
Effect of the Peri-Annulated Dichalcogenide Bridge on the Bipolar Character of Naphthalimide Derivatives Used as Organic Electrode Materials

 ,
,  , , , , , and
, , , , , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
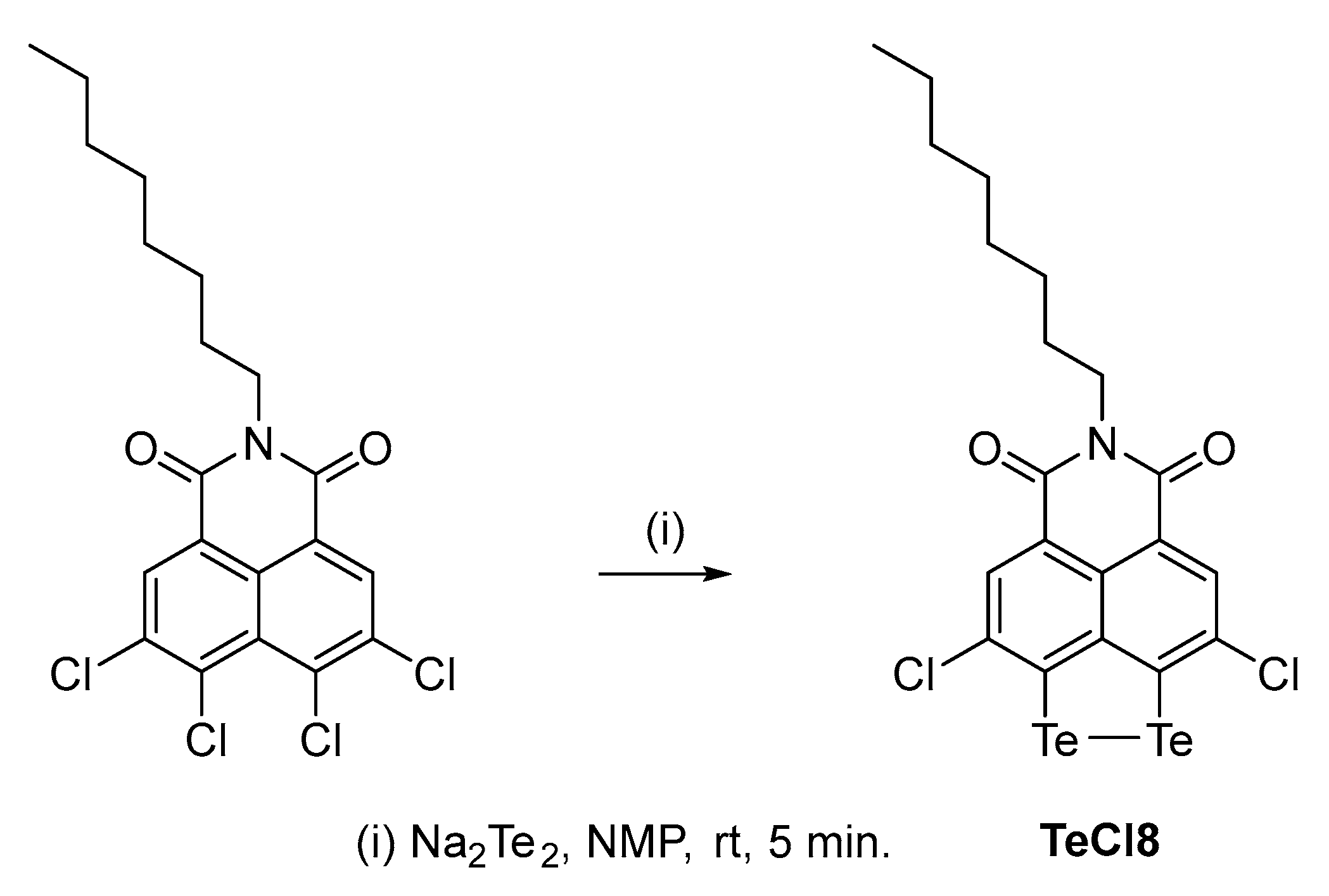
2.1. Synthesis
2.2. Preparation of Composites of NI Derivatives with rGO and Electrode Fabrication
2.3. Characterization Techniques
2.4. Computational Protocol
3. Results and Discussions
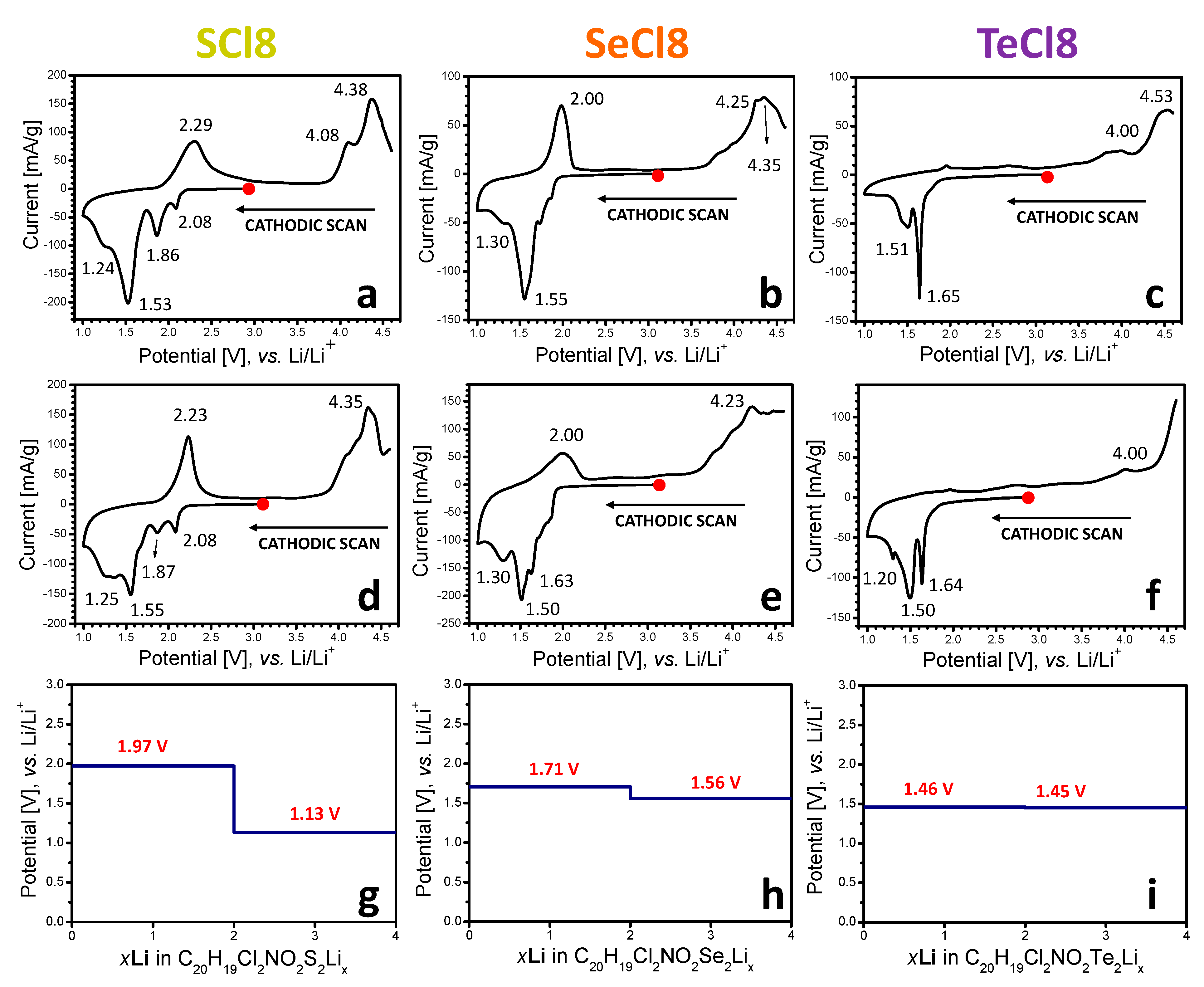
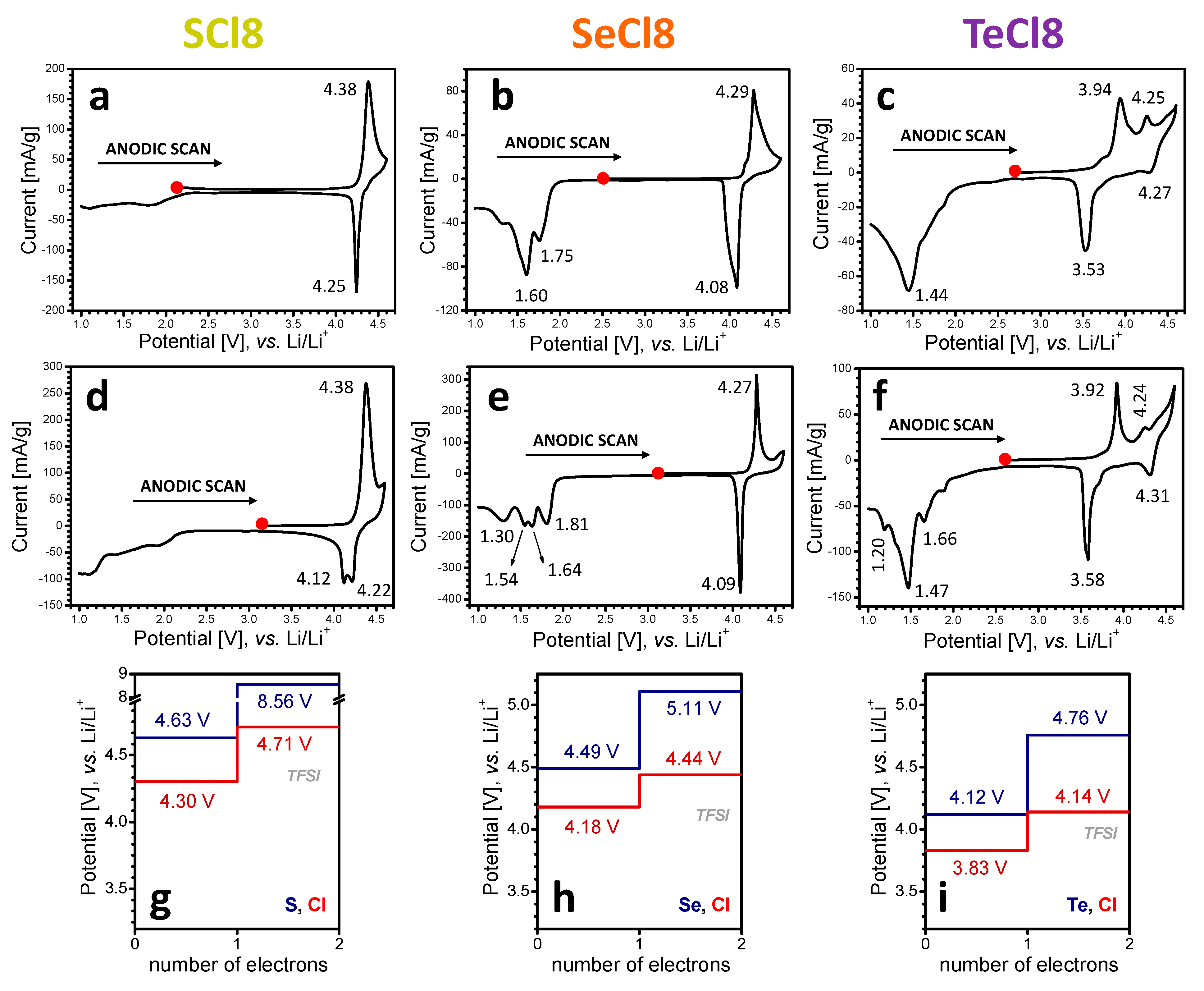
3.1. Electrochemical Redox Reactions at SCl8, SeCl8, and TeCl8 Electrodes
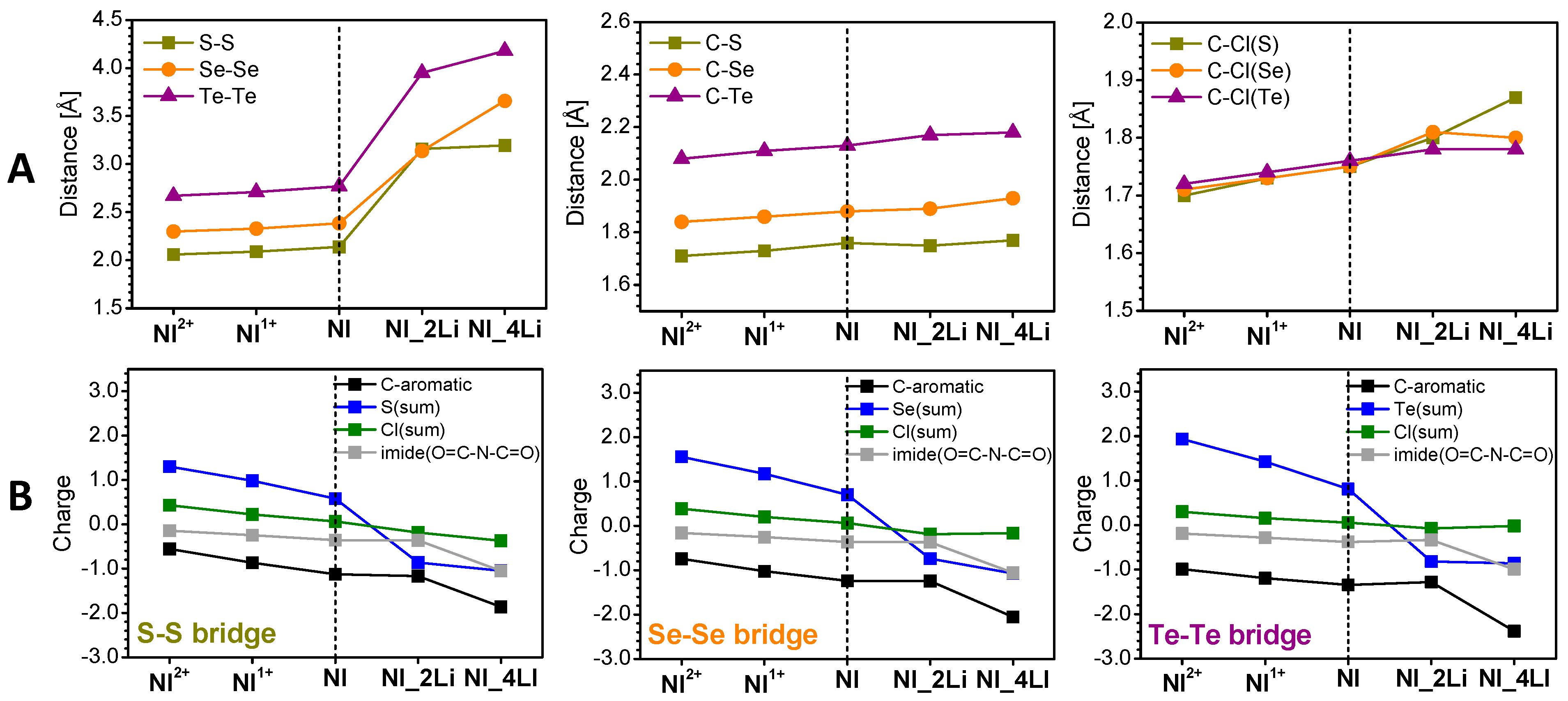
3.2. Mechanism of the Redox Reactions in SCl8, SeCl8, and TeCl8
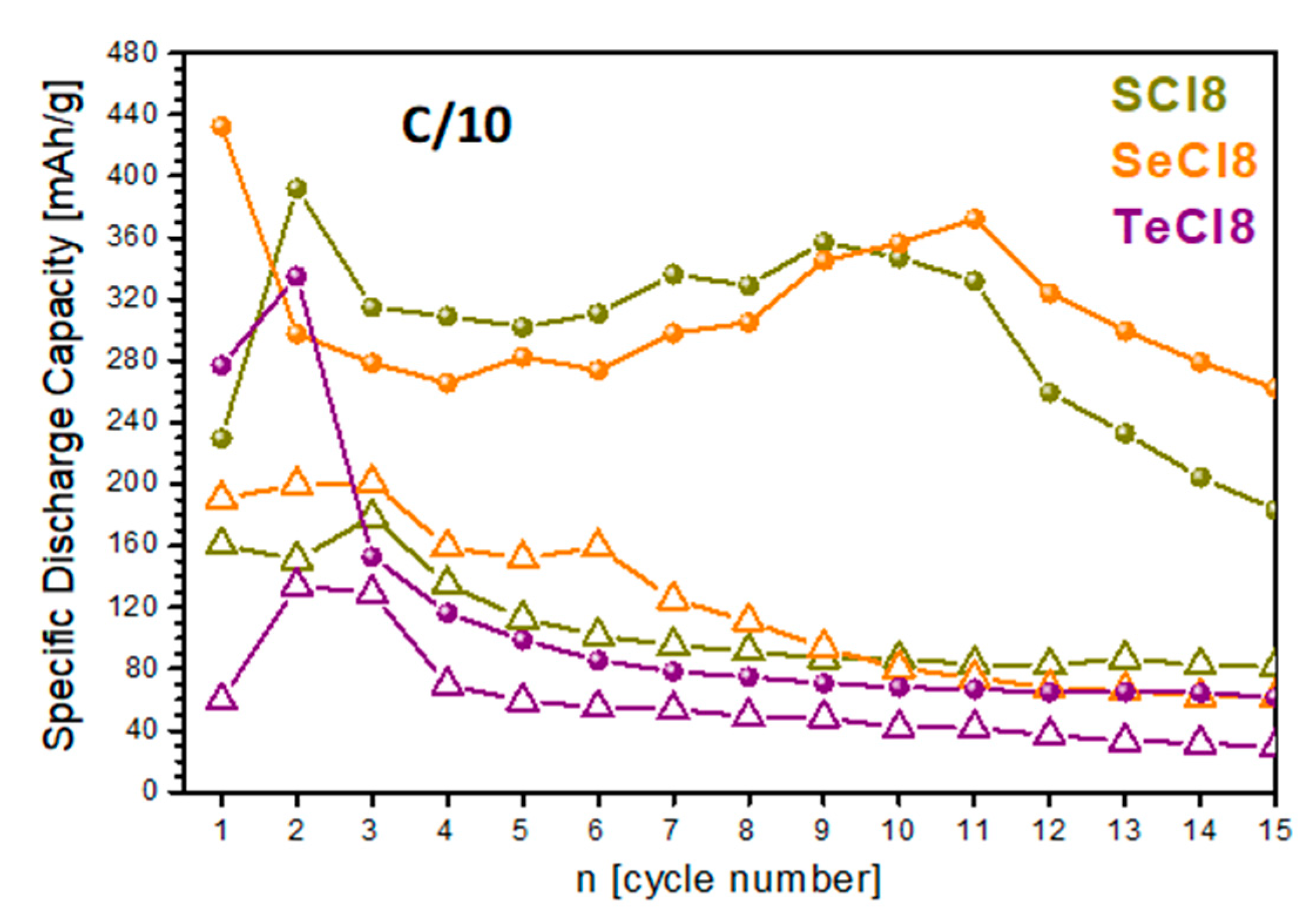
3.3. Storage Performance of SCl8, SeCl8, and TeCl8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Q.; O’Carroll, T.; Shi, F.; Huang, Y.; Chen, G.; Yang, X.; Nevar, A.; Dudko, N.; Tarasenko, N.; Xie, J.; et al. Designing organic material electrodes for lithium-ion batteries: Progress, challenges, and perspectives. Electrochem. Energy Rev. 2024, 7, 15. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, H.; Fan, K.; Zhang, G.; Tang, M.; Gao, Y.; Zhang, C.; Guan, L.; Mao, M.; Liu, H.; et al. A recyclable and scalable high-capacity organic battery. Angew. Chem. 2023, 135, e202302539. [Google Scholar] [CrossRef]
- Banerjee, T.; Kundu, R. Mini-Review on organic electrode materials: Recent breakthroughs and advancement in metal ion batteries. Energy Fuels 2024, 38, 12487–12509. [Google Scholar] [CrossRef]
- Song, Z.; Zhou, H. Towards sustainable and versatile energy storage devices: An overview of organic electrode materials. Energy Environ. Sci. 2013, 6, 2280–2301. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.; Yoo, J.; Kwon, G.; Ko, Y.; Kang, K. Organic batteries for a greener rechargeable world. Nat. Rev. Mater. 2022, 8, 54–70. [Google Scholar] [CrossRef]
- Yuan, S.; Huang, X.; Kong, T.; Yan, L.; Wang, Y. Organic electrode materials for energy storage and conversion: Mechanism, characteristics, and applications. Acc. Chem. Res. 2024, 57, 1550–1563. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, P.; Li, J.; Zhang, C.; Jiang, J.-X.; Lv, M.; Ding, Z.; Zhang, B. A low-cost naphthaldiimide based organic cathode for rechargeable lithium-ion batteries. Front. Chem. 2022, 10, 1056244. [Google Scholar] [CrossRef]
- Shi, Y.; Tang, H.; Jiang, S.; Kayser, L.V.; Li, M.; Liu, F.; Ji, F.; Lipomi, D.J.; Ong, S.P.; Chen, Z. Understanding the electrochemical properties of naphthalene diimide: Implication for stable and high-rate lithium-ion battery electrodes. Chem. Mater. 2018, 30, 3508–3517. [Google Scholar] [CrossRef]
- Masimukku, N.; Gudeika, D.; Volyniuk, D.; Bezvikonnyi, O.; Simokaitiene, J.; Matulis, V.; Lyakhov, D.; Azovskyi, V.; Gražulevičius, J.V. Bipolar 1, 8-naphthalimides showing high electron mobility and red AIE-active TADF for OLED applications. Phys. Chem. Chem. Phys. 2022, 24, 5070–5082. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, X.; Li, H.B.; Gan, F.; Zhang, J.; Dong, J.; Zhang, Q. Naphthalene-based polyimide derivatives as organic electrode materials for lithium-ion batteries. Electrochim. Acta 2017, 229, 387–395. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Hao, R.; Zhou, J.; Hussain, I.; Qin, N.; Li, M.; Chen, J.; Wang, Z.; Zheng, W.; et al. Redox of naphthalenediimide radicals in a 3D polyimide for stable Li-ion batteries. Chem. Commun. 2021, 57, 7810–7813. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chandrasekhar, N.; Biswal, B.P.; Becker, D.; Paasch, S.; Brunner, E.; Addicoat, M.; Yu, M.; Berger, R.; Feng, X. A crystalline, 2D Polyarylimide cathode for ultrastable and Ultrafast Li storage. Adv. Mater. 2019, 31, 1901478. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Wei, B.; Tang, W.; Hong, Y.; Guo, M.; He, X.; Hu, J.; Jia, S.; Fan, C. Carbon-coating small-molecule organic bipolar electrodes for symmetric Li-dual-ion batteries. Chem. Eng. J. 2023, 474, 145114. [Google Scholar] [CrossRef]
- Baskoro, F.; Lubis, A.L.; Wong, H.Q.; Liou, G.S.; Yen, H.J. Redox-active polynaphthalimides as versatile electrode materials for high-voltage, high-rate and long-cycle-life organic Li-ion batteries. J. Mater. Chem. A 2023, 11, 11210–11221. [Google Scholar] [CrossRef]
- Cho, Y.; Jang, D.; Park, J.J.; Kye, H.; Kwon, J.E.; Kim, B.G. Bipolar-type organic electrode material composed of a viologen–naphthalene diimide–viologen triad for Li-organic batteries. ACS Appl. Energy Mater. 2023, 7, 7615–7623. [Google Scholar] [CrossRef]
- Mutovska, M.; Simeonova, N.; Stoyanov, S.; Zagranyarski, Y.; Stanchovska, S.; Marinova, D. Naphthalene monoimides with peri-annulated disulfide bridge-synthesis and electrochemical redox activity. Materials 2023, 16, 7471. [Google Scholar] [CrossRef]
- Marinova, D.; Borislavov, L.; Stanchovska, S.; Kukeva, R.; Mutovska, M.; Simeonova, N.; Stoyanov, S.; Zagranyarski, Y.; Mondeshki, M.; Danchovski, Y.; et al. Peri-diselenolo-substituted 1, 8-naphthalimide derivatives as bipolar matrices for redox reactions in a non-aqueous electrolyte. Mater. Adv. 2025, 6, 788–804. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, C.; Li, Y. Molecular engineering the naphthalimide compounds as High-Capacity anolyte for nonaqueous redox flow batteries. Chem. Eng. J. 2022, 439, 135766. [Google Scholar] [CrossRef]
- Li, M.; Hicks, R.P.; Chen, Z.; Luo, C.; Guo, J.; Wang, C.; Xu, Y. Electrolytes in organic batteries. Chem. Rev. 2023, 123, 1712–1773. [Google Scholar] [CrossRef]
- Gambe, Y.; Kobayashi, H.; Honma, I. Acetonitrile-based highly concentrated electrolytes for high-power organic sodium-ion batteries. ACS Appl. Mater. Interfaces 2025, 17, 3316–3323. [Google Scholar] [CrossRef]
- Gannett, C.N.; Melecio-Zambrano, L.; Theibault, M.J.; Peterson, B.M.; Fors, B.P.; Abruña, H.D. Organic electrode materials for fast-rate, high-power battery applications. Mater. Rep. Energy 2021, 1, 100008. [Google Scholar] [CrossRef]
- Werner, D.; Apaydin, D.H.; Wielend, D.; Geistlinger, K.; Saputri, W.D.; Griesser, U.J.; Dražević, E.; Hofer, T.S.; Portenkirchner, E. Analysis of the ordering effects in anthraquinone thin films and its potential application for sodium ion batteries. J. Phys. Chem. C 2021, 125, 3745–3757. [Google Scholar] [CrossRef] [PubMed]
- Naz, N.; Manzoor, M.H.; Naqvi, S.M.G.; Ehsan, U.; Aslam, M.; Verpoort, F. Porous organic polymers; An emerging material applied in energy, environmental and biomedical applications. Appl. Mater. Today 2024, 38, 102198. [Google Scholar] [CrossRef]
- Jiao, X.; Kirianova, A.V.; Xu, X.; Kapitanova, O.O.; Krivchenko, V.A.; Napolskiy, F.S.; Volkov, V.S.; Gallyamov, M.O.; Liu, Y. Conductive additives for improving the rate capability of cathode materials in secondary lithium batteries. ACS Appl. Energy Mater. 2023, 6, 2855–2862. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, M.; Diao, C.; Song, Z.; Qin, J.; Zhan, H. Electrode engineering toward organic electrode materials. Sustain. Mater. Technol. 2025, 43, e01310. [Google Scholar] [CrossRef]
- Zhu, Z.; Chen, J. Advanced carbon-supported organic electrode materials for lithium (sodium)-ion batteries. J. Electrochem. Soc. 2015, 162, A2393–A2405. [Google Scholar] [CrossRef]
- Tong, Z.; Lv, C.; Bai, G.D.; Yin, Z.W.; Zhou, Y.; Li, J.T. A review on applications and challenges of carbon nanotubes in lithium-ion battery. Carbon Energy 2024, 7, e643. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, K.; Hu, H.; Wang, S.; Ye, D.; Monteiro, M.J.; Jia, Z.; Wang, L. Molecular-level anchoring of polymer cathodes on carbon nanotubes towards rapid-rate and long-cycle sodium-ion storage. Mater. Chem. Front. 2018, 2, 1805–1810. [Google Scholar] [CrossRef]
- Guo, W.; Yin, Y.X.; Xin, S.; Guo, Y.G.; Wan, L.J. Superior radical polymer cathode material with a two-electron process redox reaction promoted by graphene. Energy Environ. Sci. 2012, 5, 5221–5225. [Google Scholar] [CrossRef]
- Kumar, S.R.A.; Mary, D.V.; Josephine, G.S.; Ahamed, M.A.R. Graphene/GO/rGO based nanocomposites: Emerging energy and environmental application–review. Hybrid Adv. 2024, 5, 100168. [Google Scholar] [CrossRef]
- Li, A.; Feng, Z.; Sun, Y.; Shang, L.; Xu, L. Porous organic polymer/RGO composite as high performance cathode for half and full sodium ion batteries. J. Power Sources 2017, 343, 424–430. [Google Scholar] [CrossRef]
- Becke, A. Density-Functional Thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Francl, M.; Pietro, W.; Hehre, W.; Binkley, J.; DeFrees, D.; Pople, J.; Gordon, M. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* Basis set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Hellweg, A.; Rappoport, D. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. Phys. Chem. Chem. Phys. 2015, 17, 1010–1017. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Lipparini, F.; Scalmani, G.; Mennucci, B.; Cances, E.; Caricato, M.; Frisch, M.J. A variational formulation of the polarizable continuum model. J. Chem. Phys. 2010, 133, 014106. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F.; Foster, J. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO; Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; Version 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Gonçalves, R.; Lanceros-Méndez, S.; Costa, C.M. Electrode fabrication process and its influence in lithium-ion battery performance: State of the art and future trends. Electrohem. Commun. 2022, 135, 107210. [Google Scholar] [CrossRef]
- Harizanova, S.; Vulchev, V.; Stoyanova, R. Graphene-based composites for thermoelectric applications at room temperature. Materials 2023, 16, 7262. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marinova, D.; Borislavov, L.; Stanchovska, S.; Konstantinov, K.; Mutovska, M.; Stoyanov, S.; Zagranyarski, Y.; Danchovski, Y.; Rasheev, H.; Tadjer, A.; et al. Effect of the Peri-Annulated Dichalcogenide Bridge on the Bipolar Character of Naphthalimide Derivatives Used as Organic Electrode Materials. Materials 2025, 18, 2066. https://doi.org/10.3390/ma18092066
Marinova D, Borislavov L, Stanchovska S, Konstantinov K, Mutovska M, Stoyanov S, Zagranyarski Y, Danchovski Y, Rasheev H, Tadjer A, et al. Effect of the Peri-Annulated Dichalcogenide Bridge on the Bipolar Character of Naphthalimide Derivatives Used as Organic Electrode Materials. Materials. 2025; 18(9):2066. https://doi.org/10.3390/ma18092066
Chicago/Turabian StyleMarinova, Delyana, Lyuben Borislavov, Silva Stanchovska, Konstantin Konstantinov, Monika Mutovska, Stanimir Stoyanov, Yulian Zagranyarski, Yanislav Danchovski, Hristo Rasheev, Alia Tadjer, and et al. 2025. "Effect of the Peri-Annulated Dichalcogenide Bridge on the Bipolar Character of Naphthalimide Derivatives Used as Organic Electrode Materials" Materials 18, no. 9: 2066. https://doi.org/10.3390/ma18092066
APA StyleMarinova, D., Borislavov, L., Stanchovska, S., Konstantinov, K., Mutovska, M., Stoyanov, S., Zagranyarski, Y., Danchovski, Y., Rasheev, H., Tadjer, A., & Stoyanova, R. (2025). Effect of the Peri-Annulated Dichalcogenide Bridge on the Bipolar Character of Naphthalimide Derivatives Used as Organic Electrode Materials. Materials, 18(9), 2066. https://doi.org/10.3390/ma18092066