biomed. Eng. 1999, 28, 667–707. [Google Scholar] [CrossRef]
- Geesink, R.G. Osteoconductive coatings for total joint arthroplasty. Clin. Orthop. Rel. Res. 2002, 395, 53–65. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics: from concept to clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics. J. Am. Ceram. Soc. 1998, 81, 1705–1728. [Google Scholar] [CrossRef]
- Mangano, C.; Piattelli, A.; Perrotti, V.; Iezzi, G. Dense hydroxyapatite inserted into postextraction sockets: a histologic and histomorphometric 20-year case report. J. Periodontol. 2008, 79, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, W. Chromatography of insulin on calcium phosphate columns. Nature 1956, 178, 994–995. [Google Scholar] [CrossRef]
- Bernardi, G. Chromatography of nucleic acids on hydroxyapatite. Nature 1965, 206, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Duan, X.; Locklin, R.M.; Quijano, M.; Dobson, R.L.; Triffitt, J.T.; Ebetino, F.H.; Russell, G.R. Evaluation of the relative mineral-binding affinities of clinically-relevant bisphosphonates by using hydroxyapatite-column chromatography and adsorption isotherms combined with mass spectrometric analysis. Bone 2008, 42 (Suppl. 1), S90–S91. [Google Scholar] [CrossRef]
- Brand, M.; Rampalli, S.; Chaturvedi, C.P.; Dilworth, F.J. Analysis of epigenetic modifications of chromatin at specific gene loci by native chromatin immunoprecipitation of nucleosomes isolated using hydroxyapatite chromatography. Nat. Protoc. 2008, 3, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, T.; Kobayashi, S.; Ogawa, T.; Okuyama, T. Hydroxyapatite chromatography of guanidine denatured proteins: 1. Guanidine containing phosphate buffer system. Chromatography 2006, 27, 19–26. [Google Scholar]
- Smith, G.P.; Gingrich, T.R. Hydroxyapatite chromatography of phage-display virions. Biotechniques 2005, 39, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Jungbauer, A.; Hahn, R.; Deinhofer, K.; Luo, P. Performance and characterization of a nanophased porous hydroxyapatite for protein chromatography. Biotechnol. Bioeng. 2004, 87, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Doonan, S. Chromatography on hydroxyapatite. Methods Mol. Biol. 2004, 244, 191–194. [Google Scholar] [PubMed]
- Liu, T.Y.; Chen, S.Y.; Liu, D.M.; Liou, S.C. On the study of BSA-loaded calcium-deficient hydroxyapatite nano-carriers for controlled drug delivery. J. Control. Release 2005, 107, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, B.; Sidoti, M.C.; Roveri, N.; Tampieri, A.; Sandri, M.; Bertolazzi, L.; Galbusera, F.; Dubini, G.; Vena, P.; Contro, R. Controlled drug delivery from porous hydroxyapatite grafts: an experimental and theoretical approach. Mater. Sci. Eng., C 2005, 25, 207–213. [Google Scholar] [CrossRef]
- Pietrasik, J.; Szustakiewicz, K.; Zaborski, M.; Haberko, K. Hydroxyapatite: an environmentally friendly filler for elastomers. Mol. Cryst. Liq. Cryst. 2008, 483, 172–178. [Google Scholar] [CrossRef]
- Corami, A.; Mignardi, S.; Ferrini, V. Cadmium removal from single- and multi-metal (Cd plus Pb plus Zn plus Cu) solutions by sorption on hydroxyapatite. J. Colloid Interface Sci. 2008, 317, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Phonthammachai, N.; Ziyi, Z.; Jun, G.; Fan, H.Y.; White, T.J. Synthesis of high performance hydroxyapatite-gold catalysts for CO oxidation. Gold Bull. 2008, 41, 42–50. [Google Scholar] [CrossRef]
- Chen, W.; Huang, Z.L.; Liu, Y.; He, Q.J. Preparation and characterization of a novel solid base catalyst hydroxyapatite loaded with strontium. Catal. Commun. 2008, 9, 516–521. [Google Scholar] [CrossRef]
- de Groot, K.; Wolke, J.G.C.; Jansen, J.A. Calcium phosphate coatings for medical implants. Proc. Inst. Mech. Eng. Part Η: J. Eng. Med. 1998, 212, 137–147. [Google Scholar] [CrossRef]
- Gross, K.A.; Berndt, C.C. Biomedical application of apatites. In Phosphates: geochemical, geobiological and materials importance; Series: Reviews in Mineralogy and Geochemistry; Vol. 48, Hughes, J.M., Kohn, M., Rakovan, J., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2002; pp. 631–672. [Google Scholar]
- Ferraz, M.P.; Monteiro, F.J.; Manuel, C.M. Hydroxyapatite nanoparticles: a review of preparation methodologies. J. Appl. Biomater. Biomech. 2004, 2, 74–80. [Google Scholar] [PubMed]
- Damien, E.; Revell, P.A. Coralline hydroxyapatite bone graft substitute: a review of experimental studies and biomedical applications. J. Appl. Biomater. Biomech. 2004, 2, 65–73. [Google Scholar] [PubMed]
- Aoki, H. Science and medical applications of hydroxyapatite; JAAS: Tokyo, Japan, 1991; p. 245. [Google Scholar]
- Barinov, S.M.; Komlev, V.S. Calcium phosphate based bioceramics for bone tissue engineering; Materials Science Foundations (monograph series); Vol. 48, Trans Tech Publ.: Switzerland, 2008; p. 170. [Google Scholar]
- Kniep, R.; Busch, S. Biomimetic growth and self-assembly of fluorapatite aggregates by diffusion into denatured collagen matrices. Angew. Chem. Int. Ed. Engl. 1996, 35, 2624–2626. [Google Scholar] [CrossRef]
- Busch, S.; Dolhaine, H.; Duchesne, A.; Heinz, S.; Hochrein, O.; Laeri, F.; Podebrad, O.; Vietze, U.; Weiland, T.; Kniep, R. Biomimetic morphogenesis of fluorapatite-gelatin composites: fractal growth, the question of intrinsic electric fields, core / shell assemblies, hollow spheres and reorganization of denatured collagen. Eur. J. Inorg. Chem. 1999, 1643–1653. [Google Scholar] [CrossRef]
- GÖBEL, C.; Simon, P.; Buder, J.; Tlatlik, H.; Kniep, R. Phase formation and morphology of calcium phosphate-gelatine-composites grown by double diffusion technique: the influence of fluoride. J. Mater. Chem. 2004, 14, 2225–2230. [Google Scholar] [CrossRef]
- Prymak, O.; Sokolova, V.; Peitsch, T.; Epple, M. The crystallization of fluoroapatite dumbbells from supersaturated aqueous solution. Cryst. Growth Des. 2006, 6, 498–506. [Google Scholar] [CrossRef]
- Tlatlik, H.; Simon, P.; Kawska, A.; Zahn, D.; Kniep, R. Biomimetic fluorapatite-gelatine nanocomposites: pre-structuring of gelatine matrices by ion impregnation and its effect on form development. Angew. Chem. Int. Ed. Engl. 2006, 45, 1905–1910. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Zahn, D.; Lichte, H.; Kniep, R. Intrinsic electric dipole fields and the induction of hierarchical form developments in fluorapatite-gelatine nanocomposites: a general principle for morphogenesis of biominerals? Angew. Chem. Int. Ed. Engl. 2006, 45, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. A hierarchical structure for apatite crystals. J. Mater. Sci. Mater. Med. 2007, 18, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Mehmel, M. On the structure of apatite: I. Z. Kristallogr. 1930, 75, 323–331. [Google Scholar]
- Naray-Szabo, S. The structure of apatite (CaF)Ca4(PO4)3. Z. Kristallogr. 1930, 75, 387–398. [Google Scholar]
- Sudarsanan, K.; Mackie, P.E.; Young, R.A. Comparison of synthetic and mineral fluorapatite, Ca5(PO4)3F, in crystallographic detail. Mater. Res. Bull. 1972, 7, 1331–1337. [Google Scholar] [CrossRef]
- Barinov, S.M.; Shvorneva, L.I.; Ferro, D.; Fadeeva, I.V.; Tumanov, S.V. Solid solution formation at the sintering of hydroxyapatite-fluorapatite ceramics. Sci. Technol. Adv. Mater. 2004, 5, 537–541. [Google Scholar] [CrossRef]
- Nikcevic, I.; Jokanovic, V.; Mitric, M.; Nedic, Z.; Makovec, D.; Uskokovic, D. Mechanochemical synthesis of nanostructured fluorapatite / fluorhydroxyapatite and carbonated fluorapatite / fluorhydroxyapatite. J. Solid State Chem. 2004, 177, 2565–2574. [Google Scholar] [CrossRef]
- Cheng, K.; Weng, W.; Qu, H.; Du, P.; Shen, G.; Han, G.; Yang, J.; Ferreira, J.M.F. Sol-gel preparation and in vitro test of fluorapatite / hydroxyapatite films. J. Biomed. Mater. Res. B Appl.Biomater. 2004, 69B, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lorenzo, L.M.; Hart, J.N.; Gross, K.A. Influence of fluorine in the synthesis of apatites. Synthesis of solid solutions of hydroxy-fluorapatite. Biomaterials 2003, 24, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Evans, J.H.; Bostrom, T.; Grondahl, L. Synthesis and characterization of hydroxyapatite, fluoride-substituted hydroxyapatite and fluorapatite. J. Mater. Sci. Mater. Med. 2003, 14, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G.; Kerebel, L.M. Ultrastructural study and comparative analysis of fluoride content of enameloid in sea-water and fresh-water sharks. Arch. Oral Biol. 1980, 25, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G.; Kerebel, L.M.; Kerebel, B. Effects of fluoride on human enamel and selachian enameloid in vitro: a high-resolution TEM and electron diffraction study. Calcif. Tissue Int. 1981, 33, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Lowenstam, H.A.; Weiner, S. On biomineralization; Oxford University Press: Oxford, UK, 1989; p. 324. [Google Scholar]
- Weiner, S.; Dove, P.M. An overview of biomineralization processes and the problem of the vital effect. In Biomineralization; Series: Reviews in Mineralogy and Geochemistry; Vol. 54, Dove, P.M., de Yoreo, J.J., Weiner, S., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2003; pp. 1–29. [Google Scholar]
- Dahm, S.; Risnes, S. A comparative infrared spectroscopic study of hydroxide and carbonate absorption bands in spectra of shark enameloid, shark dentin, and a geological apatite. Calcif. Tissue Int. 1999, 65, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Kemp, A.; Tibbetts, I.; Truss, R.; Drennan, J. Microstructure of pharyngeal tooth enameloid in the parrotfish Scarus rivulatus (Pisces: Scaridae). J. Microscopy 2006, 221, 8–16. [Google Scholar] [CrossRef]
- Leveque, I.; Cusack, M.; Davis, S.A.; Mann, S. Promotion of fluorapatite crystallization by soluble-matrix proteins from Lingula Anatina shells. Angew. Chem. Int. Ed. Engl. 2004, 43, 885–888. [Google Scholar] [CrossRef] [PubMed]
- The amount of fluorides on the very surface of dental enamel might be increased by using fluoride-containing toothpastes and mouthwashes [317,318,319,320]. Fluoride-containing toothpastes and mouthwashes are widely used in practice due to the well-known anti-cariogenic effect of fluorides that is related to the solubility decreasing [321,322].
- Fonteles, C.S.R.; Zero, D.T.; Moss, M.E.; Fu, J. Fluoride concentrations in enamel and dentin of primary teeth after pre- and postnatal fluoride exposure. Caries Res. 2005, 39, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, A.; Krieger, C.; Attin, R.; Hellwig, E.; Attin, T. Fluoride uptake and resistance to further demineralisation of demineralised enamel after application of differently concentrated acidulated sodium fluoride gels. Clin. Oral. Investig. 2005, 9, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Waszkiel, D.; Opalko, K.; Lagocka, R.; Chlubek, D. Fluoride and magnesium content in superficial enamel layers of teeth with erosions. Fluoride 2004, 37, 271–277. [Google Scholar]
- Vieira, A.P.G.F.; Hancock, R.; Dumitriu, M.; Schwartz, M.; Limeback, H.; Grynpas, M.D. How does fluoride affect dentin microhardness and mineralization? J. Dental Res. 2005, 84, 951–957. [Google Scholar] [CrossRef]
- Driessens, F.C.M. Relation between apatite solubility and anti-cariogenic effect of fluoride. Nature 1973, 243, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.C.; Kresak, M.; Zahradnik, R.T. Fluoridated hydroxyapatite solubility and caries formation. Nature 1974, 247, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Heling, L.; Heindel, R.; Merin, B. Calcium-fluorapatite. A new material for bone implants. J. Oral Implantol. 1981, 9, 548–555. [Google Scholar] [PubMed]
- Bibby, J.K.; Bubb, N.L.; Wood, D.J.; Mummery, P.M. Fluorapatite-mullite glass sputter coated Ti6Al4V for biomedical applications. J. Mater. Sci. Mater. Med. 2005, 16, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.H.; Kim, H.W.; Lee, S.H.; Bae, C.J.; Koh, Y.H.; Kong, Y.M.; Kim, H.E. Stability and cellular responses to fluorapatite-collagen composites. Biomaterials 2005, 26, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Gineste, L.; Gineste, M.; Ranz, X.; Ellefterion, A.; Guilhem, A.; Rouquet, N.; Frayssinet, P. Degradation of hydroxylapatite, fluorapatite and fluorhydroxyapatite coatings of dental implants in dogs. J. Biomed. Mater. Res. 1999, 48, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Bhadang, K.A.; Gross, K.A. Influence of fluorapatite on the properties of thermally sprayed hydroxyapatite coatings. Biomaterials 2004, 25, 4935–4945. [Google Scholar] [CrossRef]
- Gross, K.A.; Bhadang, K.A. Sintered hydroxyfluorapatites. Part III: Sintering and resultant mechanical properties of sintered blends of hydroxyapatite and fluorapatite. Biomaterials 2004, 25, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Barinov, S.M.; Rustichelli, F.; Orlovskii, V.P.; Lodini, A.; Oscarsson, S.; Firstov, S.A.; Tumanov, S.V.; Millet, P.; Rosengren, A. Influence of fluorapatite minor additions on behavior of hydroxyapatite ceramics. J. Mater. Sci. Mater. Med. 2004, 15, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Agathopoulos, S.; Tulyaganov, D.U.; Marques, P.A.A.P.; Ferro, M.C.; Fernandes, M.H.; Correia, R.N. The fluorapatite – anorthite system in biomedicine. Biomaterials 2003, 24, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Wei, M. The effect of fluoride contents in fluoridated hydroxyapatite on osteoblast behavior. Acta Biomater. 2006, 2, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Theiszova, M.; Jantova, S.; Letasiova, S.; Palou, M.; Cipak, L. Cytotoxicity of hydroxyapatite, fluorapatite and fluor-hydroxyapatite: a comparative in vitro study. Neoplasma 2008, 55, 312–316. [Google Scholar] [PubMed]
- To honor Gustav Hilgenstock (1844-1913), a German metallurgist, who first discovered it in Thomas slags nearly 120 years ago [334,335].
- Hilgenstock, G. Eine neue Verbindung von P2O5 und CaO. Stahl Eisen 1883, 3, 498. [Google Scholar]
- Hilgenstock, G. Das vierbasische Kalkphosphat und die Basicitätsstufe des Silicats in der Thomas-Schlacxke. Stahl Eisen 1887, 7, 557–560. [Google Scholar]
- Brown, W.E.; Epstein, E.F. Crystallography of tetracalcium phosphate. J. Res. Nat. Bur. Stand. A 1965, 69A, 547–551. [Google Scholar] [CrossRef]
- Romeo, H.E.; Fanovich, M.A. Synthesis of tetracalcium phosphate from mechanochemically activated reactants and assessment as a component of bone cements. J. Mater. Sci. Mater. Med. 2008, 19, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Gil, F.J.; Ginebra, M.P.; Driessens, F.C.M.; Planell, J.A.; Best, S.M. Calcium phosphate bone cements for clinical applications. Part II: precipitate formation during setting reactions. J. Mater. Sci. Mater. Med. 1999, 10, 177–183. [Google Scholar] [CrossRef] [PubMed]
- McConnell, D.; Posner, A.S. Carbonate in apatites. Science 1961, 134, 213–215. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Effect of carbonate on the lattice parameters of apatite. Nature 1965, 206, 403–404. [Google Scholar] [CrossRef]
- LeGeros, R.Z.; Trautz, O.R.; LeGeros, J.P.; Klein, E.; Shirra, W.P. Apatite crystallites: effects of carbonate on morphology. Science 1967, 155, 1409–1411. [Google Scholar] [CrossRef]
- Astala, R.; Stott, M.J. First principles investigation of mineral component of bone: CO3 substitutions in hydroxyapatite. Chem. Mater. 2005, 17, 4125–4133. [Google Scholar] [CrossRef]
- Lafon, J.P.; Champion, E.; Bernache-Assollant, D. Processing of AB-type carbonated hydroxyapatite Ca10-x(PO4)(6-x)(CO3)(x)(OH)(2-x-2y)(CO3)(y) ceramics with controlled composition. J. Eur. Ceram. Soc. 2008, 28, 139–147. [Google Scholar] [CrossRef]
- Kannan, S.; Rebelo, A.; Lemos, A.F.; Barba, A.; Ferreira, J.M.F. Synthesis and mechanical behaviour of chlorapatite and chlorapatite / β-TCP composites. J. Eur. Ceram. Soc. 2007, 27, 2287–2294. [Google Scholar] [CrossRef]
- Teshima, K.; Yubuta, K.; Ooi, S.; Suzuki, T.; Shishido, T.; Oishi, S. Environmentally friendly growth of calcium chlorapatite whiskers from a sodium chloride flux. Cryst. Growth Des. 2006, 6, 2538–2542. [Google Scholar] [CrossRef]
- Kannan, S.; Goetz-Neunhoeffer, F.; Neubauer, J.; Ferreira, J.M.F. Ionic substitutions in biphasic hydroxyapatite and β-tricalcium phosphate mixtures: structural analysis by Rietveld refinement. J. Am. Ceram. Soc. 2008, 91, 1–12. [Google Scholar] [CrossRef]
- Pushpakanth, S.; Srinivasan, B.; Sastry, T.P.; Mandal, A.B. Biocompatible and antibacterial properties of silver-doped hydroxyapatite. J. Biomed. Nanotechnol. 2008, 4, 62–66. [Google Scholar]
- Kim, T.N.; Feng, Q.L.; Kim, J.O.; Wu, J.; Wang, H.; Chen, G.C.; Cui, F.Z. Antimicrobial effects of metal ions (Ag+, Cu2+, Zn2+) in hydroxyapatite. J. Mater. Sci. Mater. Med. 1998, 9, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Assi, P.; Marycel, B.; Correa, M.; Liberato, W.; Brito, V. Yttrium 90-hydroxyapatite, a new radioisotope for chronic synovitis in hemophilia. Haemophilia 2008, 14, 77. [Google Scholar] [CrossRef]
- Chinol, M.; Vallabhajosula, S.; Goldsmith, S.J.; Klein, M.J.; Deutsch, K.F.; Chinen, L.K.; Brodack, J.W.; Deutsch, E.A.; Watson, B.A.; Tofe, A.J. Chemistry and biological behavior of samarium-153 and rhenium-186-labeled hydroxyapatite particles: potential radiopharmaceuticals for radiation synovectomy. J. Nucl. Med. 1993, 34, 1536–1542. [Google Scholar] [PubMed]
- Argüelles, M.G.; Berlanga, I.S.L.; Torres, E.A. Preparation of 153Sm-particles for radiosynovectomy. J. Radioanal. Nucl. Chem. 1999, 240, 509–511. [Google Scholar] [CrossRef]
- O’Duffy, E.; Clunie, G.; Lui, D.; Edwards, J.; Ell, P. Double blind glucocorticoid controlled trial of samarium-153 particulate hydroxyapatite radiation synovectomy for chronic knee synovitis. Ann. Rheum. Dis. 1999, 58, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.C.; Newcomb, C.J.; Kaltz, S.R.; Spoerke, E.D.; Stupp, S.I. Biomimetic systems for hydroxyapatite mineralization inspired by bone and enamel. Chem. Rev. 2008, 108, 4754–4783. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L. Assessment of bone mineral and matrix using backscatter electron imaging and FTIR imaging. Curr. Osteoporos. Rep. 2006, 4, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regi, M.; González-Calbet, J.M. Calcium phosphates as substitution of bone tissues. Progr. Solid State Chem. 2004, 32, 1–31. [Google Scholar] [CrossRef]
- Holt, L.E.; la Mer, V.K.; Chown, H.B. Studies in calcification. I. The solubility product of secondary and tertiary calcium phosphate under various conditions. J. Biol. Chem. 1925, 64, 509–565. [Google Scholar]
- Holt, L.E.; la Mer, V.K.; Chown, H.B. Studies in calcification. II. Delayed equilibrium between the calcium phosphates and its biological significance. J. Biol. Chem. 1925, 64, 567–578. [Google Scholar]
- Gassmann, T. The preparation of a complex salt corresponding to apatite-typus and its relations to the constitution of bones. H.-S. Z. Physiol. Chem. 1913, 83, 403–408. [Google Scholar] [CrossRef]
- de Jong, W.F. La substance minérale dans les os. Recl. Trav. Chim. Pays-Bas 1926, 45, 445–449. [Google Scholar] [CrossRef]
- Bredig, M.A. The apatite structure of inorganic bone and tooth substance. H.-S. Z. Physiol. Chem. 1933, 216, 239–243. [Google Scholar] [CrossRef]
- Taylor, N.W.; Sheard, C. Microscopic and X-ray investigations on the calcification of tissue. J. Biol. Chem. 1929, 81, 479–493. [Google Scholar]
- Weiner, S.; Wagner, H.D. Material bone: structure-mechanical function relations. Ann. Rev. Mater. Sci. 1998, 28, 271–298. [Google Scholar] [CrossRef]
- Weiner, S.; Traub, W.; Wagner, H.D. Lamellar bone: structure-function relations. J. Struct. Biol. 1999, 126, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Limeback, H. Molecular mechanisms in dental hard tissue mineralization. Curr. Opin. Dent. 1991, 1, 826–835. [Google Scholar] [PubMed]
- Matkovic, V. Calcium metabolism and calcium requirements during skeletal modeling and consolidation of bone mass. Am. J. Clin. Nutr. 1991, 54, 245S–260S. [Google Scholar] [PubMed]
- Power, M.L.; Heaney, R.P.; Kalkwarf, H.J.; Pitkin, R.M.; Repke, J.T.; Tsang, R.C.; Schulkin, J. The role of calcium in health and disease. Am. J. Obstetrics Gynecol. 1999, 181, 1560–1569. [Google Scholar] [CrossRef]
- Elliott, J.C. Calcium phosphate biominerals. In Phosphates: geochemical, geobiological and materials importance; Series: Reviews in Mineralogy and Geochemistry; Vol. 48, Hughes, J.M., Kohn, M., Rakovan, J., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2002; pp. 13–49. [Google Scholar]
- Jones, F.H. Teeth and bones: application of Surface Science to dental materials and related biomaterials. Surf. Sci. Rep. 2001, 42, 75–205. [Google Scholar] [CrossRef]
- Loveridge, N. Bone: more than a stick. J. Anim. Sci. 1999, 77 (Suppl. 2), 190–196. [Google Scholar] [PubMed]
- Nightingale, J.P.; Lewis, D. Pole figures of the orientation of apatite in bones. Nature 1971, 232, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Currey, J.D. Bones: structure and mechanics; Princeton Univercity Press: Princeton, USA, 2002; p. 456. [Google Scholar]
- Rho, J.Y.; Kuhn-Spearing, L.; Zioupos, P. Mechanical properties and the hierarchical structure of bone. Med. Eng. Phys. 1998, 20, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Mann, S. Biomimetic materials chemistry; VCH: Weinheim, Germany, 1996; p. 400. [Google Scholar]
- Hancox, N.M. Biology of bone; Cambridge University Press: Cambridge, MA, USA, 1972; p. 199. [Google Scholar]
- Martin, R.B. Bone as a ceramic composite material. Mater. Sci. Forum 1999, 7, 5–16. [Google Scholar] [CrossRef]
- Tzaphlidou, M. Bone architecture: collagen structure and calcium / phosphorus maps. J. Biol. Phys. 2008, 34, Spec. Iss.. 1-2, 39-49. [Google Scholar] [CrossRef]
- Davison, K.S.; Siminoski, K.; Adachi, J.D.; Hanley, D.A.; Goltzman, D.; Hodsman, A.B.; Josse, R.; Kaiser, S.; Olszynski, W.P.; Papaioannou, A.; Ste-Marie, L.G.; Kendler, D.L.; Tenenhouse, A.; Brown, J.P. Bone strength: the whole is greater than the sum of its parts. Semin. Arthritis Rheum. 2006, 36, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.H.; Burr, D.B. Basic biomechanical measurements of bone: a tutorial. Bone 1993, 14, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Currey, J.D. Tensile yield in compact bone is determined by strain, post-yield behaviour by mineral content. J. Biomech. 2004, 37, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Currey, J.D.; Brear, K.; Zioupos, P. Notch sensitivity of mammalian mineralized tissues in impact. Proc. R. Soc. London, Ser. B 2004, 217, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.C.; Eriksson, C. Piezoelectric properties of dry and wet bone. Nature 1970, 227, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.B. Pyroelectric effect in bone and tendon. Nature 1966, 212, 704–705. [Google Scholar] [CrossRef]
- Haynes, V. Radiocarbon: analysis of inorganic carbon of fossil bone and enamel. Science 1968, 161, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Rensberger, J.M.; Watabe, M. Fine structure of bone in dinosaurs, birds and mammals. Nature 2000, 406, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Kolodny, Y.; Luz, B.; Sander, M.; Clemens, W.A. Dinosaur bones: fossils or pseudomorphs? The pitfalls of physiology reconstruction from apatitic fossils. Palaeo 1996, 126, 161–171. [Google Scholar] [CrossRef]
- Trueman, C.N.; Tuross, N. Trace elements in recent and fossil bone apatite, In Phosphates: geochemical, geobiological and materials importance; Series: Reviews in Mineralogy and Geochemistry; Vol. 48, Hughes, J.M., Kohn, M., Rakovan, J., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2002; pp. 489–522. [Google Scholar]
- Currey, J.D.; Brear, K. Hardness, Young’s modulus and yield stress in mammalian mineralized tissues. J. Mater. Sci. Mater. Med. 1990, 1, 14–20. [Google Scholar] [CrossRef]
- Brown, W.E.; Chow, L.C. Chemical properties of bone mineral. Ann. Rev. Mater. Sci. 1976, 6, 213–236. [Google Scholar] [CrossRef]
- Lakes, R. Materials with structural hierarchy. Nature 1993, 361, 511–515. [Google Scholar] [CrossRef]
- Meyers, M.A.; Lin, A.Y.M.; Seki, Y.; Chen, P.Y.; Kad, B. K.; Bodde, S. Structural biological composites: an overview. JOM 2006, 58, 36–43. [Google Scholar]
- Wahl, D.A.; Czernuszka, J.T. Collagen – hydroxyapatite composites for hard tissue repair. Eur. Cell Mater. 2006, 11, 43–56. [Google Scholar] [PubMed]
- Watt, J.C. The deposition of calcium phosphate and calcium carbonate in bone and in areas of calcification. Arch. Surg. Chicago 1925, 10, 983–990. [Google Scholar] [CrossRef]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: a new perspective. Mater. Sci. Eng. R 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Boskey, A.L. Mineralization of bones and teeth. Elements 2007, 3, 385–391. [Google Scholar] [CrossRef]
- Glimcher, M.J. Bone: nature of the calcium phosphate crystals and cellular, structural and physical chemical mechanisms in their formation. In Medical Mineralogy and Geochemistry; Series: Reviews in Mineralogy and Geochemistry; Vol. 64, Sahai, N., Schoonen, M.A.A., Eds.; Mineralogical Society of America: Washington, D.C., USA, 2006; pp. 223–282. [Google Scholar]
- Boskey, A.L.; Roy, R. Cell culture systems for studies of bone and tooth mineralization. Chem. Rev. 2008, 108, 4716–4733. [Google Scholar] [CrossRef] [PubMed]
- McKee, M.D.; Addison, W.N.; Kaartinen, M.T. Hierarchies of extracellular matrix and mineral organization in bone of the craniofacial complex and skeleton. Cell Tiss. Organ 2005, 181, 176–188. [Google Scholar] [CrossRef]
- Currey, J.D. Hierarchies in biomineral structures. Science 2005, 309, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Fantner, G.E.; Rabinovych, O.; Schitter, G.; Thurner, P.; Kindt, J.H.; Finch, M.M.; Weaver, J.C.; Golde, L.S.; Morse, D.E.; Lipman, E.A.; Rangelow, I.W.; Hansma, P.K. Hierarchical interconnections in the nano-composite material bone: fibrillar cross-links resist fracture on several length scales. Composite Sci. Technol. 2006, 66, 1202–1208. [Google Scholar] [CrossRef]
- Meyers, M.A.; Chen, P.Y.; Lin, A.Y.M.; Seki, Y. Biological materials: structure and mechanical properties. Prog. Mater. Sci. 2008, 53, 1–206. [Google Scholar] [CrossRef]
- Athanasiou, K.A.; Zhu, C.; Lanctot, D.R.; Agrawal, C.M.; Wang, X. Fundamentals of biomechanics in tissue engineering of bone. Tissue Eng. 2000, 6, 361–381. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, S.; Raabe, D. Hierarchical modeling of the elastic properties of bone at submicron scales: the role of extrafibrillar mineralization. Biophys. J. 2008, 94, 4220–4232. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.S.; Wagermaier, W.; Zickler, G.A.; Aroush, D.R.B.; Funari, S.S.; Roschger, P.; Wagner, H.D.; Fratzl, P. Nanoscale deformation mechanisms in bone. Nano Lett. 2005, 5, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Peterlik, H.; Roschger, P.; Klaushofer, K.; Fratzl, P. From brittle to ductile fracture of bone. NatureMater 2006, 5, 52–55. [Google Scholar]
- Ruppel, M.E.; Miller, L.M.; Burr, D.B. The effect of the microscopic and nanoscale structure on bone fragility. Osteop. Int. 2008, 19, 1251–1265. [Google Scholar] [CrossRef]
- Fratzl, P.; Gupta, H.S.; Paschalis, E.P.; Roschger, P. Structure and mechanical quality of the collagen-mineral nano-composite in bone. J. Mater. Chem. 2004, 14, 2115–2123. [Google Scholar] [CrossRef]
- Marino, A.A.; Becker, R.O. Evidence for direct physical bonding between the collagen fibres and apatite crystals in bone. Nature 1967, 213, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Eppell, S.J.; Tong, W.; Katz, J.L.; Kuhn, L.; Glimcher, M.J. Shape and size of isolated bone mineralites measured using atomic force microscopy. J. Orthop. Res. 2001, 19, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.M.; Iball, J. Orientation of apatite crystals in bone. Nature 1954, 174, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Jasiuk, I.; Taylor, J.; Rubin, J.; Ganey, T.; Apkarian, R.P. TEM analysis of the nanostructure of normal and osteoporotic human trabecular bone. Bone 2003, 33, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Sun, K.; Cui, F.Z.; Landis, W.J. Organization of apatite crystals in human woven bone. Bone 2003, 32, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Due to the nanoscopic dimensions, biological apatite is occasionally called “nano-apatite” [362].
- Self-assembling is the autonomous organization of components into patterns or structures without human intervention. It is considered that self-assembling processes are common throughout nature and technology [414].
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Fratzl, P.; Fratzl-Zelman, N.; Klaushofer, K.; Vogl, G.; Koller, K. Nucleation and growth of mineral crystals in bone studied by small-angle X-ray scattering. Calcif. Tissue Int. 1991, 48, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J.; Hodgens, K.J.; Arena, J.; Song, M.J.; McEwen, B.F. Structural relations between collagen and mineral in bone as determined by high voltage electron microscopic tomography. Microsc. Res. Techn. 1996, 33, 192–202. [Google Scholar] [CrossRef]
- Rosen, V.B.; Hobbs, L.W.; Spector, M. The ultrastructure of anorganic bovine bone and selected synthetic hyroxyapatites used as bone graft substitute materials. Biomaterials 2002, 23, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.Z.; Li, Y.; Ge, J. Self-assembly of mineralized collagen composites. Mater. Sci. Eng. R 2007, 57, 1–27. [Google Scholar] [CrossRef]
- Sato, K. Inorganic-organic interfacial interactions in hydroxyapatite mineralization processes. Topics Curr. Chem. 2006, 270, 127–153. [Google Scholar]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Burger, C.; Zhou, H.; Wang, H.; Sics, I.; Hsiao, B.S.; Chu, B.; Graham, L.; Glimcher, M. Lateral packing of mineral crystals in bone collagen fibrils. Biophys. J. 2008, 95, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Veis, A. Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition. Chem. Rev. 2008, 108, 4670–4693. [Google Scholar] [CrossRef] [PubMed]
- Cartilage: structure, function and biochemistry; Hall, B.K. (Ed.) Academic Press: New York, USA, 1983; p. 385.
- Robins, S.P.; Bilezikian, J.P.; Seibel, M.J. Dynamics of bone and cartilage metabolism; Academic Press: New York, USA, 1999; p. 672. [Google Scholar]
- Bones and cartilage: developmental skeletal biology; Hall, B.K. (Ed.) Academic Press: New York, USA, 2005; p. 792.
- Marino, A.A.; Becker, R.O. Evidence for epitaxy in the formation of collagen and apatite. Nature 1970, 226, 652–653. [Google Scholar] [CrossRef] [PubMed]
- Jodaikin, A.; Weiner, S.; Talmon, Y.; Grossman, E.; Traub, W. Mineral-organic matrix relations in tooth enamel. Int. J. Biol. Macromol. 1988, 10, 349–352. [Google Scholar] [CrossRef]
- Fincham, A.G.; Moradian-Oldak, J.; Diekwisch, T.G.H.; Lyaruu, D.M.; Wright, J.T.; Bringas, P.; Slavkin, H.C. Evidence for amelogenin “nanospheres” as functional components of secretory-stage enamel matrix. J. Struct. Biol. 1995, 115, 50–59. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; Pan, C.M.; Suga, S.; Watabe, N. Crystallo-chemical properties of apatite in atremate brachiopod shells. Calcif. Tissue Int. 1985, 37, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Moriwaki, Y. Orientation of apatite and organic matrix in Lingula unguis shell. Calcif. Tissue Int. 1990, 47, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Cusack, M.; Buckman, J.O.; Stachel, T. Siliceous tablets in the larval shells of apatitic discinid Brachiopods. Science 1998, 297, 2094–2096. [Google Scholar] [CrossRef]
- Rohanizadeh, R.; LeGeros, R.Z. Mineral phase in linguloid brachiopod shell: Lingula adamsi. Lethaia 2007, 40, 61–68. [Google Scholar] [CrossRef]
- Nakano, T.; Ishimoto, T.; Lee, J.W.; Umakoshi, Y. Preferential orientation of biological apatite crystallite in original, regenerated and diseased cortical bones. J. Ceram. Soc. Jpn. 2008, 116, 313–315. [Google Scholar] [CrossRef]
- Nakano, T.; Ishimoto, T.; Umakoshi, Y.; Tabata, Y. Variation in bone quality during regenerative process. Mater. Sci. Forum 2007, 539-543, 675–680. [Google Scholar] [CrossRef]
- Suvorova, E.I.; Petrenko, P.P.; Buffat, P.A. Scanning and transmission electron microscopy for evaluation of order / disorder in bone structure. Scanning 2007, 29, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.R.; Blackburn, D.; Gradassi, M.; Simon, H.G. Bone formation during forelimb regeneration: a microtomography (microct) analysis. Dev. Dyn. 2003, 226, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A.; Martin, T.J. Therapeutic approaches to bone diseases. Science 2000, 289, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Schilling, A.F.; Filke, S.; Brink, S.; Korbmacher, H.; Amling, M.; Rueger, J.M. Osteoclasts and biomaterials. Eur. J. Trauma 2006, 32, 107–113. [Google Scholar] [CrossRef]
- Bonar, L.C.; Shimizu, M.; Roberts, J.E.; Griffin, R.G.; Glimcher, M.J. Structural and composition studies on the mineral of newly formed dental enamel: a chemical, X-ray diffraction, and 31P and proton nuclear magnetic resonance study. J. Bone Miner. Res. 1991, 6, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Biltz, R.M.; Pellegrino, E.D. The hydroxyl content of calcified tissue mineral. Calcif. Tissue Res. 1971, 7, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Renugopalakrishnan, V.; Collins, B.; Glimcher, M.J. Fourier transform infrared spectroscopic study of the carbonate ions in bone mineral during aging. Calcif. Tissue Int. 1991, 49, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Miquel, J.L.; Facchini, L.; Legrand, A.P.; Glimcher, M.J. Hydroxyl groups in bone mineral. Bone 1995, 16, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Loong, C.K.; Rey, C.; Kuhn, L.T.; Combes, C.; Wu, Y.; Chen, S.H.; Glimcher, M.J. Evidence of hydroxyl-ion deficiency in bone apatites: an inelastic neutron scattering study. Bone 2000, 26, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.; Wu, Y.; Ackerman, J.L. Detection of hydroxyl ions in bone mineral by solid-state NMR spectroscopy. Science 2003, 300, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Grynpas, M.D.; Omelon, S. Transient precursor strategy or very small biological apatite crystals? Bone 2007, 41, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.S.; Brown, W.E. An intermediate state in hydrolysis of amorphous calcium phosphate. Calcif. Tissue Int. 1983, 35, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.S.; Brown, W.E. The role of octacalcium phosphate in subcutaneous heterotopic calcification. Calcif. Tissue Int. 1985, 37, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.E.; Eidelman, N.; Tomazic, B.B. Octacalcium phosphate as a precursor in biomineral formation. Adv. Dent. Res. 1987, 1, 306–313. [Google Scholar] [PubMed]
- Siew, C.; Gruninger, S.E.; Chow, L.C.; Brown, W.E. Procedure for the study of acidic calcium phosphate precursor phases in enamel mineral formation. Calcif. Tissue Int. 1992, 50, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Crane, N.J.; Popescu, V.; Morris, M.D.; Steenhuis, P.; Ignelzi, J.M.A. Raman spectroscopic evidence for octacalcium phosphate and other transient mineral species deposited during intramembranous mineralization. Bone 2006, 39, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Mou, C.Y.; Chan, J.C.C. Solid-state NMR study of the transformation of octacalcium phosphate to hydroxyapatite: a mechanistic model for central dark line formation. J. Am. Chem. Soc. 2006, 128, 6909–6918. [Google Scholar] [CrossRef] [PubMed]
- Eanes, E.D.; Gillessen, I.H.; Posner, A.S. Intermediate states in the precipitation of hydroxyapatite. Nature 1965, 208, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Termine, J.D.; Posner, A.S. Infrared analysis of rat bone: age dependency of amorphous and crystalline mineral fractions. Science 1966, 153, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Termine, J.D.; Posner, A.S. Infra-red determination of the percentage of crystallinity in apatitic calcium phosphates. Nature 1966, 211, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.A.; Posner, A.S. Measurement of non-crystalline calcium phosphate in bone mineral. Proc. Soc. Exptl. Biol. Med. 1966, 122, 137–142. [Google Scholar] [CrossRef]
- Posner, A.S. Crystal chemistry of bone mineral. Physiol. Rev. 1969, 49, 760–792. [Google Scholar] [PubMed]
- Posner, A.S. Bone mineral on the molecular level. Fed. Proc. 1973, 32, 1933–1937. [Google Scholar] [PubMed]
- Boskey, A.L.; Posner, A.S. Formation of hydroxyapatite at low supersaturation. J. Phys. Chem. 1976, 80, 40–44. [Google Scholar] [CrossRef]
- Posner, A.S. The chemistry of bone mineral. Bull. Hosp. Joint Dis. 1978, 39, 126–144. [Google Scholar] [PubMed]
- Glimcher, M.J.; Bonar, L.C.; Grynpas, M.D.; Landis, W.J.; Roufosse, A.H. Recent studies of bone mineral: is the amorphous calcium phosphate theory valid? J. Cryst. Growth 1981, 53, 100–119. [Google Scholar] [CrossRef]
- Meyer, J.L.; Eanes, E.D. A thermodynamic analysis of the amorphous to crystalline calcium phosphate transformation. Calcif. Tissue Res. 1978, 25, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.L.; Eanes, E.D. A thermodynamic analysis of the secondary transition in the spontaneous precipitation of calcium phosphate. Calcif. Tissue Res. 1978, 25, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Politi, Y.; Arad, T.; Klein, E.; Weiner, S.; Addadi, L. Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase. Science 2004, 306, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Sagi, I.; Addadi, L. Choosing the crystallization path less traveled. Science 2005, 309, 1027–1028. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S. Transient precursor strategy in mineral formation of bone. Bone 2006, 39, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Pekounov, Y.; Petrov, O.E. Bone resembling apatite by amorphous-to-crystalline transition driven self-organisation. J. Mater. Sci. Mater. Med. 2008, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, J.; Sharir, A.; Addadi, L.; Weiner, S. Amorphous calcium phosphate is a major component of the forming fin bones of zebrafish: indications for an amorphous precursor phase. Proc. Natl. Acad. Sci. USA 2008, 105, 12748–12753. [Google Scholar] [CrossRef] [PubMed]
- Sahar, N.D.; Hong, S.I.; Kohn, D.H. Micro- and nano-structural analyses of damage in bone. Micron 2005, 36, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Bilezikian, J.P.; Raisz, L.G.; Martin, T.J. Principles of Bone Biology, 3rd Ed. ed; Academic Press: New York, USA, 2008; p. 1900. [Google Scholar]
- Burnell, J.M.; Teubner, E.J.; Miller, A.G. Normal maturational changes in bone matrix, mineral, and crystal size in the rat. Calcif. Tissue Int. 1980, 31, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Traub, W. Organization of hydroxyapatite crystals within collagen fibrils. FEBS Lett. 1986, 206, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, E.D.; Blitz, R.M. Mineralization in the chick embryo I. Monohydrogen phosphate and carbonate relationships during maturation of the bone crystal complex. Calcif. Tissue Res. 1972, 10, 128–135. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; Balmain, N.; Bonel, G. Age-related changes in mineral of rat and bovine cortical bone. Calcif. Tissue Int. 1987, 41, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Hina, A.; Tofighi, A.; Glimcher, M.J. Maturation of poorly crystalline apatites: chemical and structural aspect in vivo and in vitro behavior. Cell Mater. 1995, 5, 345–356. [Google Scholar]
- Verdelis, K.; Lukashova, L.; Wright, J.T.; Mendelsohn, R.; Peterson, M.G.E.; Doty, S.; Boskey, A.L. Maturational changes in dentin mineral properties. Bone 2007, 40, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Plate, U.; Tkotz, T.; Wiesmann, H.P.; Stratmann, U.; Joos, U.; Höhling, H.J. Early mineralization of matrix vesicles in the epiphyseal growth plate. J. Microsc. 1996, 183, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, U.; Schaarschmidt, K.; Wiesmann, H.P.; Plate, U.; Höhling, H.J. Mineralization during matrix-vesicle-mediated mantle dentine formation in molars of albino rats: a microanalytical and ultrastructural study. Cell Tiss. Res. 1996, 284, 223–230. [Google Scholar] [CrossRef]
- Jahnen-Dechent, W.; Schinke, T.; Trindl, A.; Muller-Esterl, W.; Sablitzky, F.; Kaiser, S.; Blessing, M. Cloning and targeted deletion of the mouse fetuin gene. J. Biol. Chem. 1997, 272, 31496–31503. [Google Scholar] [CrossRef] [PubMed]
- Schinke, T.; McKnee, M.D.; Karsenty, G. Extracellular matrix calcification: where is the action? Nat.Genet. 1999, 21, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Jahnen-Dechent, W.; Schäfer, G.; Heiss, A.; Grötzinger, J. Systemic inhibition of spontaneous calcification by the serum protein a2-HS glycoprotein / fetuin. Z. Kardiol. 2001, 90 (Suppl. 3), III/47-III/56. [Google Scholar] [CrossRef]
- Avery, J.K. Oral development and histology, 3rd Ed. ed; Thieme Medical Publishers Inc.: New York, USA, 2001; p. 435. [Google Scholar]
- ten Cate, A.R. Oral histology: development, structure, and function, 5th Ed. ed; Mosby-Year Book: Saint Louis, USA, 1998; p. 497. [Google Scholar]
- Xue, J.; Zhang, L.; Zou, L.; Liao, Y.; Li, J.; Xiao, L.; Li, W. High-resolution X-ray microdiffraction analysis of natural teeth. J. Synchrotron Rad. 2008, 15, 235–238. [Google Scholar] [CrossRef]
- Gaft, M.; Shoval, S.; Panczer, G.; Nathan, Y.; Champagnon, B.; Garapon, C. Luminescence of uranium and rare-earth elements in apatite of fossil fish teeth. Palaeogeogr. Palaeocl. 1996, 126, 187–193. [Google Scholar] [CrossRef]
- Huang, C.M.; Zhang, Q.; Bai, S.; Wang, C.S. FTIR and XRD analysis of hydroxyapatite from fossil human and animal teeth in Jinsha Relict, Chengdu. Spectrosc. Spect. Anal. 2007, 27, 2448–2452. [Google Scholar]
- Ho, S.P.; Yu, B.; Yun, W.; Marshall, G.W.; Ryder, M.I.; Marshall, S.J. Structure, chemical composition and mechanical properties of human and rat cementum and its interface with root dentin. Acta Biomater. 2009, 5, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Strictly speaking, there are some differences among these biological materials. For example, the hardness of live dentin is less than that of enamel but is greater than that of bone or cementum [488]. When pulp of the tooth dies or is removed by a dentist, the properties of dentin change: it becomes brittle, liable to fracture and looses a reparative capability.
- Margolis, H.C.; Beniash, E.; Fowler, C.E. Role of macromolecular assembly of enamel matrix proteins in enamel formation. J. Dent. Res. 2006, 86, 775–793. [Google Scholar] [CrossRef]
- Vieira, A.; Hancock, R.; Limeback, H.; Schwartz, M.; Grynpas, M.D. How does fluoride concentration in the tooth affect apatite crystal size? J. Dent. Res. 2003, 82, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.Z.; Ge, J. New observations of the hierarchical structure of human enamel, from nanoscale to microscale. J. Tissue Eng. Regener. Med. 2007, 1, 185–191. [Google Scholar] [CrossRef]
- Jandt, K.D. Probing the future in functional soft drinks on the nanometre scale – towards tooth friendly soft drinks. Trends Food Sci. Technol. 2006, 17, 263–271. [Google Scholar] [CrossRef]
- Chen, H.; Chen, Y.; Orr, B.G.; Banaszak-Holl, M.; Majoros, I.; Clarkson, B.H. Nanoscale probing of the enamel nanorod surface using polyamidoamine dendrimers. Langmuir 2004, 20, 4168–4171. [Google Scholar] [CrossRef] [PubMed]
- Rönnholm, E. The amelogenesis of human teeth as revealed by electron microscopy. II. The development of the enamel crystallites. J. Ultrastruct. Res. 1962, 6, 249–303. [Google Scholar] [CrossRef] [PubMed]
- Nylen, M.U.; Evans, E.D.; Omnel, K.A. Crystal growth in rat enamel. J. Cell. Biol. 1963, 18, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Miake, Y.; Shimoda, S.; Fukae, M.; Aoba, T. Epitaxial overgrowth of apatite crystals on the thin-ribbon precursor at early stages of porcine enamel mineralization. Calcif. Tissue Int. 1993, 53, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G.; Menanteau, J.; Kerebel, L.M.; Mitre, D. Length and shape of enamel crystals. Calcif. Tissue Int. 1984, 36, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Jodaikin, A.; Traub, W.; Weiner, S. Enamel rod relations in the developing rat incisor. J. Ultrastruct. Res. 1984, 89, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Bres, E.F.; Hutchison, J.L. Surface structure study of biological calcium phosphate apatite crystals from human tooth enamel. J. Biomed. Mater. Res. 2002, 63, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, I.; Frank, R.M. High-resolution transmission electron microscopy of adult human peritubular dentine. Cell Tissue Res. 1985, 242, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Brès, E.F.; Voegel, J.C.; Frank, R.M. High-resolution electron microscopy of human enamel crystals. J. Microsc. 1990, 160, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Connell, S.; Kirkham, J.; Shorea, R.; Smith, A. Dental enamel – a biological ceramic: regular substructures in enamel hydroxyapatite crystals revealed by atomic force microscopy. J. Mater. Chem. 2004, 14, 2242–2248. [Google Scholar] [CrossRef]
- Warf, R.D.; Watson, R.R. Calcium phosphate – nutrition in prevention of early childhood dental caries. In Wild-type food in health promotion and disease prevention: the Columbus concept; de Meester, F., Watson, R.R., Eds.; Humana Press: Totowa, NJ, USA, 2008; pp. 343–353. [Google Scholar]
- Simmer, J.P.; Fincham, A.G. Molecular mechanisms of dental enamel formation. Crit. Rev. Oral Biol. Med. 1995, 6, 84–108. [Google Scholar] [CrossRef] [PubMed]
- Diekwisch, T.G.; Berman, B.J.; Genters, S.; Slavkin, H.C. Initial enamel crystals are not spatially associated with mineralized dentine. Cell Tissue Res. 1995, 279, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Aoba, T. Recent observations on enamel crystal formation during mammalian amelogenesis. Anat. Rec. 1996, 245, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.E. Cellular and chemical events during enamel maturation. Crit. Rev. Oral. Biol. Med. 1998, 9, 128–161. [Google Scholar] [CrossRef] [PubMed]
- Sydney-Zax, M.; Mayer, I.; Deutsch, D. Carbonate content in developing human and bovine enamel. J. Dent. Res. 1991, 70, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Renugopalakrishnan, V.; Shimizu, M.; Collins, B.; Glimcher, M.J. A resolution-enhanced Fourier Transform Infrared spectroscopic study of the environment of the CO32- ion in the mineral phase of enamel during its formation and maturation. Calcif. Tissue Int. 1991, 49, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Ogasawara, T.; Tagami, J.; Akao, M.; Kuboki, Y.; Nagai, N.; LeGeros, R.Z. pH and carbonate levels in. developing enamel. Connect. Tissue Res. 1998, 38, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Lowenstam, H.A.; Weiner, S. Transformation of amorphous calcium phosphate to crystalline dahillite in the radular teeth of chitons. Science 1985, 227, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Selvig, K.A. Periodic lattice images of hydroxyapatite crystals in human bone and dental hard tissues. Calcif. Tissue Res. 1970, 6, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Selvig, K.A. Electron microscopy of dental enamel: analysis of crystal lattice images. Cell Tissue Res. 1973, 137, 271–280. [Google Scholar]
- Cuisinier, F.J.G.; Steuer, P.; Senger, B.; Voegel, J.C.; Frank, R.M. Human amelogenesis: high resolution electron microscopy of nanometer-sized particles. Cell Tissue Res. 1993, 273, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cuisinier, F.J.G.; Steuer, P.; Brisson, A.; Voegel, J.C. High resolution electron microscopy study of crystal growth mechanisms in chicken bone composites. J. Cryst. Growth 1995, 156, 443–453. [Google Scholar] [CrossRef]
- Houllé, P.; Voegel, J.C.; Schultz, P.; Steuer, P.; Cuisinier, F.J.G. High resolution electron microscopy: structure and growth mechanisms of human dentin crystals. J. Dent. Res. 1997, 76, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Mann, S. Molecular tectonics in biomineralization and biomimetic materials chemistry. Nature 1993, 365, 499–505. [Google Scholar] [CrossRef]
- Kirkham, J.; Zhang, J.; Brookes, S.J.; Shore, R.C.; Wood, S.R.; Smith, D.A.; Wallwork, M.L.; Ryu, O.H.; Robinson, C. Evidence for charge domains on developing enamel crystal surfaces. J. Dent. Res. 2000, 79, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.M.; Tafforeau, P. New visions of dental tissue research: tooth development, chemistry, and structure. Evol. Anthropol. 2008, 17, 213–226. [Google Scholar] [CrossRef]
- Warshawsky, H.; Nanci, A. Stereo electron microscopy of enamel crystallites. J. Dent. Res. 1982, 61, 1504–1514. [Google Scholar]
- Warshawsky, H. Organization of crystals in enamel. Anat. Rec. 1989, 224, 242–262. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yao, X.; Walker, M.P.; Wang, Y. Chemical / molecular structure of the dentin-enamel junction is dependent on the intratooth location. Calcif. Tiss. Int. 2009, 84, 221–228. [Google Scholar] [CrossRef]
- Arsenault, A.L.; Robinson, B.W. The dentino-enamel junction: a structural and microanalytical study of early mineralization. Calcif. Tissue Int. 1989, 45, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y. High resolution electron microscopy in the dentino-enamel junction. J. Electron Microsc. (Tokyo) 1992, 41, 387–391. [Google Scholar]
- Hayashi, Y. High resolution electron microscopic study on the human dentine crystal. J. Electron Microsc. (Tokyo) 1993, 42, 141–146. [Google Scholar]
- Bodier-Houllé, P.; Steuer, P.; Meyer, J.M.; Bigeard, L.; Cuisinier, F.J.G. High-resolution electron-microscopic study of the relationship between human enamel and dentin crystals at the dentino-enamel junction. Cell Tissue Res. 2000, 301, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Hanaizumi, Y.; Oshima, H. Occurrence of amorphous and crystalline mineral deposits at the epithelial-mesenchymal interface of incisors in the calcium-loaded rat: Implication of novel calcium binding domains. Anat. Rec. 1996, 245, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Warshawsky, H. Lattice fringe continuity in the absence of crystal continuity in enamel. Adv. Dent. Res. 1996, 10, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hu, Y.; Ng, C. Microstructure and interfacial fracture at the cementum-enamel junctions in equine and bovine teeth. J. Mater. Res. 2006, 21, 2146–2155. [Google Scholar] [CrossRef]
- Ho, S.P.; Goodis, H.; Balooch, M.; Nonomura, G.; Marshall, S.J.; Marshall, G.W. The effect of sample preparation technique on determination of structure and nanomechanical properties of human cementum hard tissue. Biomaterials 2004, 25, 4847–4857. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.L.; White, S.N.; Luo, W.; Fong, H.; Sarikaya, M.; Snead, M.L. Regulated gene expression dictates enamel structure and tooth function. Matrix Biol. 2001, 20, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Finke, M.; Parker, D.M.; Jandt, K.D. Influence of soft drinks on the thickness and morphology of in situ acquired pellicle layer on enamel. J. Colloid Interface Sci. 2002, 251, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Barbour, M.E.; Parker, D.M.; Jandt, K.D. Enamel dissolution as a function of solution degree of saturation with respect to hydroxyapatite: a nanoindentation study. J. Colloid Interface Sci. 2003, 265, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lippert, F.; Parker, D.M.; Jandt, K.D. Susceptibility of deciduous and permanent enamel to dietary acid-induced erosion studied with atomic force microscopy nanoindentation. Eur. J. Oral Sci. 2004, 112, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Barbour, M.E.; Parker, D.M.; Allen, G.C.; Jandt, K.D. Human enamel erosion in constant composition citric acid solutions as a function of degree of saturation with respect to hydroxyapatite. J. Oral Rehabil. 2005, 32, 16–21. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Calcium phosphates in demineralization and remineralization processes. J. Clin. Dent. 1999, 10, 65–73. [Google Scholar]
- Walker, G.; Cai, F.; Shen, P.; Reynolds, C.; Ward, B.; Fone, C.; Honda, S.; Koganei, M.; Oda, M.; Reynolds, E. Increased remineralization of tooth enamel by milk containing added casein phosphopeptide-amorphous calcium phosphate. J. Dairy Res. 2006, 73, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Lippert, F.; Parker, D.M.; Jandt, K.D. In situ remineralisation of surface softened human enamel studied with AFM nanoindentation. Surface Sci. 2004, 553, 105–114. [Google Scholar] [CrossRef]
- Lippert, F.; Parker, D.M.; Jandt, K.D. In vitro demineralization / remineralization cycles at human tooth enamel surfaces investigated by AFM and nanoindentation. J. Colloid Interface Sci. 2004, 280, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Lippert, F.; Parker, D.M.; Jandt, K.D. Toothbrush abrasion of surface softened enamel studied with tapping mode AFM and AFM nanoindentation. Caries Res. 2004, 38, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Onuma, K.; Yamagishi, K.; Oyane, A. Nucleation and growth of hydroxyapatite nanocrystals for nondestructive repair of early caries lesions. J. Cryst. Growth 2005, 282, 199–207. [Google Scholar] [CrossRef]
- Cochrane, N.J.; Saranathan, S.; Cai, F.; Cross, K.J.; Reynolds, E.C. Enamel subsurface lesion remineralisation with casein phosphopeptide stabilised solutions of calcium, phosphate and fluoride. Caries Res. 2008, 42, 88–97. [Google Scholar] [CrossRef] [PubMed]
- McClendon, J.F. Fluorapatite and teeth. Science 1966, 151, 151. [Google Scholar] [CrossRef] [PubMed]
- Price, J.; Faucheux, C.; Allen, S. Deer antlers as a model of mammalian regeneration. Curr. Top. Dev. Biol. 2005, 67, 2–49. [Google Scholar]
- Yue, Z.; Deng, X.; Feng, H. The mechanisms of deer antlers development and regeneration. J. Econ. Animal 2005, 9, 46–49. [Google Scholar]
- Zhao, L.; Yue, Z.; Zhang, X.; Deng, X. The deer antlers endochondral ossification and its regulation mechanisms. J. Econ. Animal 2006, 10, 238–241. [Google Scholar]
- Landete-Castilleijos, T.; Garcia, A.; Gallego, L. Body weight, early growth and antler size influence antler bone mineral composition of Iberian Red Deer (Cervus elaphus hispanicus). Bone 2007, 40, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Huxley, J. The relative size of antlers of deer. Proc. Zool. Soc. London 1931, 72, 819–864. [Google Scholar]
- Chen, P.Y.; Stokes, A.G.; McKittrick, J. Comparison of the structure and mechanical properties of bovine femur bone and antler of the North American elk (Cervus elaphus canadensis). Acta Biomater. 2009, 5, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Zioupos, P.; Wang, X.T.; Currey, J.D. Experimental and theoretical quantification of the development of damage in fatigue tests of bone and antler. J. Biomech. 1996, 29, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Landete-Castilleijos, T.; Currey, J.D.; Estevez, J.A.; Gaspar-Lopez, E.; Garcia, A.; Gallego, L. Influence of physiological effort of growth and chemical composition on antler bone mechanical properties. Bone 2007, 41, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.A.; McCutcheon, A.L.; Dennis, G.R.; Mulley, R.C.; Wilson, M.A. Pore size analysis of fallow deer (Dama dama) antler bone. J. Mater. Sci. 2005, 40, 5733–5739. [Google Scholar] [CrossRef]
- Akhtar, R.; Daymond, M. R.; Almer, J. D.; Mummery, P. M. Elastic strains in antler trabecular bone determined by synchrotron X-ray diffraction. Acta Biomater. 2008, 4, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.J. Deer antlers regeneration, function and evolution; Academic Press: New York, USA, 1983; p. 316. [Google Scholar]
- Horns, pronghorns, and antlers: evolution, morphology, physiology, and social significance; Bubenik, G.A.; Bubenik, A.B. (Eds.) Springer: New York, USA, 1990; p. 562.
- Kierdorf, U.; Kierdorf, H. Antlers as biomonitors of environmental pollution by lead and fluoride: a review. Eur. J. Wildlife Res. 2005, 51, 137–150. [Google Scholar] [CrossRef]
- Barling, P.M.; Gupta, D.K.; Lim, C.E.L. Involvement of phosphodiesterase I in mineralization: histochemical studies using antler from red deer (Cervus elaphus) as a model. Calcif. Tissue Int. 1999, 65, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Barling, P.M.; Chong, K.W. The involvement of phosphohydrolases in mineralization: studies on enzymatic activities extracted from red deer antler. Calcif. Tissue Int. 1999, 65, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, U.; Kierdorf, H. The fluoride content of antlers as an indicator of fluoride exposure in red deer (Cervus elaphus): a historical biomonitoring study. Fluoride 2000, 33, 92–94. [Google Scholar]
- Pathak, N.N.; Pattanaik, A.K.; Patra, R.C.; Arora, B.M. Mineral composition of antlers of three deer species reared in captivity. Small Rumin. Res. 2001, 42, 61–65. [Google Scholar] [CrossRef]
- Yuxia, Y.; Rui, D.; Wang, Y.; Wang, S. Calcium and phosphors contents of three-branched and two-branched antler and ossificational antler from sika deer. J. Econ. Animal 2002, 6, 6–8. [Google Scholar]
- Kim, H.Y.; Rhyu, M.R. Sectional composition of minerals in domestic deer antler. Korean J. Food Sci. Technol. 2000, 32, 31–36. [Google Scholar]
- Li, C.; Suttie, J.M.; Clarck, D.E. Histological examination of antler regeneration in red deer (Cervus elaphus). Anat. Rec. 2005, 282, 163–174. [Google Scholar] [CrossRef]
- Meister, W. Changes in biological structure of the long bones of white-tailed deer during the growth of antlers. Anat. Rec. 1956, 124, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Muir, P.D.; Sykes, A.R.; Barrell, G.K. Calcium metabolism in red deer (Cervus elaphus) offered herbages during antlerogenesis: kinetic and stable balance studies. J. Agric. Sci. Camb. 1987, 109, 357–364. [Google Scholar] [CrossRef]
- Baxter, B.J.; Andrews, R.N.; Barrell, G.K. Bone turnover associated with antler growth in red deer (Cervus elaphus). Anat. Rec. 1999, 256, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Landete-Castilleijos, T.; Estevez, J.A.; Martinez, A.; Ceacero, F.; Garcia, A.; Gallego, L. Does chemical composition of antler bone reflect the physiological effort made to grow it? Bone 2007, 40, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Baciut, M.; Baciut, G.; Simon, V.; Albon, C.; Coman, V.; Prodan, P.; Florian, S.; Bran, S. Investigation of deer antler as a potential bone regenerating biomaterial. J. Optoelectronics Adv. Mater. 2007, 9, 2547–2550. [Google Scholar]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Kazama, J.J.; Amizuka, N.; Fukagawa, M. Ectopic calcification as abnormal biomineralization. Ther. Apher. Dial. 2006, 10 (Suppl. 1), S34–S38. [Google Scholar] [CrossRef]
- Mackarell, W.W.; Moore, B.; Thomas, W.T. On the presence of insoluble salts of calcium (oxalate and phosphate) in renal calculi in large amount in a preponderating number of cases, and the bearing of this finding upon calcium metabolism in gout and allied conditions. Biochem. J. 1911, 5, 161–180. [Google Scholar] [PubMed]
- Frondel, C.; Prien, E.L. Carbonate-apatite and hydroxylapatite in urinary calculi. Science 1942, 95, 431. [Google Scholar] [CrossRef] [PubMed]
- Frondel, C.; Prien, E.L. Deposition of calcium phosphates accompanying senile degeneration and disease. Science 1946, 103, 326. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, D.; Cozzolino, M. The mechanism of calcium deposition in soft tissues. Contrib. Nephrol. 2005, 149, 279–286. [Google Scholar] [PubMed]
- Goff, A.K.; Reichard, R. A soft-tissue calcification: differential diagnosis and pathogenesis. J. Forensic Sci. 2006, 51, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Bittmann, S.; Gunther, M.W.; Ulus, H. Tumoral calcinosis of the gluteal region in a child: case report with overview of different soft-tissue calcifications. J. Pediatric Surg. 2003, 38, E4–E7. [Google Scholar] [CrossRef]
- Molloy, E.S.; McCarthy, G.M. Basic calcium phosphate crystals: pathways to joint degeneration. Curr. Opin. Rheumatol. 2006, 18, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Kazama, J.J.; Amizuka, N.; Fukagawa, M. The making of a bone in blood vessels: from the soft shell to the hard bone. Kidney Int. 2007, 72, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Achilles, W. Crystallization in gel matrices: a new experimental model of calcium stone formation. Contrib. Nephrol. 1987, 58, 59–64. [Google Scholar] [PubMed]
- Achilles, W.; Jockel, U.; Schaper, A.; Burk, M.; Riedmiller, H. In vitro formation of “urinary stones”: generation of spherulites of calcium phosphate in gel and overgrowth with calcium oxalate using a new flow model of crystallization. Scanning Microsc. 1995, 9, 577-585, discussion 585-586. [Google Scholar] [PubMed]
- Ross, A.E.; Handa, S.; Lingeman, J.E.; Matlaga, B.R. Kidney stones during pregnancy: an investigation into stone composition. Urol. Res. 2008, 36, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Ringdén, I.; Tiselius, H.G. Composition and clinically determined hardness of urinary tract stones. Scand. J. Urol. Nephrol. 2007, 41, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Le May, O.; Kaqueler, J.C.; Kodaka, T. Electron probe micro-analysis of human dental pulp stones. Scan. Microscc. 1993, 7, 267–272. [Google Scholar]
- Kodaka, T.; Hirayama, A.; Mori, R.; Sano, T. Spherulitic brushite stones in the dental pulp of a cow. J. Electron Microsc. 1998, 47, 57–65. [Google Scholar] [CrossRef]
- Hayashizaki, J.; Ban, S.; Nakagaki, H.; Okumura, A.; Yoshii, S.; Robinson, C. Site specific mineral composition and microstructure of human supra-gingival dental calculus. Arch. Oral Biol. 2008, 53, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Zelentsov, E.L.; Moroz, T.N.; Kolmogorov, Yu.P.; Tolmachev, V.E.; Dragun, G.N.; Palchik, N.A.; Grigorieva, T.N. The elemental SRXRF analysis and mineral composition of human salivary stones. Nucl. Instrum. Methods Phys. Res., Sect. A 2001, 470, 417–421. [Google Scholar] [CrossRef]
- Ortlepp, J.R.; Schmitz, F.; Mevissen, V.; Weiß, S.; Huster, J.; Dronskowski, R.; Langebartels, G.; Autschbach, R.; Zerres, K.; Weber, C.; Hanrath, P.; Hoffmann, R. The amount of calcium-deficient hexagonal hydroxyapatite in aortic valves is influenced by gender and associated with genetic polymorphisms in patients with severe calcific aortic stenosis. Eur. Heart J. 2004, 25, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M. Cellular mechanism of calcification and its prevention in glutaraldehyde treated vascular tissue. Z. Kardiol. 2001, 90 (Suppl. 3), III99–III105. [Google Scholar] [CrossRef]
- Tomazic, B.B. Physicochemical principles of cardiovascular calcification. Z. Kardiol. 2001, 90 (Suppl. 3), III68–III80. [Google Scholar] [CrossRef]
- Marra, S.P.; Daghlian, C.P.; Fillinger, M.F.; Kennedy, F.E. Elemental composition, morphology and mechanical properties of calcified deposits obtained from abdominal aortic aneurysms. Acta Biomater. 2006, 2, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, L.A.; Turner, R.T.; Ritman, E.R. Endochondral bone formation in the heart: a possible mechanism of coronary calcification. Endocrinology 2003, 144, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Suvorova, E.I.; Buffat, P.A. Pathological mineralization of cardiac valves: causes and mechanism. J. Long Term Eff. Med. Implants 2005, 15, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C. Ectopic calcification: new concepts in cellular regulation. Z. Kardiol. 2001, 90 (Suppl. 3), III31–III37. [Google Scholar] [CrossRef]
- Glasmacher, B.; Nellen, E.; Reul, H.; Rau, G. In vitro hemocompatibility testing of new materials for mechanical heart valves. Mat.-Wiss. u. Werkstofftech. 1999, 30, 806–808. [Google Scholar] [CrossRef]
- Deiwick, M.; Glasmacher, B.; Pettenazzo, E.; Hammel, D.; Castellon, W.; Thiene, G.; Reul, H.; Berendes, E.; Scheld, H.H. Primary tissue failure of bioprostheses: new evidence from in vitro tests. Thorac. Cardiovasc. Surg. 2001, 49, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Pettenazzo, E.; Deiwick, M.; Thiene, G.; Molin, G.; Glasmacher, B.; Martignago, F.; Bottio, T.; Reul, H.; Valente, M. Dynamic in vitro calcification of bioprosthetic porcine valves: evidence of apatite crystallization. J. Thorac. Cardiovasc. Surg. 2001, 121, 500–509. [Google Scholar] [CrossRef]
- Schoen, F.J.; Levy, R.J. Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann. Thorac. Surg. 2005, 79, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Sensui, K.; Saitoh, S.; Kametani, K.; Makino, K.; Ohira, M.; Kimura, T.; Cheng, G.A.; Hata, Y. Property analysis of ectopic calcification in the carpal tunnel identification of apatite crystals: a case report. Arch. Orthop. Trauma Surg. 2003, 123, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Choi, S.K. Analysis of aqueous humor calcium and phosphate from cataract eyes with and without diabetes mellitus. KJO 2007, 21, 90–94. [Google Scholar] [PubMed]
- McCarty, D.J.; Hogan, H.M.; Gatter, R.A.; Grossmann, M. Studies on pathological calcifications in human cartilage. J. Bone Joint Surg. 1966, 48, 309–325. [Google Scholar] [PubMed]
- Laird, D.F.; Mucalo, M.R.; Yokogawa, Y. Growth of calcium hydroxyapatite (Ca-HAp) on cholesterol and cholestanol crystals from a simulated body fluid: a possible insight into the pathological calcifications associated with atherosclerosis. J. Colloid Interface Sci. 2006, 295, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.R.; Ignatiev, K.; Lee, P.L.; Abbott, K.; Pachman, L.M. Pathological calcification in juvenile dermatomyositis (JDM): microCT and synchrotron X-ray diffraction reveal hydroxyapatite with varied microstructures. Connect. Tissue Res. 2004, 45, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Pachman, L.M.; Boskey, A.L. Clinical manifestations and pathogenesis of hydroxyapatite crystal deposition in juvenile dermatomyositis. Curr. Rheumatol. Rep. 2006, 8, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Hale, E.K. Metastatic calcification. Dermatology Online J. 2003, 9, p. 2. http://dermatology.cdlib.org/94/NYU/Nov2001/3.html accessed in April 2009.
- Grech, P.; Ell, P.J.; Martin, T.J.; Barrington, N.A.; Martin, T.J. Diagnosis in metabolic bone disease; Hodder Arnold H&S: USA, 1998; p. 300. [Google Scholar]
- Mohr, W. Apatite diseases. The pathological substrate in dependence of tissue processing and in consideration of pathogenesis. Aktuel Rheumatol. 2003, 28, 53–58. [Google Scholar] [CrossRef]
- Tomazic, B.B. Characterization of mineral phases in cardiovascular calcification. In Hydroxyapatite and related materials; Brown, P.W., Constantz, B., Eds.; CRC Press: Boca Raton, FL, USA, 1994; pp. 93–116. [Google Scholar]
- Lee, R.S.; Kayser, M.V.; Ali, S.Y. Calcium phosphate microcrystal deposition in the human intervertebral disc. J. Anat. 2006, 208, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.K. Update in calcium deposition diseases. Curr. Opin. Rheumatol. 2007, 19, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Lagier, R.; Baud, C.A. Magnesium whitlockite, a calcium phosphate crystal of special interest in pathology. Pathol. Res. Pract. 2003, 199, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Wesson, J.A.; Ward, M.D. Pathological biomineralization of kidney stones. Elements 2007, 3, 415–421. [Google Scholar] [CrossRef]
- LeGeros, R.Z.; Orly, I.; LeGeros, J.P.; Gomez, C.; Kazimiroff, J.; Tarpley, T.; Kerebel, B. Scanning electron microscopy and electron probe microanalyses of the crystalline components of human and animal dental calculi. Scan. Microsc. 1988, 2, 345–356. [Google Scholar]
- Kirsch, T. Determinants of pathological mineralization. Curr. Opin. Rheumatol. 2006, 18, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Ectopic calcification: gathering hard facts about soft tissue mineralization. Am. J. Pathol. 1999, 154, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T. Physiological and pathological mineralization: a complex multifactorial process. Curr. Opin. Orthop. 2007, 18, 425–427. [Google Scholar] [CrossRef]
- Speer, M.Y.; Giachelli, C.M. Regulation of cardiovascular calcification. Cardiovasc. Pathol. 2004, 13, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Inducers and inhibitors of biomineralization: lessons from pathological calcification. Orthod. Craniofac. Res. 2005, 8, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Speer, M.Y.; Li, X.; Rajachar, R.M.; Yang, H. Regulation of vascular calcification: roles of phosphate and osteopontin. Circ. Res. 2005, 96, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.P.; Odenwald, T.; Ritz, E. Calcium, calcium regulatory hormones, and calcimimetics: impact on cardiovascular mortality. J. Am. Soc. Nephrol. 2006, 17, 78–80. [Google Scholar] [CrossRef]
- Azari, F.; Vali, H.; Guerquin-Kern, J.L.; Wu, T.D.; Croisy, A.; Sears, S.K.; Tabrizian, M.; McKee, M.D. Intracellular precipitation of hydroxyapatite mineral and implications for pathologic calcification. J. Struct. Biology 2008, 162, 468–479. [Google Scholar] [CrossRef]
- Stayton, P.S.; Drobny, G.P.; Shaw, W.J.; Long, J.R.; Gilbert, M. Molecular recognition at the protein-hydroxyapatite interface. Crit. Rev. Oral. Biol. Med. 2003, 14, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.M.; Fitzpatrick, L.A.; Inoue, D.; Qiao, J.H.; Fishbein, M.C.; Detrano, R.C.; Shah, P.K. Rajavashisth, T.B. Molecular, endocrine and genetic mechanisms of arterial calcification. Endocr. Rev. 2004, 25, 629–672. [Google Scholar] [CrossRef] [PubMed]
- Lamas, G.A.; Ackermann, A. Clinical evaluation of chelation therapy: is there any wheat amidst the chaff? Am. Heart J. 2000, 140, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Knudtson, M.L.; Wyse, D.G.; Galbraith, P.D.; Brant, R.; Hildebrand, K.; Paterson, D.; Richardson, D.; Burkart, C.; Burgess, E. Chelation therapy for ischemic heart disease: a randomized controlled trial. JAMA 2002, 287, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Koutsoukos, P.G.; Demadis, K.D.; Pokrovsky, O.S. Principles of demineralization: modern strategies for the isolation of organic frameworks. Part I. Common definitions and history. Micron 2008, 39, 1062–1091. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Koutsoukos, P.G.; Demadis, K.D.; Pokrovsky, O.S. Principles of demineralization: modern strategies for the isolation of organic frameworks. Part II. Decalcification. Micron 2009, 40, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Porter, A.E.; Thian, E.S.; Huang, J. Bioceramics: past, present and for the future. J. Eur. Ceram. Soc. 2008, 28, 1319–1327. [Google Scholar] [CrossRef]
- Jandt, K.D. Evolutions, revolutions and trends in biomaterials science – a perspective. Adv. Eng. Mater. 2007, 9, 1035–1050. [Google Scholar] [CrossRef]
- Salinas, A.J.; Vallet-Regí, M. Evolution of ceramics with medical applications. Z. Anorg. Allg. Chem. 2007, 633, 1762–1773. [Google Scholar] [CrossRef]
- Ring, M.E. Dentistry: an illustrated history; Harry N. Abrams, Inc.: New York, USA, 1992; p. 320. [Google Scholar]
- Albee, F.H. Studies in bone growth – triple calcium phosphate as stimulus to osteogenesis. Ann. Surg. 1920, 71, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Balazsi, C.; Weber, F.; Kover, Z.; Horvath, E.; Nemeth, C. Preparation of calcium-phosphate bioceramics from natural resources. J. Eur. Ceram. Soc. 2007, 27, 1601–1606. [Google Scholar] [CrossRef]
- Murugan, R.; Ramakrishna, S. Crystallographic study of hydroxyapatite bioceramics derived from various sources. Cryst. Growth Des. 2005, 5, 111–112. [Google Scholar] [CrossRef]
- Kokubo, T.; Kim, H.M.; Kawashita, M. Novel bioactive materials with different mechanical properties. Biomaterials 2003, 24, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Bioceramics and their clinical applications; Kokubo, T. (Ed.) Woodhead Publishing Ltd.: Abington, Cambridge, MA, USA, 2008; p. 784.
- Greenspan, D.C. Bioactive ceramic implant materials. Curr. Opin. Solid State Mater. Sci. 1999, 4, 389–393. [Google Scholar] [CrossRef]
- Blokhuis, T.J.; Termaat, M.F.; den Boer, F.C.; Patka, P.; Bakker, F.C.; Haarman, H.J.T.M. Properties of calcium phosphate ceramics in relation to their in vivo behavior. J. Trauma 2000, 48, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M. Bioactive ceramics: challenges and perspectives. J. Ceram. Soc. Jpn. 2001, 109, S49–S57. [Google Scholar] [CrossRef]
- Cao, W.; Hench, L.L. Bioactive materials. Ceram. Intern. 1996, 22, 493–507. [Google Scholar] [CrossRef]
- Okuda, T.; Ioku, K.; Yonezawa, I.; Minagi, H.; Gonda, Y.; Kawachi, G.; Kamitakahara, M.; Shibata, Y.; Murayama, H.; Kurosawa, H.; Ikeda, T. The slow resorption with replacement by bone of a hydrothermally synthesized pure calcium-deficient hydroxyapatite. Biomaterials 2008, 29, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Seeley, Z.; Bandyopadhyay, A.; Bose, S. Tricalcium phosphate based resorbable ceramics: influence of NaF and CaO addition. Mater. Sci. Eng., C 2008, 28, 11–17. [Google Scholar] [CrossRef]
- Cushnie, E.K.; Khan, Y.M.; Laurencin, C.T. Amorphous hydroxyapatite-sintered polymeric scaffolds for bone tissue regeneration: physical characterization studies. J. Biomed. Mater. Res. A 2008, 84A, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Linhart, W.; Briem, D.; Amling, M.; Rueger, J.M.; Windolf, J. Mechanical failure of porous hydroxyapatite ceramics 7.5 years after implantation in the proximal tibial. Unfallchirurg. 2004, 107, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Burger, E.L.; Patel, V. Calcium phosphates as bone graft extenders. Orthopedics 2007, 30, 939–942. [Google Scholar] [PubMed]
- He, L.H.; Standard, O.C.; Huang, T.T.; Latella, B.A.; Swain, M.V. Mechanical behaviour of porous hydroxyapatite. Acta Biomater. 2008, 4, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.; Malik, K.R.; Darr, J.A.; Rehman, I. Recent developments in processing and surface modification of hydroxyapatite. Adv. Appl. Ceram. 2006, 105, 113–139. [Google Scholar] [CrossRef]
- LeGeros, R.Z.; LeGeros, J.P. Calcium phosphate bioceramics: past, present, future. Key Eng. Mater. 2003, 240-242, 3–10. [Google Scholar] [CrossRef]
- Jones, A.C.; Arns, C.H.; Sheppard, A.P.; Hutmacher, D.W.; Milthorpe, B.K.; Knackstedt, M.A. Assessment of bone ingrowth into porous biomaterials using MICRO-CT. Biomaterials 2007, 28, 2491–2504. [Google Scholar] [CrossRef] [PubMed]
- McAfee, P.C.; Cunningham, B.W.; Orbegoso, D.O.; Sefter, J.C.; Dmitriev, A.E.; Fedder, I.L. Analysis of porous ingrowth in intervertebral disc prostheses. Spine 2003, 28, 332–340. [Google Scholar] [PubMed]
- Mastrogiacomo, M.; Scaglione, S.; Martinetti, R.; Dolcini, L.; Beltrame, F.; Cancedda, R.; Quarto, R. Role of scaffold internal structure on in vivo bone formation in macroporous calcium phosphate bioceramics. Biomaterials 2006, 27, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Dohi, Y.; Ohgushi, H.; Shimaoka, H.; Ikeuchi, M.; Matsushima, A.; Yonemasu, K.; Hosoi, H. Influence of the porosity of hydroxyapatite ceramics on in vitro and in vivo bone formation by cultured rat bone marrow stromal cells. J. Mater. Sci. Mater. Med. 2006, 17, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Chow, L.C. Formation of macropores in calcium phosphate cement implants. J. Biomed. Mater. Res. 2001, 12, 135–139. [Google Scholar]
- Walsh, D.; Tanaka, J. Preparation of a bone-like apatite foam cement. J. Mater. Sci. Mater. Med. 2001, 12, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, E.; Chulia, D.; Pouget, C.; Viana, M. Fabrication of porous substrates: a review of processes using pore forming agents in the biomaterial field. J. Pharm. Sci. 2008, 97, 1135–1154. [Google Scholar] [CrossRef] [PubMed]
- Komlev, V.S.; Barinov, S.M. Porous hydroxyapatite ceramics of bi-modal pore size distribution. J. Mater. Sci. Mater. Med. 2002, 13, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Turner, I.G.; Miles, A.W. Mechanical characterization of dense calcium phosphate bioceramics with interconnected porosity. J. Mater. Sci. Mater. Med. 2007, 18, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Kasuga, T.; Abe, Y. Preparation and compressive strength behaviour of porous ceramics with β-Ca3(PO3)2 fiber skeletons. J. Am. Ceram. Soc. 1997, 80, 225–231. [Google Scholar] [CrossRef]
- von Doernberg, M.C.; von Rechenberg, B.; Bohner, M.; Grünenfelder, S.; van Lenthe, G.H.; Müller, R.; Gasser, B.; Mathys, R.; Baroud, G.; Auer, J. In vivo behavior of calcium phosphate scaffolds with four different pore sizes. Biomaterials 2006, 27, 5186–5198. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Hopwood, J.D.; Mann, S. Crystal tectonics: construction of reticulated calcium phosphate frameworks in bicontinuous reverse microemulsions. Science 1994, 264, 1576–1578. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tan, S.; Jiang, D. Improving the properties of porous hydroxyapatite ceramics by fabricating methods. J. Mater. Sci. 2005, 40, 4939–4942. [Google Scholar] [CrossRef]
- Zhang, J.; Fujiwara, M.; Xu, Q.; Zhu, Y.; Iwasa, M.; Jiang, D. Synthesis of mesoporous calcium phosphate using hybrid templates. Micropor. Mesopor. Mat. 2008, 111, 411–416. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, L.; Li, Y.; Shi, T. Preparation of irregular mesoporous hydroxyapatite. Mater. Res. Bull. 2008, 43, 1607–1614. [Google Scholar] [CrossRef]
- Cyster, L.A.; Grant, D.M.; Howdle, S.M.; Rose, F.R.A.J.; Irvine, D.J.; Freeman, D.; Scotchford, C.A.; Shakesheff, K.M. The influence of dispersant concentration on the pore morphology of hydroxyapatite ceramics for bone tissue engineering. Biomaterials 2005, 26, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Koh, Y.H.; Yoon, B.H.; Kim, H.E.; Kim, H.W. Highly porous hydroxyapatite bioceramics with interconnected pore channels using camphene-based freeze casting. Mater. Lett. 2007, 61, 2270–2273. [Google Scholar] [CrossRef]
- Jones, J.R.; Hench, L.L. Regeneration of trabecular bone using porous ceramics. Curr. Opin. Solid State Mater. Sci. 2003, 7, 301–307. [Google Scholar] [CrossRef]
- Tas, A.C. Preparation of porous apatite granules from calcium phosphate cement. J. Mater. Sci. Mater. Med. 2008, 19, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Rahaman, M.N.; Dogan, F.; Bal, B.S. Freeze casting of porous hydroxyapatite scaffolds. I. Processing and general microstructure. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 86B, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Tang, Y.F.; Lv, C.T.; Zhou, Z.H. Preparation of uniform porous hydroxyapatite biomaterials by a new method. Appl. Surf. Sci. 2008, 254, 5359–5362. [Google Scholar] [CrossRef]
- Fu, Y.C.; Ho, M.L.; Wu, S.C.; Hsieh, H.S.; Wang, C.K. Porous bioceramic bead prepared by calcium phosphate with sodium alginate gel and PE powder. Mater. Sci. Eng., C 2008, 28, 1149–1158. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Turner, I.G.; Miles, A.W. Fabrication of porous bioceramics with porosity gradients similar to the bimodal structure of cortical and cancellous bone. J. Mater. Sci. Mater. Med. 2007, 18, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Khalil, K.A.; Kim, S.W.; Dharmaraj, N.; Kim, K.W.; Kim, H.Y. Novel mechanism to improve toughness of the hydroxyapatite bioceramics using high-frequency induction heat sintering. J. Mater. Process. Technol. 2007, 187-188, 417–420. [Google Scholar] [CrossRef]
- Bioactivity is defined as the property of the material to develop a direct, adherent and strong bonding with bone [271,272].
- Yan, X.; Yu, C.; Zhou, X.; Tang, J.; Zhao, D. Highly ordered mesoporous bioactive glasses with superior in vitro bone-forming bioactivities. Angew. Chem. Int. Ed. Engl. 2004, 43, 5980–5984. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Barba, I.; Ruiz-González, L.; Doadrio, J.C.; González-Calbet, J.M.; Vallet-Regí, M. Tissue regeneration: a new property of mesoporous materials. Solid State Sci. 2005, 7, 983–989. [Google Scholar] [CrossRef]
- da Silva, R.V.; Bertran, C.A.; Kawachi, E.Y.; Camilli, J.A. Repair of cranial bone defects with calcium phosphate ceramic implant or autogenous bone graft. J. Craniofac. Surg. 2007, 18, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Jordan, D.R.; Gilberg, S.; Bawazeer, A. Coralline hydroxyapatite orbital implant (Bio-Eye): experience with 158 patients. Ophthal. Plast. Reconstr. Surg. 2004, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Lew, H.; Kim, S.J.; Lee, S.Y. Exposure rate of hydroxyapatite orbital implants a 15-year experience of 802 cases. Ophthalmology 2008, 115, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Schnettler, R.; Stahl, J.P.; Alt, V.; Pavlidis, T.; Dingeldein, E.; Wenisch, S. Calcium phosphate-based bone substitutes. Eur. J. Trauma 2004, 4, 219–229. [Google Scholar]
- Zyman, Z.Z.; Glushko, V.; Dedukh, N.; Malyshkina, S.; Ashukina, N. Porous calcium phosphate ceramic granules and their behaviour in differently loaded areas of skeleton. J. Mater. Sci. Mater. Med. 2008, 19, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Tadic, D.; Epple, M. A thorough physicochemical characterization of 14 calcium phosphate-based bone substitution materials in comparison to natural bone. Biomaterials 2004, 25, 987–994. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Properties of osteoconductive biomaterials: calcium phosphates. Clin. Orthop. Rel. Res. 2002, 395, 81–98. [Google Scholar] [CrossRef]
- Ruan, J.M.; Zou, J.P.; Zhou, J.N.; Hu, J.Z. Porous hydroxyapatite – tricalcium phosphate bioceramics. Powder Metall. 2006, 49, 66–69. [Google Scholar] [CrossRef]
- Bohner, M. Calcium orthophosphates in medicine: from ceramics to calcium phosphate cements. Injury 2000, 31 (Suppl. 4), D37–D47. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphate cements and concretes. Materials 2009, 2, 221–291. [Google Scholar] [CrossRef]
- Wilson, C.E.; Kruyt, M.C.; de Bruijn, J.; van Blitterswijk, C.A.; Oner, F.C.; Verbout, A.J.; Dhert, W.J.A. A new in vivo screening model for posterior spinal bone formation: comparison of ten calcium phosphate ceramic material treatments. Biomaterials 2006, 27, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.D.; Sachs, M.A.; Woodard, C.R. Calcium phosphate bone cements: a comprehensive review. J. Long Term Eff. Med. Implants 2003, 13, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ambard, A.J.; Mueninghoff, L. Calcium phosphate cement: review of mechanical and biological properties. J. Prosthodont. 2006, 15, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Costantino, P.D.; Friedman, C.D.; Jones, K.; Chow, L.C.; Pelzer, H.J.; Sisson, G.A. Hydroxyapatite cement. I. Basic chemistry and histologic properties. Arch. Otolaryngol. Head Neck Surg. 1991, 117, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Costantino, P.D.; Friedman, C.D.; Jones, K.; Chow, L.C.; Pelzer, H.J.; Sisson, G.A. Hydroxyapatite cement. II. Obliteration and reconstruction of the cat frontal sinus. Arch. Otolaryngol. Head Neck Surg. 1991, 117, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Park, Y.K.; Kim, J.H.; Lee, S.H.; Ryu, K.N.; Park, C.M.; Kim, Y.S. The biomechanical evaluation of calcium phosphate cements for use in vertebroplasty. J. Neurosurg. Spine 2006, 4, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, S.; Chen, T.; Liu, C.; Lin, S.; Tian, X. Calcium phosphate cement reinforced by polypeptide copolymers. J. Biomed. Mater. Res. B Appl. Biomater. 2006, 76B, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Iooss, P.; Leray, A.M.; Grimandi, G.; Daculsi, G.; Merle, C. A new injectable bone substitute combining poly(ε-caprolactone) microparticles with biphasic calcium phosphate granules. Biomaterials 2001, 22, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, O.; Goyenvalle, E.; Bouler, J.M.; Guicheux, J.; Pilet, P.; Weiss, P.; Daculsi, G. Macroporous biphasic calcium phosphate ceramics versus injectable bone substitute: a comparative study 3 and 8 weeks after implantation in rabbit bone. J. Mater. Sci. Mater. Med. 2001, 12, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Weiss, P.; Bourges, X.; Amadordel-Valle, G.; Daculsi, G. Crystallization at the polymer / calcium-phosphate interface in a sterilized injectable bone substitute IBS. Biomaterials 2002, 23, 2789–2794. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, O.; Khairoun, I.; Bosco, J.; Obadia, L.; Bourges, X.; Rau, C.; Magne, D.; Bouler, J.M.; Aguado, E.; Daculsi, G. Noninvasive bone replacement with a new injectable calcium phosphate biomaterial. J. Biomed. Mater. Res. A 2003, 66A, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Trojani, C.; Boukhechba, F.; Scimeca, J.C.; Vandenbos, F.; Michiels, J.F.; Daculsi, G.; Boileau, P.; Weiss, P.; Carle, G.F.; Rochet, N. Ectopic bone formation using an injectable biphasic calcium phosphate / Si-HPMC hydrogel composite loaded with undifferentiated bone marrow stromal cells. Biomaterials 2006, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Lerouxel, E.; Weiss, P.; Giumelli, B.; Moreau, A.; Pilet, P.; Guicheux, J.; Corre, P.; Bouler, J.M.; Daculsi, G.; Malard, O. Injectable calcium phosphate scaffold and bone marrow graft for bone reconstruction in irradiated areas: an experimental study in rats. Biomaterials 2006, 27, 4566–4572. [Google Scholar] [CrossRef] [PubMed]
- Blouin, S.; Moreau, M.F.; Weiss, P.; Daculsi, G.; Basle, M.F.; Chappard, D. Evaluation of an injectable bone substitute (β-TCP / hydroxyapatite / hydroxy-propyl-methyl-cellulose) in severely osteopenic and aged rats. J. Biomed. Mater. Res. A 2006, 78A, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Is there a chemical interaction between calcium phosphates and hydroxypropylmethylcellulose (HPMC) in organic / inorganic composites? J. Biomed. Mater. Res. 2001, 54, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Chris Arts, J.J.; Verdonschot, N.; Schreurs, B.W.; Buma, P. The use of a bioresorbable nano-crystalline hydroxyapatite paste in acetabular bone impaction grafting. Biomaterials 2006, 27, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Laschke, M.W.; Witt, K.; Pohlemann, T.; Menger, M.D. Injectable nanocrystalline hydroxyapatite paste for bone substitution: in vivo analysis of biocompatibility and vascularization. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 82B, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Huber, F.X.; Berger, I.; McArthur, N.; Huber, C.; Kock, H.P.; Hillmeier, J.; Meeder, P.J. Evaluation of a novel nanocrystalline hydroxyapatite paste and a solid hydroxyapatite ceramic for the treatment of critical size bone defects (CSD) in rabbits. J. Mater. Sci. Mater. Med. 2008, 19, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.H.; Weir, M.D.; Burguera, E.F.; Fraser, A.M. Injectable and macroporous calcium phosphate cement scaffold. Biomaterials 2006, 27, 4279–4287. [Google Scholar] [CrossRef] [PubMed]
- Kruyt, M.C.; Persson, C.; Johansson, G.; Dhert, W.J.; de Bruijn, J. Towards injectable cell-based tissue-engineered bone: the effect of different calcium phosphate microparticles and pre-culturing. Tissue Eng. 2006, 12, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Trojani, C.; Boukhechba, F.; Scimeca, J.C.; Vandenbos, F.; Michiels, J.F.; Daculsi, G.; Boileau, P.; Weiss, P.; Carle, G.F.; Rochet, N. Ectopic bone formation using an injectable biphasic calcium phosphate / Si-HPMC hydrogel composite loaded with undifferentiated bone marrow stromal cells. Biomaterials 2006, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Zyman, Z.Z.; Ivanov, I.; Rochmistrov, D.; Glushko, V.; Tkachenko, N.; Kijko, S. Sintering peculiarities for hydroxyapatite with different degrees of crystallinity. J. Biomed. Mater. Res. 2001, 54, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Willmann, G. Coating of implants with hydroxyapatite – material connections between bone and metal. Adv. Eng. Mater. 1999, 1, 95–105. [Google Scholar] [CrossRef]
- Epinette, J.A.M.D.; Geesink, R.G.T. Hydroxyapatite coated hip and knee arthroplasty; Elsevier: Amsterdam, The Netherlands, 1995; p. 394. [Google Scholar]
- Schliephake, H.; Scharnweber, D.; Roesseler, S.; Dard, M.; Sewing, A.; Aref, A. Biomimetic calcium phosphate composite coating of dental implants. Int. J. Oral Max. Impl. 2006, 21, 738–746. [Google Scholar]
- Yang, Y.; Kim, K.H.; Ong, J.L. A review on calcium phosphate coatings produced using a sputtering process – an alternative to plasma spraying. Biomaterials 2005, 26, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.L.; Mano, J.F.; Reis, R.L. Nature-inspired calcium phosphate coatings: present status and novel advances in the science of mimicry. Curr. Opin. Solid State Mater. Sci. 2003, 7, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Kurella, A.; Dahotre, N.B. Surface modification for bioimplants: the role of laser surface engineering. J. Biomater. Appl. 2005, 20, 5–50. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.C.; Yen, S.K. The process of electrochemical deposited hydroxyapatite coatings on biomedical titanium at room temperature. Mater. Sci. Eng., C 2002, 20, 153–160. [Google Scholar] [CrossRef]
- Eliaz, N.; Sridhar, T.M.; Mudali, U.K.; Raj, B. Electrochemical and electrophoretic deposition of hydroxyapatite for orthopaedic applications. Surf. Eng. 2005, 21, 238–242. [Google Scholar] [CrossRef]
- Ma, M.; Ye, W.; Wang, X.X. Effect of supersaturation on the morphology of hydroxyapatite crystals deposited by electrochemical deposition on titanium. Mater. Lett. 2008, 62, 3875–3877. [Google Scholar] [CrossRef]
- Ong, J.L.; Appleford, M.; Oh, S.; Yang, Y.; Chen, W.H.; Bumgardner, J.D.; Haggard, W.O. The characterization and development of bioactive hydroxyapatite coatings. JOM 2006, 58, 67–69. [Google Scholar] [CrossRef]
- Qu, H.; Wei, M. The effect of temperature and initial pH on biomimetic apatite coating. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 87, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Seshadri, S.K.; Kwon, T.Y.; Kim, K.H. Calcium phosphate-based coatings on titanium and its alloys. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 85, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.K.; Chen, C.H.; Huang, K.Y.; Wang, G.J. Eight-year results of hydroxyapatite-coated hip arthroplasty. J. Arthroplasty 2006, 21, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.; Tindall, A.; Shetty, A.A.; James, K.D.; Rand, C. 15-year follow-up results of the hydroxyapatite ceramic-coated femoral stem. J. Orthop. Surg. 2006, 14, 151–154. [Google Scholar]
- Buchanan, J.M. 17 year review of hydroxyapatite ceramic coated hip implants – a clinical and histological evaluation. Key Eng. Mater. 2006, 309-311, 1341–1344. [Google Scholar] [CrossRef]
- Buchanan, J.M.; Goodfellow, S. Nineteen years review of hydroxyapatite ceramic coated hip implants: a clinical and histological evaluation. Key Eng. Mater. 2008, 361-363, 1315–1318. [Google Scholar] [CrossRef]
- Binahmed, A.; Stoykewych, A.; Hussain, A.; Love, B.; Pruthi, V. Long-term follow-up of hydroxyapatite-coated dental implants – a clinical trial. Int. J. Oral Max. Impl. 2007, 22, 963–968. [Google Scholar]
- Iezzi, G.; Scarano, A.; Petrone, G.; Piattelli, A. Two human hydroxyapatite-coated dental implants retrieved after a 14-year loading period: a histologic and histomorphometric case report. J. Periodontol. 2007, 78, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Osteoconductivity is the ability to provide a scaffold or template for the formation of new bone on its surface by attachment, migration, proliferation, and differentiation of bone-forming cells [649].
- Geesink, R.G.T. Osteoconductive coating for total joint arthroplasty. Clin. Orthop. Related Res. 2002, 395, 53–65. [Google Scholar] [CrossRef]
- Zheng, X.; Zhou, S.; Li, X.; Weng, J. Shape memory properties of poly(D,L-lactide) / hydroxyapatite composites. Biomaterials 2006, 27, 4288–4295. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, W.; Grynpas, M.D.; Tully, A.E.; Bowman, J.; Abram, J. Hydroxyapatite reinforced polyethylene – a mechanically compatible implant material for bone replacement. Biomaterials 1981, 2, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Epple, M. Composites of calcium phosphate and polymers as bone substitution materials. Eur. J. Trauma 2006, 32, 125–131. [Google Scholar] [CrossRef]
- Mickiewicz, R.A.; Mayes, A.M.; Knaack, D. Polymer – calcium phosphate cement composites for bone substitutes. J. Biomed. Mater. Res. 2002, 61, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Ma, P.X. Structure and properties of nano-hydroxyapatite / polymer composite scaffolds for bone tissue engineering. Biomaterials 2004, 25, 4749–4757. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Berndt, C.C.; Gross, K.A. Hydroxyapatite / polymer composite flame sprayed coatings for orthopedic application. J. Biomater. Sci. Polym. Ed. 2002, 13, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Lewandrowski, K.U.; Bondre, S.P.; Shea, M.; Untch, C.M.; Hayes, W.C.; Hile, D.D.; Wise, D.L.; Trantolo, D.J. Composite resorbable polymer / hydroxylapatite composite screws for fixation of osteochondral osteotomies. Biomed. Mater. Eng. 2002, 12, 423–438. [Google Scholar] [PubMed]
- Salernitano, E.; Migliares, C. Composite materials for biomedical applications: a review. J. Appl. Biomater. Biomech. 2003, 1, 3–18. [Google Scholar] [PubMed]
- Kusmanto, F.; Walker, G.; Gan, Q.; Walsh, P.; Buchanan, F.; Dickson, G.; McCaigue, M.; Maggs, C.; Dring, M. Development of composite tissue scaffolds containing naturally sourced mircoporous hydroxyapatite. Chem. Eng. J. 2008, 139, 398–407. [Google Scholar] [CrossRef]
- Roeder, R.K.; Converse, G.L.; Kane, R.J.; Yue, W.M. Hydroxyapatite-reinforced polymer biocomposites for synthetic bone substitutes. JOM 2008, 60, 38–45. [Google Scholar] [CrossRef]
- O’Donnell, J.N.R.; Antonucci, J.M.; Skrtic, D. Amorphous calcium phosphate composites with improved mechanical properties. J. Bioact. Comp. Polym. 2006, 21, 169–184. [Google Scholar] [CrossRef]
- Baji, A.; Wong, S.C.; Srivatsan, T.S.; Njus, G.O.; Mathur, G. Processing methodologies for polycaprolactone-hydroxyapatite composites: a review. Mater. Manufac. Proc. 2006, 21, 211–218. [Google Scholar] [CrossRef]
- Ruhe, P.Q.; Hedberg-Dirk, E.L.; Padron, N.T.; Spauwen, P.H.; Jansen, J.A.; Mikos, A.G. Porous poly(D,L-lactic-co-glycolic acid) / calcium phosphate cement composite for reconstruction of bone defects. Tissue Eng. 2006, 12, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Basu, B. Designing materials for hard tissue replacement. J. Korean Ceram. Soc. 2008, 45, 1–29. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphate-based biocomposites and hybrid biomaterials. J. Mater. Sci. 2009, 44, 2343–2387. [Google Scholar] [CrossRef]
- Orly, I.; Gregoire, M.; Menanteau, J.; Heughebaert, M.; Kerebel, B. Chemical changes in hydroxyapatite biomaterial under in vivo and in vitro biological conditions. Calcif. Tissue Int. 1989, 45, 20–26. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Calcium phosphate-based osteoinductive materials. Chem. Rev. 2008, 108, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- The term “biomimetics” (“the mimicry of life”) was coined by an American inventor, engineer and biophysicist Otto Herbert Schmitt (1913 – 1998) in the 1950-s.
- Benyus, J.M. Biomimicry: innovation inspired by nature; William Morrow: New York, USA, 1997; p. 308. [Google Scholar]
- Watt, J.C. The behavior of calcium phosphate and calcium carbonate (bone salts) precipitated in various media, with applications to bone formation. Biol. Bull. 1923, 44, 280–288. [Google Scholar] [CrossRef]
- Dorozhkin, S.V.; Dorozhkina, E.I.; Epple, M. A model system to provide a good in vitro simulation of biological mineralization. Cryst. Growth Des. 2004, 4, 389–395. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Salinas, A.J.; Vallet-Regi, M. Effect of the continuous solution exchange on the in vitro reactivity of a CaO – SiO2 sol-gel glass. J. Biomed. Mater. Res. 2000, 51, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regi, M.; Pėrez-Pariente, J.; Izquierdo-Barba, I.; Salinas, A.J. Compositional variations in the calcium phosphate layer growth on gel glasses soaked in a simulated body fluid. Chem. Mater. 2000, 12, 3770–3775. [Google Scholar] [CrossRef]
- Koutsoukos, P.; Amjad, Z.; Tomson, M.B.; Nancollas, G.H. Crystallization of calcium phosphates: a constant composition study. J. Am. Chem. Soc. 1980, 102, 1553–1557. [Google Scholar] [CrossRef]
- Tomson, M.B.; Nancollas, G.H. Mineralization kinetics: a constant composition approach. Science 1978, 200, 1059–1060. [Google Scholar] [CrossRef] [PubMed]
- Manjubala, I.; Scheler, S.; Bössert, J.; Jandt, K.D. Mineralisation of chitosan scaffolds with nano-apatite formation by double diffusion technique. Acta Biomater. 2006, 2, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V.; Dorozhkina, E.I. The influence of bovine serum albumin on the crystallization of calcium phosphates from a revised simulated body fluid. Colloids Surf., A 2003, 215, 191–199. [Google Scholar] [CrossRef]
- Dorozhkina, E.I.; Dorozhkin, S.V. In vitro crystallization of carbonateapatite on cholesterol from a modified simulated body fluid. Colloids Surf., A 2003, 223, 231–237. [Google Scholar] [CrossRef]
- Dorozhkin, S.V.; Dorozhkina, E.I.; Epple, M. Precipitation of carbonateapatite from a revised simulated body fluid in the presence of glucose. J. Appl. Biomater. Biomech. 2003, 1, 200–208. [Google Scholar] [PubMed]
- Dorozhkin, S.V.; Dorozhkina, E.I. In vitro simulation of vascular calcification by the controlled crystallization of amorphous calcium phosphates onto porous cholesterol. J. Mater. Sci. 2005, 40, 6417–6422. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. In vitro mineralization of silicon containing calcium phosphate bioceramics. J. Am. Ceram. Soc. 2007, 90, 244–249. [Google Scholar] [CrossRef]
- Becker, A.; Epple, M. A high-throughput crystallisation device to study biomineralisation in vitro. Mater. Res. Soc. Symp. Proc. 2005, 873E, K12.1.1–K12.1.10. [Google Scholar]
- Krings, M.; Kanellopoulou, D.; Mavrilas, D.; Glasmacher, B. In vitro pH-controlled calcification of biological heart valve prostheses. Mat.-Wiss. u. Werkstofftech. 2006, 37, 432–435. [Google Scholar] [CrossRef]
- Wang, L.J.; Guan, X.Y.; Yin, H.Y.; Moradian-Oldak, J.; Nancollas, G.H. Mimicking the self-organized microstructure of tooth enamel. J. Phys. Chem. C 2008, 112, 5892–5899. [Google Scholar] [CrossRef]
- Howland, J.; Kramer, B. Calcium and phosphorus in the serum in relation to rickets. Am. J. Dis. Child. 1921, 22, 105–119. [Google Scholar]
- Tisdall, F.F. The effects of ultra violet rays on the calcium and inorganic phosphate content of the blood serum of rachitic infants. Can. Med. Assoc. J. 1922, 7, 536–538. [Google Scholar]
- Hanks, J.H.; Wallace, R.E. Relation of oxygen and temperature in the preservation of tissues by refrigeration. Proc. Soc. Exp. Biol. Med. 1949, 71, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Takashima, H.; Yamamoto, H.; Miyazaki, T. Functionally gradient bonelike hydroxyapatite coating on a titanium metal substrate created by a discharging method in HBSS without organic molecules. Int. J. Oral Max. Impl. 2004, 19, 177–183. [Google Scholar]
- Marques, P.A.A.P.; Serro, A.P.; Saramago, B.J.; Fernandes, A.C.; Magalhães, M.C.F.; Correia, R.N. Mineralisation of two phosphate ceramics in HBSS: role of albumin. Biomaterials 2003, 24, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Gopikrishna, V.; Baweja, P.S.; Venkateshbabu, N.; Thomas, T.; Kandaswamy, D. Comparison of coconut water, propolis, HBSS, and milk on PDL cell survival. J. Endod. 2008, 34, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, T.; Asami, K.; Asaoka, K. Repassivation of titanium and surface oxide film regenerated in simulated bioliquid. J. Biomed. Mater. Res. 1998, 40, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.A.; Anza, E.; Costa, A.M.; Oliveira, F.J.; Silva, R.F. Electrochemical behavior of Ti / Al2O3 interfaces produced by diffusion bonding. Mater. Res. 2003, 6, 439–444. [Google Scholar] [CrossRef]
- Named after Harry Eagle (1905 – 1992), an American physician and pathologist.
- Named after Renato Dulbecco (born February 22, 1914), an Italian virologist who won the 1975 Nobel Prize in Physiology or Medicine for his work on reverse transcriptase.
- Meuleman, N.; Tondreau, T.; Delforge, A.; Dejeneffe, M.; Massy, M.; Libertalis, M.; Bron, D.; Lagneaux, L. Human marrow mesenchymal stem cell culture: serum-free medium allows better expansion than classical α-MEM medium. Eur. J. Haematol. 2006, 76, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Gotoh, E. Corrosion fatigue properties of metallic biomaterials in Eagle’s medium. Mater. Trans. 2002, 43, 2949–2955. [Google Scholar] [CrossRef]
- Coelho, M.J.; Cabral, A.T.; Fernandes, M.H. Human bone cell cultures in biocompatibility testing. Part I: Osteoblastic differentiation of serially passaged human bone marrow cells cultured in α-MEM and in DMEM. Biomaterials 2000, 21, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Weng, W.J.; Deng, X.L.; Cheng, K.; Liu, X.G.; Du, P.Y.; Shen, G.; Han, G.R. Surface morphology variations of porous nano-calcium phosphate / poly(L-lactic acid) composites in PBS. Key Eng. Mater. 2006, 309, 569–572. [Google Scholar] [CrossRef]
- Lewis, A.C.; Kilburn, M.R.; Papageorgiou, I.; Allen, G.C.; Case, C.P. Effect of synovial fluid, phosphate-buffered saline solution, and water on the dissolution and corrosion properties of CoCrMo alloys as used in orthopedic implants. J. Biomed. Mater. Res. A 2005, 73A, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.P.; Williamson, R.T. A review of saliva: normal composition, flow, and function. J. Prosthet. Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Preetha, A.; Banerjee, R. Comparison of artificial saliva substitutes. Trends Biomater. Artif. Organs 2005, 18, 178–186. [Google Scholar]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitsugi, T.; Yamamuro, T. Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W3. J. Biomed. Mater. Res. 1990, 24, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Leng, Y. Theoretical analysis of calcium phosphate precipitation in simulated body fluid. Biomaterials 2005, 26, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Tas, A.C. Synthesis of biomimetic Ca-hydroxyapatite powders at 37°C in synthetic body fluids. Biomaterials 2000, 21, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Landi, E.; Tampieri, A.; Celotti, G.; Langenati, R.; Sandri, M.; Sprio, S. Nucleation of biomimetic apatite in synthetic body fluids: dense and porous scaffold development. Biomaterials 2005, 26, 2835–2845. [Google Scholar] [CrossRef] [PubMed]
- Jalota, S.; Bhaduri, S.B.; Tas, A.C. Using a synthetic body fluid (SBF) solution of 27 mM HCO3- to make bone substitutes more osteointegrative. Mater. Sci. Eng., C 2008, 28, 129–140. [Google Scholar] [CrossRef]
- Kim, H.M.; Miyazaki, T.; Kokubo, T.; Nakamura, T. Revised simulated body fluid. In Bioceramics 13; Giannini, S., Moroni, A., Eds.; Trans Tech Publ.: Switzerland, 2001; Volume 192-195, pp. 47–50. [Google Scholar]
- Oyane, A.; Kim, H.M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. A 2003, 65A, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Bohner, M.; Lemaitre, J. Can bioactivity be tested in vitro with SBF solution? Biomaterials 2009, 30, 2175–2179. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M. Ceramic bioactivity and related biomimetic strategy. Curr. Opin. Solid State Mater. Sci. 2003, 7, 289–299. [Google Scholar] [CrossRef]
- Marques, P.A.A.P.; Magalhães, M.C.F.; Correia, R.N. Inorganic plasma with physiological CO2 / HCO3− buffer. Biomaterials 2003, 24, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkina, E.I.; Dorozhkin, S.V. Surface mineralisation of hydroxyapatite in modified simulated body fluid (mSBF) with higher amounts of hydrogencarbonate ions. Colloids Surf., A 2002, 210, 41–48. [Google Scholar] [CrossRef]
- Miyaji, F.; Kim, H.M.; Handa, S.; Kokubo, T.; Nakamura, T. Bonelike apatite coating on organic polymers: novel nucleation process using sodium silicate solution. Biomaterials 1999, 20, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Kishimoto, K.; Miyaji, F.; Kokubo, T.; Yao, T.; Suetsugu, Y.; Tanaka, J.; Nakamura, T. Composition and structure of apatite formed on organic polymer in simulated body fluid with a high content of carbonate ion. J. Mater. Sci. Mater. Med. 2000, 11, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Barrere, F.; van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. Influence of ionic strength and carbonate on the Ca-P coating formation from SBF×5 solution. Biomaterials 2002, 23, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Barrere, F.; van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. Nucleation of biomimetic Ca-P coatings on Ti6Al4V from a SBF×5 solution: influence of magnesium. Biomaterials 2002, 23, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Tas, A.C.; Bhaduri, S.B. Rapid coating of Ti6Al4V at room temperature with a calcium phosphate solution similar to 10x simulated body fluid. J. Mater. Res. 2004, 19, 2742–2749. [Google Scholar] [CrossRef]
- Dorozhkina, E.I.; Dorozhkin, S.V. Structure and properties of the precipitates formed from condensed solutions of the revised simulated body fluid. J. Biomed. Mater. Res. A 2003, 67A, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Pompe, W.; Lampenscherf, S.; Rößler, S.; Scharnweber, D.; Weis, K.; Worch, H.; Hofinger, J. Functionally graded bioceramics. Mater. Sci. Forum 1999, 308-311, 325–330. [Google Scholar] [CrossRef]
- Fan, Y.; Duan, K.; Wang, R. A composite coating by electrolysis-induced collagen self-assembly and calcium phosphate mineralization. Biomaterials 2005, 26, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, K.; Goda, T.; Takeuchi, N.; Einaga, H.; Tanabe, T. Preparation of collagen / calcium phosphate multilayer sheet using enzymatic mineralization. Biomaterials 2004, 25, 5481–5489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Z.L.; Liao, S.S.; Cui, F.Z. Nucleation sites of calcium phosphate crystals during collagen mineralization. J. Am. Ceram. Soc. 2003, 86, 1052–1054. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Chen, X.; Zhao, N. Biomimetic formation of hydroxyapatite / collagen matrix composite. Adv. Eng. Mater. 2006, 8, 97–100. [Google Scholar] [CrossRef]
- Lickorish, D.; Ramshaw, J.A.M.; Werkmeister, J.A.; Glattauer, V.; Howlett, C.R. Collagen – hydroxyapatite composite prepared by biomimetic process. J. Biomed. Mater. Res. A 2004, 68A, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yin, Y.; Lu, W.W.; Leong, J.C.; Zhang, W.; Zhang, J.; Zhang, M.; Yao, K. Preparation and histological evaluation of biomimetic three-dimensional hydroxyapatite / chitosan-gelatin network composite scaffolds. Biomaterials 2002, 23, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Kim, H.E.; Salih, V. Stimulation of osteoblast responses to biomimetic nanocomposites of gelatin – hydroxyapatite for tissue engineering scaffolds. Biomaterials 2005, 26, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Knowles, J.C.; Kim, H.E. Porous scaffolds of gelatin-hydroxyapatite nanocomposites obtained by biomimetic approach: characterization and antibiotic drug release. J. Biomed. Mater. Res. B Appl. Biomater. 2005, 74B, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Q.L.; Zen, Q.; Li, G.; Jiang, H.; Liu, L.; Darvell, B.W. Biomimetic mineralization and bioactivity of phosphorylated chitosan. Key Eng. Mater. 2005, 288-289, 429–432. [Google Scholar] [CrossRef]
- Li, Q.L.; Chen, Z.; Ou, G.; Liu, L.; Jiang, H.; Zeng, Q.; Li, G.; He, G.; Mo, A.; Darvell, B.W. Biomimetic synthesis of apatite – polyelectrolyte complex (chitosan – phosphorylated chitosan) hydrogel as an osteoblast carrier. Key Eng. Mater. 2005, 288-289, 75–78. [Google Scholar] [CrossRef]
- Stupp, S.I.; Ciegler, G.W. Organoapatites: materials for artificial bone. I. Synthesis and microstructure. J. Biomed. Mater. Res. 1992, 26, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Stupp, S.I.; Braun, P.V. Molecular manipulation of microstructures: biomaterials, ceramics, and semiconductors. Science 1997, 277, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Stupp, S.I.; Mejicano, G.C.; Hanson, J.A. Organoapatites: materials for artificial bone. II. Hardening reactions and properties. J. Biomed. Mater. Res. 1993, 27, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Stupp, S.I.; Hanson, J.A.; Eurell, J.A.; Ciegler, G.W.; Johnson, A. Organoapatites: materials for artificial bone. III. Biological testing. J. Biomed. Mater. Res. 1993, 27, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Layrolle, P.; de Bruijn, J.; van Blitterswijk, C.A.; de Groot, K. Biomimetic coprecipitation of calcium phosphate and bovine serum albumin on titanium alloy. J. Biomed. Mater. Res. 2001, 57, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Layrolle, P.; Stigter, M.; de Groot, K. Biomimetic and electrolytic calcium phosphate coatings on titanium alloy: physicochemical characteristics and cell attachment. Biomaterials 2004, 25, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Leng, Y. Electrochemical activation of titanium for biomimetic coating of calcium phosphate. Biomaterials 2005, 26, 3853–3859. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Boanini, E.; Bracci, B.; Facchini, A.; Panzavolta, S.; Segatti, F.; Sturba, L. Nanocrystalline hydroxyapatite coatings on titanium: a new fast biomimetic method. Biomaterials 2005, 26, 4085–4089. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Himeno, T.; Kawashita, M.; Lee, J.H.; Kokubo, T.; Nakamura, T. Surface potential change in bioactive titanium metal during the process of apatite formation in simulated body fluid. J. Biomed. Mater. Res. A 2003, 67A, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Allegrini, S.; Rumpel, E.; Kauschke, E.; Fanghänel, J.; König, B. Hydroxyapatite grafting promotes new bone formation and osseointegration of smooth titanium implants. Ann. Anat. 2006, 188, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Arnould, C.; Delhalle, J.; Mekhalif, Z. Multifunctional hybrid coating on titanium towards hydroxyapatite growth: electrodeposition of tantalum and its molecular functionalization with organophosphonic acids films. Electrochim. Acta 2008, 53, 5632–5638. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Kusumocahyo, S.P.; Kanamori, T.; Shinbo, T. Mineralization of hydroxyapatite on a polymer substrate in a solution supersaturated by polyelectrolyte. J. Appl. Polymer Sci. 2006, 100, 1465–1470. [Google Scholar] [CrossRef]
- Bodin, A.; Gustafsson, L.; Gatenholm, P. Surface-engineered bacterial cellulose as template for crystallization of calcium phosphate. J. Biomater. Sci. Polym. Ed. 2006, 17, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Toworfe, G.K.; Composto, R.J.; Shapiro, I.M.; Ducheyne, P. Nucleation and growth of calcium phosphate on amine-, carboxyl- and hydroxyl-silane self-assembled monolayers. Biomaterials 2006, 27, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Griffith, L.G.; Naughton, G. Tissue engineering – current challenges and expanding opportunities. Science 2002, 295, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- In 2003, the NSF published a report titled: “The emergence of tissue engineering as a research field”, which provides a thorough description of the history of this field. See http://www.nsf.gov/pubs/2004/nsf0450/start.htm accessed in April 2009.
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Schieker, M.; Seitz, H.; Drosse, I.; Seitz, S.; Mutschler, W. Biomaterials as scaffold for bone tissue engineering. Eur. J. Trauma 2006, 32, 114–124. [Google Scholar] [CrossRef]
- Fini, M.; Giardino, R.; Borsari, V.; Torricelli, P.; Rimondini, L.; Giavaresi, G.; Aldini, N.N. In vitro behaviour of osteoblasts cultured on orthopaedic biomaterials with different surface roughness, uncoated and fluorohydroxyapatite-coated, relative to the in vivo osteointegration rate. Int. J. Artif. Organs 2003, 26, 520–528. [Google Scholar] [PubMed]
- Zimmerman, R.; Ila, D.; Muntele, C.; Rodrigues, M.; Poker, D.B.; Hensley, D. Enhanced tissue adhesion by increased porosity and surface roughness of carbon based biomaterials. Nucl. Instrum. Methods Phys. Res., Sect. B 2002, 191, 825–829. [Google Scholar] [CrossRef]
- Sato, M.; Webster, T.J. Designing orthopedic implant surfaces: harmonization of nanotopographical and chemical aspects. Nanomed. 2006, 1, 351–354. [Google Scholar] [CrossRef]
- Okamoto, M.; Dohi, Y.; Ohgushi, H.; Shimaoka, H.; Ikeuchi, M.; Matsushima, A.; Yonemasu, K.; Hosoi, H. Influence of the porosity of hydroxyapatite ceramics on in vitro and in vivo bone formation by cultured rat bone marrow stromal cells. J. Mater. Sci. Mater. Med. 2006, 17, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Annaz, B.; Hing, K.A.; Kayser, M.; Buckland, T.; di Silvio, L. Porosity in phase-pure hydroxyapatite. J. Microscopy 2004, 216, 97–109. [Google Scholar] [CrossRef]
- Ebaretonbofa, E.; Evans, J.R. High porosity hydroxyapatite foam scaffolds for bone substitute. J. Porous Mater. 2002, 9, 257–263. [Google Scholar] [CrossRef]
- Specchia, N.; Pagnotta, A.; Cappella, M.; Tampieri, A.; Greco, F. Effect of hydroxyapatite porosity on growth and differentiation of human osteoblast-like cells. J. Mater. Sci. 2002, 37, 577–584. [Google Scholar] [CrossRef]
- Teixeira, C.C.; Nemelivsky, Y.; Karkia, C.; LeGeros, R.Z. Biphasic calcium phosphate: a scaffold for growth plate chondrocyte maturation. Tissue Eng. 2006, 12, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Artzi, Z.; Weinreb, M.; Givol, N.; Rohrer, M.D.; Nemcovsky, C.E.; Prasad, H.S.; Tal, H. Biomaterial resorbability and healing site morphology of inorganic bovine bone and beta tricalcium phosphate in the canine. A 24-month longitudinal histologic study and morphometric analysis. Int. J. Oral Max. Impl. 2004, 19, 357–368. [Google Scholar]
- Dellinger, J.G.; Eurell, J.A.C.; Jamison, R.D. Bone response to 3D periodic hydroxyapatite scaffolds with and without tailored microporosity to deliver bone morphogenetic protein 2. J. Biomed. Mater. Res. A 2006, 76A, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Pal, S. Development of porous hydroxyapatite scaffolds. Mater. Manuf. Process. 2006, 21, 325–328. [Google Scholar] [CrossRef]
- Bae, C.J.; Kim, H.W.; Koh, Y.H.; Kim, H.E. Hydroxyapatite (HA) bone scaffolds with controlled macrochannel pores. J. Mater. Sci. Mater. Med. 2006, 17, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wei, J.; Kong, H.; Liu, C.; Pan, K. Biocompatibility and osteogenesis of calcium phosphate cement scaffolds for bone tissue engineering. Adv. Mater. Res. 2008, 47-50 (Part 2), 1383–1386. [Google Scholar] [CrossRef]
- Guo, H.; Su, J.; Wei, J.; Kong, H.; Liu, C. Biocompatibility and osteogenicity of degradable Ca-deficient hydroxyapatite scaffolds from calcium phosphate cement for bone tissue engineering. Acta Biomater. 2009, 5, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Webster, T.J. Nanomedicine for implants: a review of studies and necessary experimental tools. Biomaterials 2007, 28, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Okazaki, M.; Nakahira, A.; Sasaki, J.; Egusa, H.; Sohmura, T. Modification of apatite materials for bone tissue engineering and drug delivery carriers. Curr. Med. Chem. 2007, 14, 2726–2733. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Koh, Y.H.; Kong, Y.M.; Kang, J.G.; Kim, H.E. Strontium substituted calcium phosphate biphasic ceramics obtained by a powder precipitation method. J. Mater. Sci. Mater. Med. 2004, 15, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regi, M.; Arcos, D. Silicon substituted hydroxyapatites. A method to upgrade calcium phosphate based implants. J. Mater. Chem. 2005, 15, 1509–1516. [Google Scholar] [CrossRef]
- Gbureck, U.; Thull, R.; Barralet, J.E. Alkali ion substituted calcium phosphate cement formation from mechanically activated reactants. J. Mater. Sci. Mater. Med. 2005, 16, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Ventura, J.M.; Ferreira, J.M.F. In situ formation and characterization of flourine-substituted biphasic calcium phosphate ceramics of varied F-HAP / β-TCP ratios. Chem. Mater. 2005, 17, 3065–3068. [Google Scholar] [CrossRef]
- Gbureck, U.; Knappe, O.; Grover, L.M.; Barralet, J.E. Antimicrobial potency of alkali ion substituted calcium phosphate cements. Biomaterials 2005, 26, 6880–6886. [Google Scholar] [CrossRef] [PubMed]
- Pietak, A.M.; Reid, J.W.; Stott, M.J.; Sayer, M. Silicon substitution in the calcium phosphate bioceramics. Biomaterials 2007, 28, 4023–4032. [Google Scholar] [CrossRef] [PubMed]
- Tadic, D.; Veresov, A.; Putlayev, V.I.; Epple, M. In vitro preparation of nanocrystalline calcium phosphates as bone substitution materials in surgery. Mat.-Wiss. u. Werkstofftech. 2003, 34, 1048–1051. [Google Scholar] [CrossRef]
- Lilley, K.J.; Gbureck, U.; Wright, A.J.; Farrar, D.F.; Barralet, J.E. Cement from nanocrystalline hydroxyapatite: effect of calcium phosphate ratio. J. Mater. Sci. Mater. Med. 2005, 16, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Meejoo, S.; Maneeprakorn, W.; Winotai, P. Phase and thermal stability of nanocrystalline hydroxyapatite prepared via microwave heating. Thermochim. Acta 2006, 447, 115–120. [Google Scholar] [CrossRef]
- Zhu, X.; Eibl, O.; Berthold, C.; Scheideler, L.; Geis-Gerstorfer, J. Structural characterization of nanocrystalline hydroxyapatite and adhesion of pre-osteoblast cells. Nanotechnology 2006, 17, 2711–2721. [Google Scholar] [CrossRef]
- Li, H.; Zhu, M.Y.; Li, L.H.; Zhou, C.R. Processing of nanocrystalline hydroxyapatite particles via reverse microemulsions. J. Mater. Sci. 2008, 43, 384–389. [Google Scholar] [CrossRef]
- Banerjee, A.; Bose, S. Effect of dopants on the properties of nanocrystalline hydroxyapatite. Ceram. Trans. 2005, 164, 91–99. [Google Scholar]
- Feng, W.; Sen, L.M.; Peng, L.Y.; Song, G.S. Synthesis of nanocrystalline hydroxyapatite powders in stimulated body fluid. J. Mater. Sci. 2005, 40, 2073–2076. [Google Scholar] [CrossRef]
- Ito, H.; Oaki, Y.; Imai, H. Selective synthesis of various nanoscale morphologies of hydroxyapatite via an intermediate phase. Cryst. Growth Des. 2008, 8, 1055–1059. [Google Scholar] [CrossRef]
- Fathi, M.H.; Hanifi, A.; Mortazavi, V. Preparation and bioactivity evaluation of bone-like hydroxyapatite nanopowder. J. Mater. Process. Technol. 2008, 202, 536–542. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Yin, Y.; Yao, F.; Yao, K. Modulation of nano-hydroxyapatite size via formation on chitosan-gelatin network film in situ. Biomaterials 2007, 28, 781–790. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Huang, Z.; Liu, Y.; Chen, W.; Xu, T. Template-directed one-step synthesis of flowerlike porous carbonated hydroxyapatite spheres. Mater. Lett. 2007, 61, 141–143. [Google Scholar] [CrossRef]
- You, C.; Miyazaki, T.; Ishida, E.; Ashizuka, M.; Ohtsuki, C.; Tanihara, M. Fabrication of poly(vinyl alcohol) – apatite hybrids through biomimetic process. J. Eur. Ceram. Soc. 2007, 27, 1585–1588. [Google Scholar] [CrossRef]
- Aizawa, M.; Porter, A.E.; Best, S.M.; Bonfield, W. Ultrastructural observation of single-crystal apatite fibres. Biomaterials 2005, 26, 3427–3433. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Ryu, S.C.; Yoon, S.Y.; Stevens, R.; Park, H.C. Preparation of whisker-shaped hydroxyapatite / β-tricalcium phosphate composite. Mater. Chem. Phys. 2008, 109, 440–447. [Google Scholar] [CrossRef]
- Morisue, H.; Matsumoto, M.; Chiba, K.; Matsumoto, H.; Toyama, Y.; Aizawa, M.; Kanzawa, N.; Fujimi, T.J.; Uchida, H.; Okada, I. A novel hydroxyapatite fiber mesh as a carrier for recombinant human bone morphogenetic protein-2 enhances bone union in rat posterolateral fusion model. Spine 2006, 31, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, A.; Ikoma, T.; Kobayashi, H.; Tanaka, J. Preparation and mechanical property of core-shell type chitosan / calcium phosphate composite fiber. Mater. Sci. Eng., C 2004, 24, 723–728. [Google Scholar] [CrossRef]
- Wu, Y.; Hench, L.L.; Du, J.; Choy, K.L.; Guo, J. Preparation of hydroxyapatite fibers by electrospinning technique. J. Am. Ceram. Soc. 2004, 87, 1988–1991. [Google Scholar] [CrossRef]
- Ramanan, S.R.; Venkatesh, R. A study of hydroxyapatite fibers prepared via sol-gel route. Mater. Lett. 2004, 58, 3320–3323. [Google Scholar] [CrossRef]
- Seo, D.S.; Lee, J.K. Synthesis of hydroxyapatite whiskers through dissolution-reprecipitation process using EDTA. J. Cryst. Growth 2008, 310, 2162–2167. [Google Scholar] [CrossRef]
- Neira, I.S.; Guitián, F.; Taniguchi, T.; Watanabe, T.; Yoshimura, M. Hydrothermal synthesis of hydroxyapatite whiskers with sharp faceted hexagonal morphology. J. Mater. Sci. 2008, 43, 2171–2178. [Google Scholar] [CrossRef]
- Ribeiro, C.C.; Barrias, C.C.; Barbosa, M.A. Preparation and characterisation of calcium-phosphate porous microspheres with a uniform size for biomedical applications. J. Mater. Sci. Mater. Med. 2006, 17, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.C.; Barrias, C.C.; Barbosa, M.A. Calcium phosphate-alginate microspheres as enzyme delivery matrices. Biomaterials 2004, 25, 4363–4373. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Y.; Wang, M.; Cheung, W.L.; Guo, B.C.; Jia, D.M. Synthesis of carbonated hydroxyapatite nanospheres through nanoemulsion. J. Mater. Sci. Mater. Med. 2008, 19, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Gu, H.J.; Lee, H.H. Microspheres of collagen-apatite nanocomposites with osteogenic potential for tissue engineering. Tissue Eng. 2007, 13, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Deville, S.; Saiz, E.; Tomsia, A.P. Freeze casting of hydroxyapatite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 5480–5489. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Jin, H.H.; Park, H.Y.; Park, I.M.; Park, H.C.; Yoon, S.Y. Preparation of porous hydroxyapatite scaffolds for bone tissue engineering. Mater. Sci. Forum 2006, 510-511, 754–757. [Google Scholar] [CrossRef]
- Leukers, B.; Gülkan, H.; Irsen, S.H.; Milz, S.; Tille, C.; Schieker, M.; Seitz, H. Hydroxyapatite scaffolds for bone tissue engineering made by 3D printing. J. Mater. Sci. Mater. Med. 2005, 16, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Deville, S.; Saiz, E.; Nalla, R.K.; Tomsia, A.P. Strong biomimetic hydroxyapatite scaffolds. Adv. Sci. Technol. 2006, 49, 148–152. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, D.; Yin, G.; Chen, H. Progress of biphasic calcium phosphate bioceramic as scaffold materials of bone tissue engineering. J. Chinese Ceram. Soc. 2004, 32, 1143–1149. [Google Scholar]
- Tampieri, A.; Celotti, G.; Sprio, S.; Delcogliano, A.; Franzese, S. Porosity-graded hydroxyapatite ceramics to replace natural bone. Biomaterials 2001, 22, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, K.; Oaki, Y.; Ichimiya, H.; Komotori, J.; Imai, H. Preparation of hierarchically organized calcium phosphate-organic polymer composites by calcification of hydrogel. Sci. Technol. Adv. Mater. 2006, 7, 219–225. [Google Scholar] [CrossRef]
- Okamoto, M.; Dohi, Y.; Ohgushi, H.; Shimaoka, H.; Ikeuchi, M.; Matsushima, A.; Yonemasu, K. Hosoi, H. Influence of the porosity of hydroxyapatite ceramics on in vitro and in vivo bone formation by cultured rat bone marrow stromal cells. J. Mater. Sci. Mater. Med. 2006, 17, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L.; Polak, J.M. Third-generation biomedical materials. Science 2002, 295, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Blawas, A.S.; Reichert, W.M. Protein patterning. Biomaterials 1998, 19, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Park, M.S.; Jeon, O.; Choi, C.Y.; Kim, B.S. Poly(lactide-co-glycolide) / hydroxyapatite composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Hu, Y.; Yan, Y.; Liu, L.; Xiong, Z.; Wei, Y.; Bai, J. Surface modification of PLGA / β-TCP scaffold for bone tissue engineering: hybridization with collagen and apatite. Surf. Coat. Technol. 2007, 201, 9549–9557. [Google Scholar] [CrossRef]
- Kim, S.S.; Ahn, K.M.; Park, M.S.; Lee, J.H.; Choi, C.Y.; Kim, B.S. A poly(lactide-co-glycolide) / hydroxyapatite composite scaffold with enhanced osteoconductivity. J. Biomed. Mater. Res. A 2007, 80A, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Beaussart, A.; Chen, Y.; Mak, A.F.T. Transfer of apatite coating from porogens to scaffolds: uniform apatite coating within porous poly(D,L-lactic-co-glycolic acid) scaffold in vitro. J. Biomed. Mater. Res. A 2007, 80, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Habraken, W.J.E.M.; Wolke, J.G.C.; Mikos, A.G.; Jansen, J.A. PLGA microsphere / calcium phosphate cement composites for tissue engineering: in vitro release and degradation characteristics. J. Biomater. Sci. Polym. Ed. 2008, 19, 1171–1188. [Google Scholar] [CrossRef] [PubMed]
- Charles-Harris, M.; Koch, M.A.; Navarro, M.; Lacroix, D.; Engel, E.; Planell, J.A. A PLA / calcium phosphate degradable composite material for bone tissue engineering: an in vitro study. J. Mater. Sci. Mater. Med. 2008, 19, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Smith, L.A.; Hu, J.; Ma, P.X. Biomimetic nanofibrous gelatin / apatite composite scaffolds for bone tissue engineering. Biomaterials 2009, 30, 2252–2258. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Salcedo, S.; Izquierdo-Barba, I.; Arcos, D.; Vallet-Regi, M. In vitro evaluation of potential calcium phosphate scaffolds for tissue engineering. Tissue Eng. 2006, 12, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Meganck, J.A.; Baumann, M.J.; Case, E.D.; McCabe, L.R.; Allar, J.N. Biaxial flexure testing of calcium phosphate bioceramics for use in tissue engineering. J. Biomed. Mater. Res. A 2005, 72A, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Case, E.D.; Smith, I.O.; Baumann, M.J. Microcracking and porosity in calcium phosphates and the implications for bone tissue engineering. Mater. Sci. Eng., A 2005, 390, 246–254. [Google Scholar] [CrossRef]
- Sibilla, P.; Sereni, A.; Aguiari, G.; Banzi, M.; Manzati, E.; Mischiati, C.; Trombelli, L.; del Senno, L. Effects of a hydroxyapatite-based biomaterial on gene expression in osteoblast-like cells. J. Dent. Res. 2006, 85, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, A.A.; Hennessy, K.M.; Bellis, S.L. The effect of adsorbed serum proteins, RGD and proteoglycan-binding peptides on the adhesion of mesenchymal stem cells to hydroxyapatite. Biomaterials 2007, 28, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiacomo, M.; Muraglia, A.; Komlev, V.; Peyrin, F.; Rustichelli, F.; Crovace, A.; Cancedda, R. Tissue engineering of bone: search for a better scaffold. Orthod. Craniofac. Res. 2005, 8, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Schek, R.M.; Taboas, J.M.; Hollister, S.J.; Krebsbach, P.H. Tissue engineering osteochondral implants for temporomandibular joint repair. Orthod. Craniofac. Res. 2005, 8, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Krylova, E.A.; Ivanov, A.A.; Krylov, S.E.; Plashchina, I.G.; Grigorjan, A.S.; Goldstein, D.V.; Pulin, A.A.; Fatkhudinov, T.H. Hydroxyapatite – alginate structure as living cells supporting system. Minerva Biotecnol. 2006, 18, 17–22. [Google Scholar]
- Hench, L.L. Biomaterials: a forecast for the future. Biomaterials 1998, 19, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Ratner, B.D.; Bryant, S.J. Biomaterials: where we have been and where we are going. Ann. Rev. Biomed. Eng. 2004, 6, 41–75. [Google Scholar] [CrossRef]
- Drouet, C.; Largeot, C.; Raimbeaux, G.; Estournès, C.; Dechambre, G.; Combes, C.; Rey, C. Bioceramics: spark plasma sintering (SPS) of calcium phosphates. Adv. Sci. Technol. 2006, 49, 45–50. [Google Scholar] [CrossRef]
- Anderson, J.M. The future of biomedical materials. J. Mater. Sci. Mater. Med. 2006, 17, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- White, A.A.; Best, S.M.; Kinloch, I.A. Hydroxyapatite – carbon nanotube composites for biomedical applications: A review. Int. J. Appl. Ceram. Technol. 2007, 4, 1–13. [Google Scholar] [CrossRef]
- Kealley, C.; Elcombe, M.; van Riessen, A.; Ben-Nissan, B. Development of carbon nanotube-reinforced hydroxyapatite bioceramics. Physica B 2006, 385-386, 496–498. [Google Scholar] [CrossRef]
- Balani, K.; Anderson, R.; Laha, T.; Andara, M.; Tercero, J.; Crumpler, E.; Agarwal, A. Plasma-sprayed carbon nanotube reinforced hydroxyapatite coatings and their interaction with human osteoblasts in vitro. Biomaterials 2007, 28, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A. Calcium phosphate nanoparticles: second-generation nonviral vectors in gene therapy. Exp. Rev. Mol. Diag. 2005, 5, 893–905. [Google Scholar] [CrossRef]
- Li, Y.H.; Cao, L.Y.; Huang, J.F.; Zeng, X.R. Characteristics and preparation of nanometer hydroxyapatite in medical science. J. Clin. Rehabil. Tiss. Eng. Res. 2008, 12, 8143–8146. [Google Scholar]
- For multiphase compositions of various calcium orthophosphates, the problem of accurate phase quantification often arises. The problem is usually solved by the Rietveld refinement and the readers are referred to a recent paper on this subject [908].
- Reid, J.W.; Hendry, J.A. Rapid, accurate phase quantification of multiphase calcium phosphate materials using Rietveld refinement. J. Appl. Cryst. 2006, 39, 536–543. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Green chemical synthesis of calcium phosphate bioceramics. J. Appl. Biomater. Biomech. 2008, 6, 104–109. [Google Scholar] [PubMed]
- Mizushima, Y.; Ikoma, T.; Tanaka, J.; Hoshi, K.; Ishihara, T.; Ogawa, Y.; Ueno, A. Injectable porous hydroxyapatite microparticles as a new carrier for protein and lipophilic drugs. J. Control. Release 2006, 110, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Ginebra, M.P.; Traykova, T.; Planell, J.A. Calcium phosphate cements as bone drug delivery systems: a review. J. Control. Release 2006, 113, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ginebra, M.P.; Traykova, T.; Planell, J.A. Calcium phosphate cements: competitive drug carriers for the musculoskeletal system? Biomaterials 2006, 27, 2171–2177. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Lei, J.; Yu, C.; Tu, B.; Zhao, D. Hard-templating synthesis of a novel rod-like nanoporous calcium phosphate bioceramics and their capacity as antibiotic carriers. Mater. Chem. Phys. 2007, 103, 489–493. [Google Scholar] [CrossRef]
- Barrère, F.; Mahmood, T.A.; de Groot, K.; van Blitterswijk, C.A. Advanced biomaterials for skeletal tissue regeneration: instructive and smart functions. Mater. Sci. Eng., R 2008, 59, 38–71. [Google Scholar] [CrossRef]
- Sudo, A.; Hasegawa, M.; Fukuda, A.; Uchida, A. Treatment of infected hip arthroplasty with antibiotic-impregnated calcium hydroxyapatite. J. Arthroplasty 2008, 23, 145–150. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dorozhkin, S.V. Calcium Orthophosphates in Nature, Biology and Medicine. Materials 2009, 2, 399-498. https://doi.org/10.3390/ma2020399
Dorozhkin SV. Calcium Orthophosphates in Nature, Biology and Medicine. Materials. 2009; 2(2):399-498. https://doi.org/10.3390/ma2020399
Chicago/Turabian StyleDorozhkin, Sergey V. 2009. "Calcium Orthophosphates in Nature, Biology and Medicine" Materials 2, no. 2: 399-498. https://doi.org/10.3390/ma2020399
APA StyleDorozhkin, S. V. (2009). Calcium Orthophosphates in Nature, Biology and Medicine. Materials, 2(2), 399-498. https://doi.org/10.3390/ma2020399