Abstract
We constructed computational models of bare Zr3C2 and surface-functionalized Zr3C2T2 (T = O, S, F, Cl), and utilized first-principles calculations to systematically explore the effects of these surface-functionalized groups on the structural stability, electronic properties, and lithium storage performance of Zr3C2T2. Compared to halogen functional groups (e.g., F, Cl), the structure and electronic properties of Zr3C2 are more profoundly influenced by oxygen group functional elements (O, S). The formation energy of Zr3C2T2 (T = O, S) functionalized by the same periodic oxygen group elements is lower than that of Zr3C2T2 (T = F, Cl) functionalized by the same periodic halogens. Regarding electronic properties, the oxygen and sulfur functional groups have strong hybridization with Zr3C2 in the valence band and generate a new band structure, which makes the DOS move toward the conduction band. The adsorption energy calculations reveal that lithium ions exhibit stable adsorption on bare Zr3C2 and O/S-functionalized Zr3C2T2 surfaces, whereas no stable adsorption occurs on Zr3C2F2 or Zr3C2Cl2. In terms of adsorbing lithium atoms, bare Zr3C2 tends to adsorb at the HCP position, while Zr3C2O2 and Zr3C2S2 tend to adsorb at the CCP position. First-principles calculations indicate distinct theoretical lithium storage capacities for Zr3C2-based materials: monolayer adsorption yields capacities of 180.13 mAh/g (bare Zr3C2), 162.64 mAh/g (Zr3C2O2), and 148.20 mAh/g (Zr3C2S2); bilayer adsorption significantly increases these values to 360.25, 325.29, and 296.41 mAh/g, respectively.
1. Introduction
With the rapid development of new energy vehicles and intelligent electronic devices, it is crucial to develop more efficient energy storage devices [1]. Among many energy storage solutions, lithium-ion batteries (LIBs) are the optimal choice due to their high energy density, rapid charging/discharging capabilities, and long cycle life [2]. The ability of LIBs to store and release energy is primarily governed by the reversible cycling of lithium ions within electrode materials during charge/discharge cycles [3]. Conventional graphite anodes are constrained by their limited theoretical capacity and poor rate performance, which fall short of the stringent requirements for next-generation LIBs [4]. Consequently, the development of advanced anode materials has become a central focus in LIB research [5]. Among the numerous candidate electrode materials, two-dimensional (2D) materials have received extensive attention due to their unique layered structures [6]. The currently reported 2D materials, including graphene [7,8,9,10], transition metal disulfide [11,12,13], phosphorene [14,15,16,17], and MXene [18,19,20,21,22,23,24], are promising electrode materials for lithium-ion batteries. Although graphene, transition metal disulfide, and phosphorene can be used as electrode materials, their low electrical conductivity and lithium-ion transport rate greatly affect the performance of the batteries [25,26,27]. Since the successful preparation of MXene in 2011 [28], it has gradually attracted significant research attention because of low energy barriers and large interlayer spaces for metal ion diffusion [29,30].
MXene is typically synthesized by the etching of “A” layers from layered ternary MAX phases using hydrofluoric acid or a solution composed of a mixture of hydrochloric acid and lithium fluoride. The chemical formula of MXene is usually expressed as Mn+1XnTx (n = 1~3) [31], where M represents a metallic element, mainly transition metals such as Ti, Zr, and Hf; X stands for either C or N; and T denotes surface functional groups, such as -OH and halogen groups (-F, -Cl) [32]. This material is composed of metal carbides or nitrides and has a two-dimensional layered structure. The applications of MXene in the field of energy storage have received increasing attention since Naguib et al. [28] reported that MXene materials can be used as electrode materials for lithium-ion batteries. Currently reported MXenes for lithium-ion battery electrodes include Ti3C2 [33,34,35], Mo2C [36,37], Nb2C [38,39], V2C [39,40,41], Mo2CS2 [42], the double-transition metal MXene Mo2TiC2 [43], MoWC [44], etc. Extensive reports have shown that the properties of MXenes strongly depend on the surface functional groups [45,46]. Li et al. [47] analyzed the effect of different groups (O, H, and OH) on the lithium storage properties of Ti3C2 using the DFT method. The results revealed that O reduced the adsorption energy of lithium atoms, while F limited the adsorption of Li atoms, and the Li atoms could not even stably adsorb on the Ti3C2H2 and Ti3C2(OH)2 surfaces. Li et al. [48] modified the lithium storage performance of Ti2C by introducing eight different doping atoms. The results showed that different doping atoms altered the d-band center of the material, thereby affecting the adsorption energy of lithium on the MXene surface. Using V as the doping atom resulted in the best adsorption energy, open circuit voltage, and minimum diffusion energy. Abdelsalam et al. [49] used DFT to calculate the adsorption of lithium on Zr2C and Zr2CO2 MXenes. The results demonstrated that lithium can be stably adsorbed on Zr2C and Zr2CO2, with a theoretical capacity higher than graphite, making it a favorable candidate for lithium-ion battery electrodes.
A new Zr3C2Tx MXene was successfully synthesized [50] by selectively etching Al3C3 from Zr3Al3C5 and was then theoretically [51] investigated. Recently, computational insights into the influence of surface functionalization groups on Zr3C2T2 (T = O, F) [52] for sodium-sulfur batteries and Zr3C2T2 (T = H, Si, P, S, Se) [53] for lithium-ion batteries were studied. Khan [54] reviewed recent advances in the synthesis, properties, and novel applications of Zr3C2T2. Although substantial progress has been made in understanding functional group effects on MXenes, the influence of F and Cl surface functional groups on the structure, electronic behavior, and other properties of Zr3C2 MXenes still needs further exploration.
In this paper, the effects of surface terminals belonging to the oxygen (T = O, S) and halogen (T = F, Cl) groups on Zr3C2 were comparatively studied. We first constructed and optimized Zr3C2 and functionalized Zr3C2T2 (T = O, S, F, Cl), and then calculated the formation energies of the surface functional terminals at potential sites. At last, the effect of the functional groups (O, S, F, Cl) on the structural, electronic, and lithium storage properties of Zr3C2 MXene was studied.
2. Computing Method
The DFT calculations were performed using the CASTEP [55] module in Materials Studio software (Version 7.0). The Ultrasoft pseudopotentials method [56] and the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) function [57] were employed during the calculations. Atomic pseudopotentials of Zr, C, O, S, F, and Cl were formed separately using electronic structures of 4d25s2, 2s22p2, 2s22p4, 3s23p4, 2s22p5, and 3s23p5. An 11 × 11 × 1 k-point grid for the Brillouin zone integration was generated by using the Monkhorst–Pack sampling method [58]. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) method [59] was used for geometry optimization. The following parameters were set: an energy cutoff value of 450 eV, an energy convergence tolerance of 5.0 × 10−6 eV, a maximum force convergence tolerance of 0.01 eV/Å, a maximum stress convergence tolerance of 0.02 GPa, and a maximum displacement deviation less than 5.0 × 10−4 Å.
3. Results and Discussion
3.1. Structural Properties
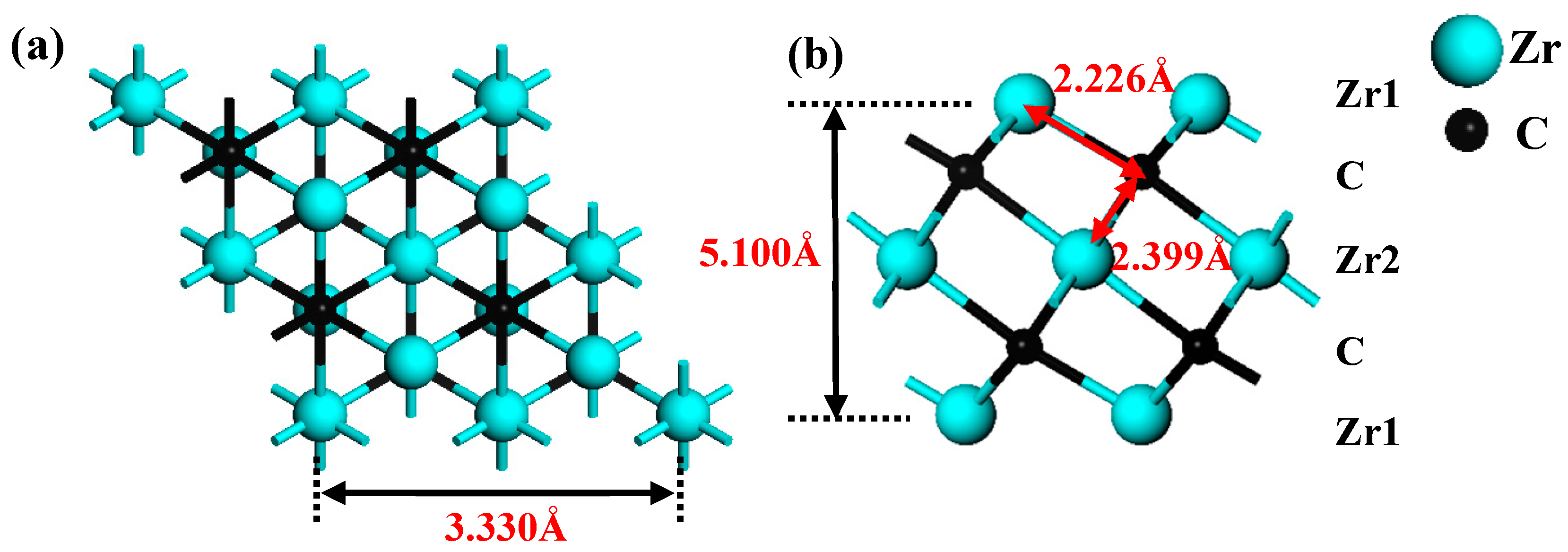
In this paper, the initial MAX material Zr3AlC2 came from the Material Project database [60]. The hexagonal Zr3C2 structure is obtained by “etching” Al atom layers from Zr3AlC2. The crystal structures of Zr3C2 are shown in Figure 1a,b. The optimized lattice parameter of bare Zr3C2 is 3.330 Å (close to 3.349 Å [61]), and the thickness of the monolayer is 5.100 Å, which is labeled in Figure 1a,b. The functional groups have three potential adsorption sites on the surface of the MXene: a top (TOP) site over the first layer of Zr1 atoms; a hexagonal close-packed (HCP) site over C atoms; and a cubic close-packed (CCP) site over the second layer of Zr2 atoms. The TOP site is usually a coordination atom that enhances reactivity and plays a crucial role in adsorption processes that require bond activation, while HCP and CCP are determined by the arrangement of sub-surface atoms and differ in coordination and symmetry: HCP is aligned with deep atoms, while CCP is not aligned with deep atoms. These changes affect the binding strength and selectivity, guiding the adsorption reaction pathway. By adjusting the distribution of these sites through crystal surface engineering or nanostructures, the application of materials in energy storage, stability, and catalysis can be optimized. According to the work of Li et al. [62], the Zr3C2T2 (T = O, S, F, Cl) models containing functional groups were constructed.
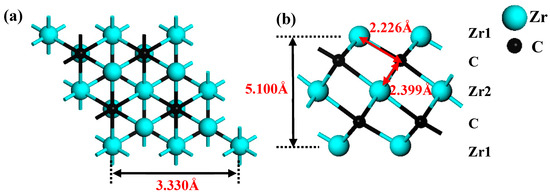
Figure 1.
Crystal structures of 2 × 2 × 1 supercell of Zr3C2: (a) top view and (b) side view.
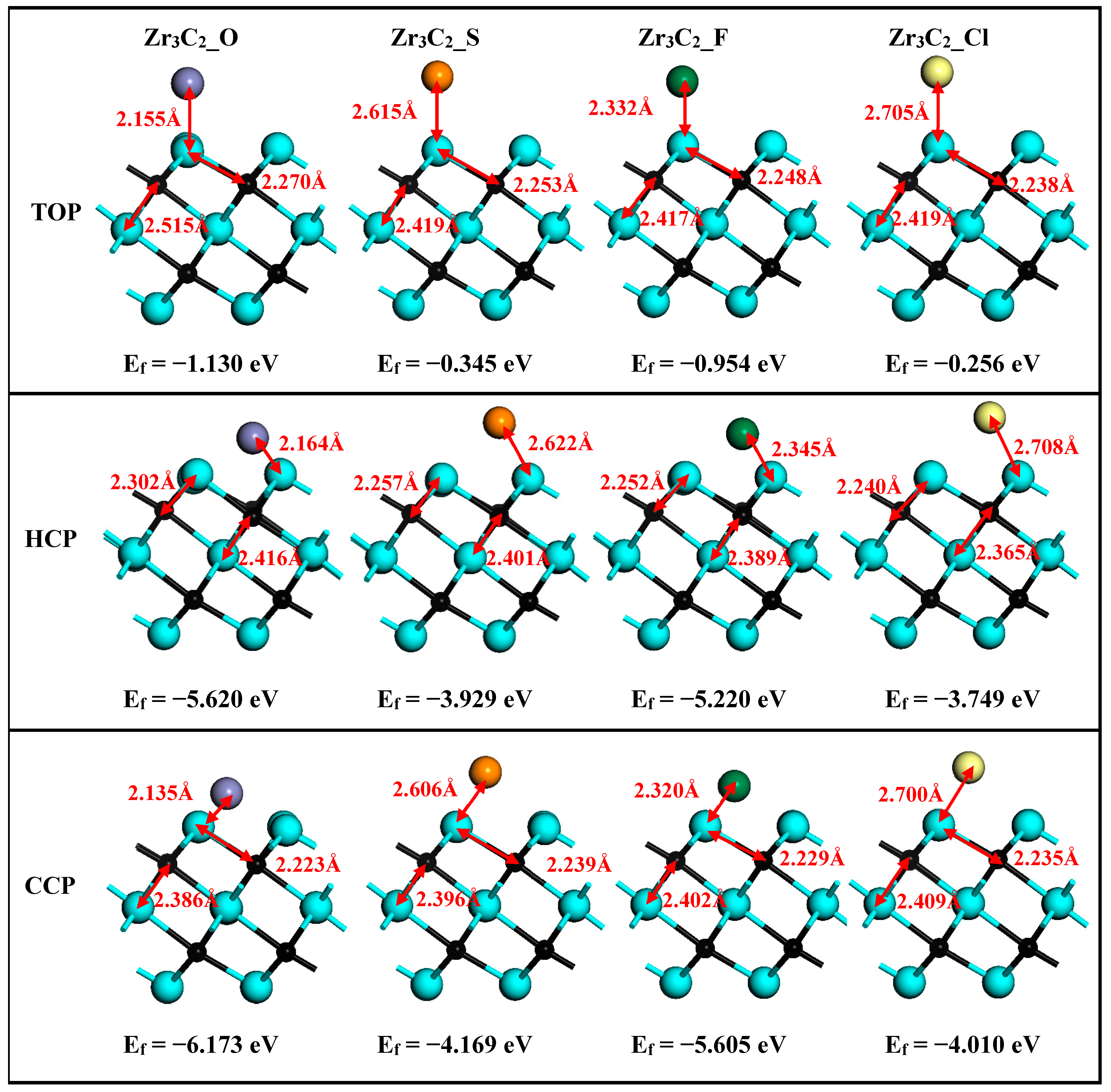
After structural relaxation, the structures, bond lengths of dZr1–T (between the surface Zr1 and functionalized terminal atoms), dZr1–C (between the surface Zr1 and C atoms), and dZr2–C (between the middle Zr2 and C atoms) of Zr3C2T2 (T = O, S, F, Cl) are shown in Figure 2. It is shown in Figure 2 that the lattice constants of Zr3C2T2 (T = O, S, F, Cl) have been slightly changed compared to bare Zr3C2 in Figure 1. Compared with dZr1–C = 2.226 Å and dZr2–C = 2.399 Å in the bare Zr3C2, the bond length dZr2–C (2.365–2.419 Å) remains nearly constant, while the dZr1–C (2.223–2.302 Å) increases significantly in Zr3C2T2 (T = O, S, F, Cl). Furthermore, comparative analysis reveals distinct chemical trends: the bond lengths between the S or Cl and Zr atoms are significantly longer than those of the O or F and Zr atoms, and the bond length between the halogen functional groups (F, Cl) and Zr3C2 is longer than the counterpart between the oxygen group elements (O, S) and Zr3C2. The atomic size and atomic electronegativity may cause these discrepancies. Compared with the HCP and TOP positions, the CCP position has the shortest bond length with Zr3C2, making it the most stable site for the functional group.
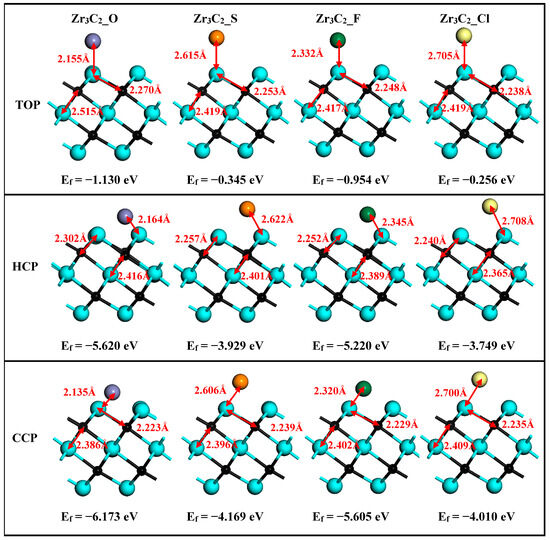
Figure 2.
The optimized structures of Zr3C2T2 (T = O, S, F, Cl) with the HCP, CCP, and TOP sites.
To analyze the thermodynamic stability of functional groups at the HCP, CCP, and TOP positions on the Zr3C2 surface, the formation energies are calculated using Equation (1) [47]:
where Etot(Zr3C2T2) refers to the total energy associated with Zr3C2T2; E(Zr3C2) and E(T2) are the isolated energies of bare Zr3C2 and the functional groups, respectively. Specifically, relative to the O2, F2, and Cl2 molecules, represents twice the energy of a single S atom. The results of the formation energies are also shown with the corresponding structure in Figure 2. It is shown in Figure 2 that all the formation energies are negative, confirming their energetic stability. Additionally, Figure 2 reveals that surface functional groups in Zr3C2T2 (T = O, S, F, Cl) preferentially occupy CCP sites, which exhibit the lowest formation energies among the three considered sites. This observation aligns with findings previously reported in the literature [63].
For the bare Zr3C2 and Zr3C2T2 with the most stable configurations (CPP sites), the optimized lattice parameters and formation energies were summarized for comparison in Table 1. It is revealed in Table 1 that the formation energies of surface-functionalized Zr3C2T2 (−6.173 eV for Zr3C2O2 and −5.605 eV for Zr3C2F2) located in the second period of the periodic table are lower than those located in the third period (−4.169 eV for Zr3C2S2 and −4.010 eV for Zr3C2Cl2).

Table 1.
The optimized structural parameters, formation energy, and work function at CPP sites of Zr3C2 and Zr3C2T2.
In addition, the work functions of Zr3C2 and Zr3C2T2 were calculated, which are also listed in Table 1. The results show that the work function is directly proportional to the formation energy of the functional groups: the greater the absolute value of formation energy, the larger the work function. The obtained work functions are in the following order: Zr3C2 < Zr3C2Cl2 < Zr3C2F2 < Zr3C2S2 < Zr3C2O2.
3.2. Electronic Properties
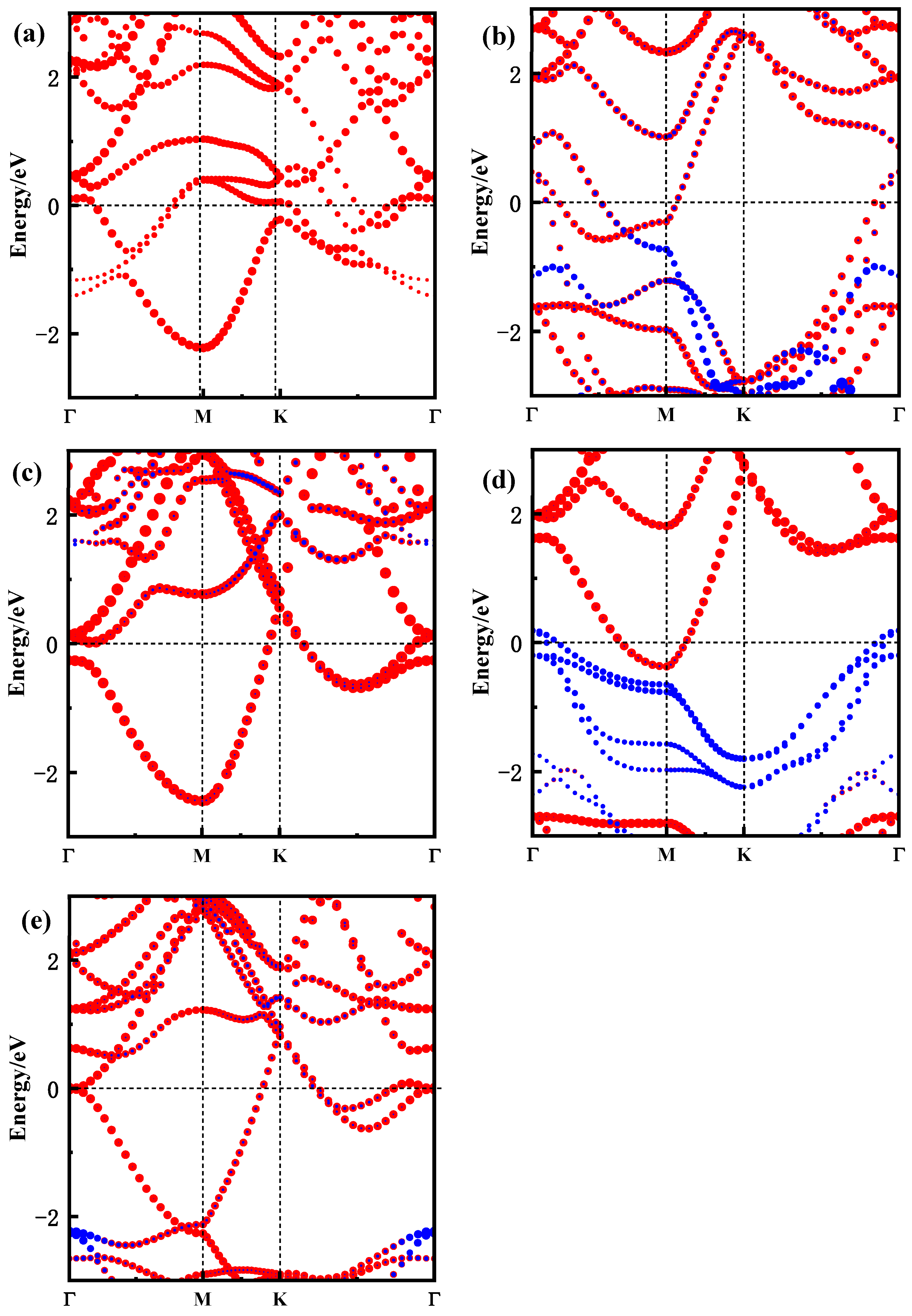
The band structure is a crucial factor in understanding the electronic properties of materials. For the Zr3C2T2 MXene, the electronic properties are closely linked to the surface functional groups. To analyze the impact of these functional groups on the electronic behavior of the MXene, the high symmetry path Γ(0 0 0)-M(0.5 0 0)-K(0.333 0.333 0)-Γ(0 0 0) was selected to calculate the band structure of Zr3C2T2 (T = O, S, F, Cl), and the results are shown in Figure 3. It is indicated that the functional groups primarily affect the valence band of Zr3C2T2, while their impact on the conduction band is minimal. Compared to the fluorine and chlorine functional groups, the band structure near the Fermi level is more affected by the O and S functional groups. When Zr3C2 is functionalized by O and S, as shown in Figure 3a,c, the band structures near the Fermi level are the result of functional group atoms, and even new band structures are generated near the Fermi level. However, in the band structure of Zr3C2F2 and Zr3C2Cl2, it is clear in Figure 3b,d that the contribution of functional groups near the Fermi level is almost invisible, and the contribution of functional groups is mainly in the valence band where the energy is much lower than the Fermi level. In addition, an interesting phenomenon was found in which the band structure of Zr3C2O2 and Zr3C2S2 had a clear tendency to move towards the conduction band, in contrast to Zr3C2F2 and Zr3C2Cl2. This is mainly due to the stronger hybridization of the O and S functional groups with Zr3C2, which enhances the expansibility of atomic orbitals of the valence band near the Fermi level.
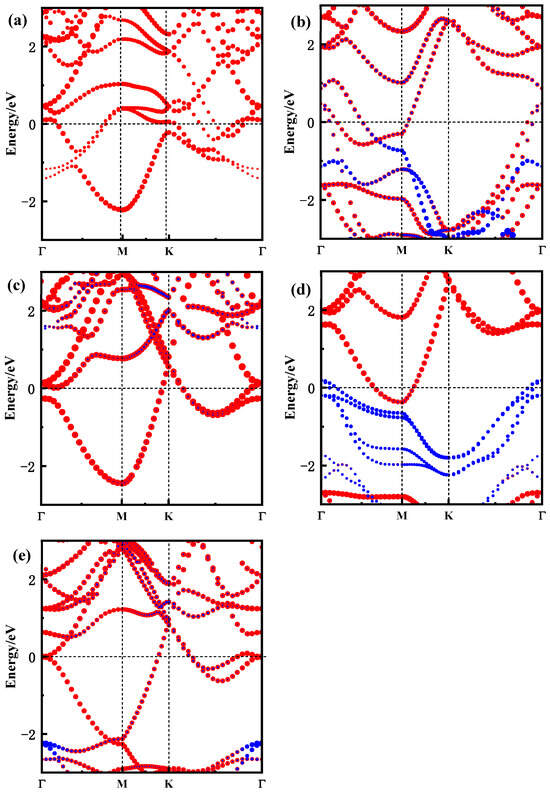
Figure 3.
Band structure of Zr3C2 and Zr3C2T2 along the high-symmetry path Γ(0 0 0)-M(0.5 0 0)-K(0.333 0.333 0)-Γ(0 0 0). (a) Zr3C2; (b) Zr3C2O2; (c) Zr3C2F2; (d) Zr3C2S2; and (e) Zr3C2Cl2. (The red and blue lines represent the energy band contributed by Zr3C2 and functional group T2, respectively).
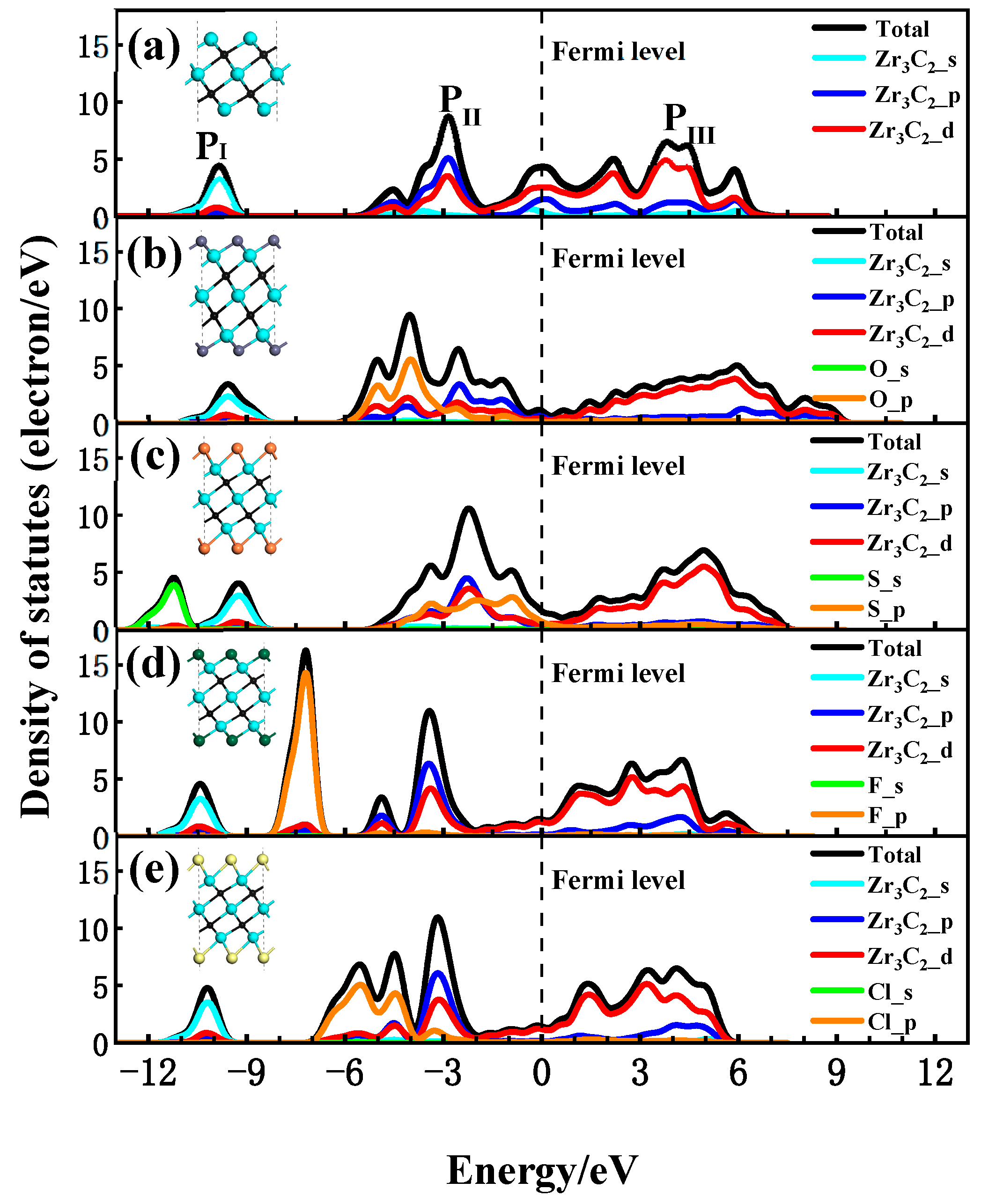
To discuss the interaction between the functional groups and Zr3C2, the total (DOS) and partial density of states (PDOS) for the bare Zr3C2 and functionalized Zr3C2T2 with the corresponding structural diagram are shown in Figure 4. In the DOS and PDOS of bare Zr3C2 in Figure 4a, a broader hybridization peak (0–6 eV) appears in the conduction band above the Fermi level due to the hybridization of Zr and C, indicating a strong Zr-C bond. There are two main peaks in the valence band, one contributed by the Zr3C2-p orbitals (−10 eV) and the other by the combined Zr3C2-p and Zr3C2-d orbitals (−3 eV). In addition, a large peak at the Fermi level may induce instabilities in bare Zr3C2, suggesting that Zr3C2 tends to be functionalized by terminal groups [61]. As can be seen in Figure 4b–e, different functional groups have different effects on the DOS of Zr3C2T2. The results show that when Zr3C2 was functionalized by O, F, S, and Cl, the DOS near the Fermi level was affected by the p orbital of the functional groups, while the s orbital of the functional groups only affected the valence band away from the Fermi level. In the PDOS of Zr3C2O2 and Zr3C2S2, as shown in Figure 4b,c, it is the hybridization between the p orbitals of the functional groups (O-p and S-p) and the Zr3C2-d orbitals that enhances the extensibility of the hybridization peak originally located at −3 eV and made the DOS near the Fermi level move towards the conduction band. This phenomenon observed in Figure 4b,c aligns with the band structures of Zr3C2O2 and Zr3C2S2, which exhibit a distinct tendency to shift toward the conduction band. In addition, the broader hybridization peaks in the conduction band are also affected by the oxygen and sulfur functional groups. Because of the effect of the functional groups, the contribution of the Zr3C2-p orbitals to the wide peaks is reduced and shows stronger localization at −2 eV~0 eV. However, it can be seen in Figure 4d,e that when the Zr3C2 MXene was functionalized by F and Cl, the hybrid peak between the functional groups and Zr3C2 was not found.
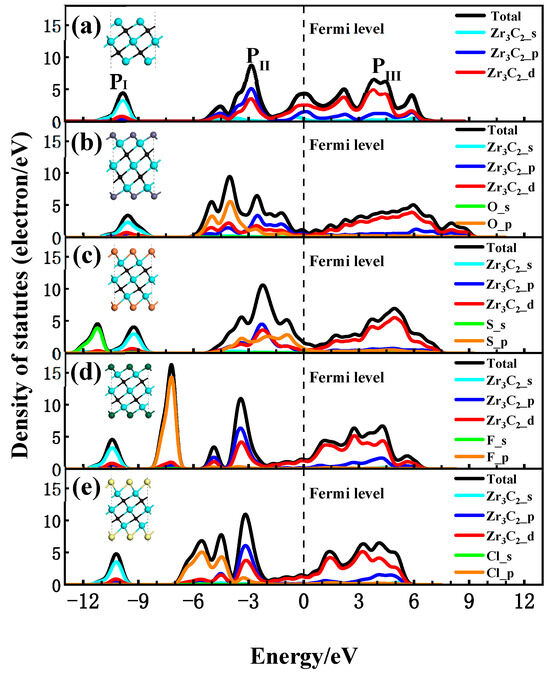
Figure 4.
Density of states of Zr3C2 and Zr3C2T2 with corresponding structural diagram: (a) bare Zr3C2; (b) Zr3C2O2; (c) Zr3C2S2; (d) Zr3C2F2; and (e) Zr3C2Cl2.
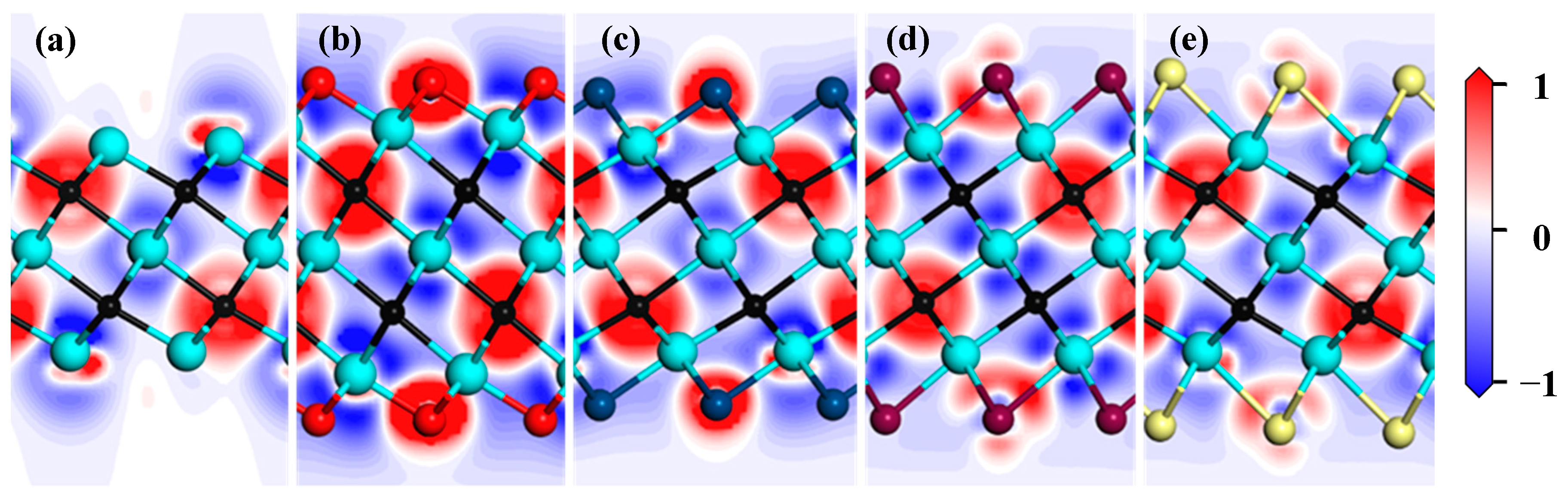
Differential charge density is an important way to analyze interatomic bonding and charge transfer. To discuss the effect of functional groups on charge transfer in the Zr3C2 MXene, differential charge density for the bare Zr3C2 and functionalized Zr3C2T2 are shown in Figure 5, in which the electron depletion and accumulation are denoted in red and blue, respectively. The result in Figure 5 shows that the charges accumulated around the C atoms, and the charges depleted around the surface Zr atoms in the bare Zr3C2. This suggests that bare Zr3C2 exhibits ionic bonding characteristics, with strong directional bonds forming between the Zr and C atoms. It is noteworthy that a small amount of charge accumulates on the surface of the low coordination Zr atoms, while the charge is consumed on the side of these Zr atoms near the C atom. When Zr3C2 is functionalized, the functional group atoms participate in charge transfer. Compared with the Zr3C2O2 and Zr3C2S2, the charge transfers between the functional group atoms of Zr3C2F2 and Zr3C2Cl2 with the surface Zr atoms are weaker.
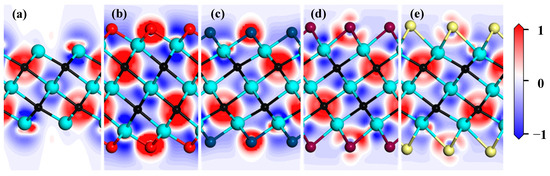
Figure 5.
Differential density charge of bare Zr3C2 and Zr3C2T2 (T = O, S, F, Cl): (a) bare Zr3C2, (b) Zr3C2O2, (c) Zr3C2F2, (d) Zr3C2S2, and (e) Zr3C2Cl2 (red represents electron depletion and blue represents accumulation).
Moreover, the Mulliken population analysis offers a quantitative measure for evaluating bonding characteristics. As shown in Table 2, the analysis reveals a charge transfer of 1.56 electrons from the Zr to C atoms in the bare Zr3C2, of which 0.9 electrons are contributed by the central Zr atom. Due to the influence of the functional groups, the charge transfers between the Zr and C atoms increased (1.60–1.66 electrons), indicating that the Zr-C bond is stronger. In addition, when Zr3C2 is functionalized by oxygen and sulfur, there is a strong charge transfer between the functional group atoms and the surface Zr atoms. However, when Zr3C2 is functionalized by fluorine and chlorine, the charge transfer between the functional group atoms and the surface Zr atoms is weak. These discrepancies may be caused by atomic electronegativity.
Table 2.
Mulliken population of bare Zr3C2 and Zr3C2T2.
3.3. Lithium Storage Performance
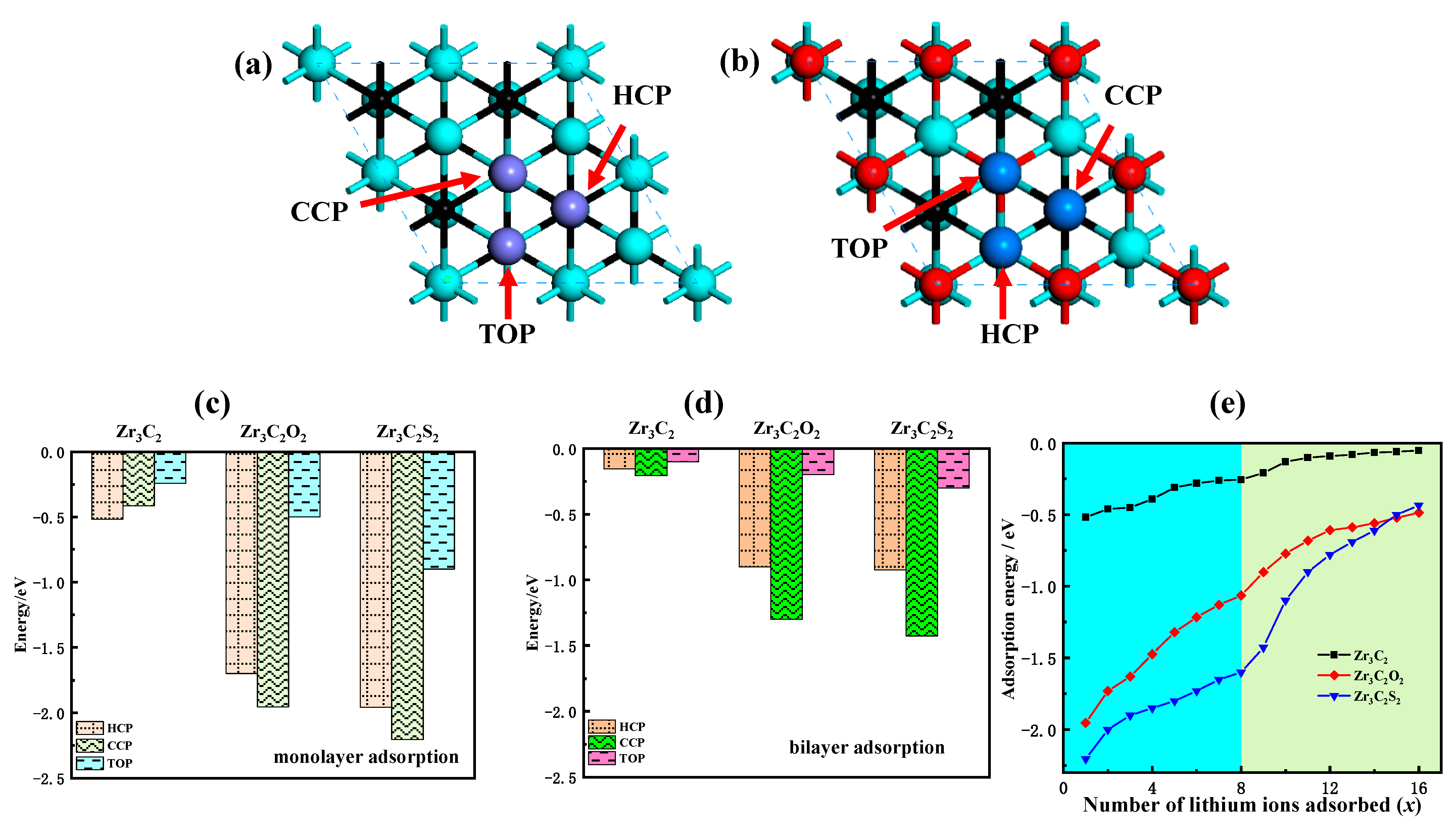
Monolayer adsorption and multilayer adsorption are studied in the study of the lithium storage performance of Zr3C2T2. The potential sites of monolayer and multilayer adsorption of lithium ions on the Zr3C2 surface are shown in Figure 6a,b.
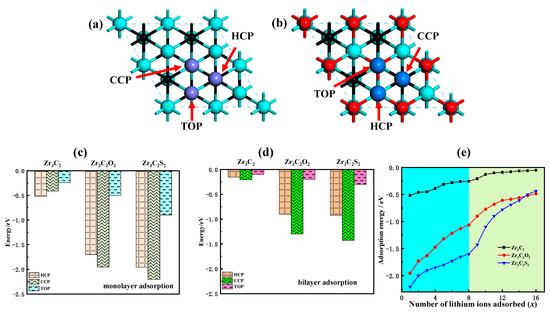
Figure 6.
Lithium storage properties of bare Zr3C2 and Zr3C2T2. (a,b) The potential adsorption sites of lithium ions on bare Zr3C2 and Zr3C2T2 surfaces, respectively; (c,d) The monolayer and bilayer adsorption energies of lithium ions at HCP, CCP, and TOP sites, respectively; (e) The relationship between the number of adsorbed lithium ions (x) and the adsorption energy.
To identify the most stable adsorption site for lithium ions on the surface of both bare Zr3C2 and functionalized Zr3C2T2, the adsorption energy at each potential site was determined using Equation (2) [47]:
where Esub+xLi denotes the total energy of bare Zr3C2 or Zr3C2T2 with lithium ions adsorbed, x represents the number of adsorbed lithium ions, Esub is the energy of isolated bare Zr3C2 or Zr3C2T2, and E(Li) corresponds to the average energy of lithium ions. Figure 6c–d present the monolayer and bilayer adsorption energies of lithium ions at various adsorption sites. For the bare Zr3C2, the monolayer adsorption energies (Figure 6d) are −0.52 eV (HCP), −0.41 eV (CCP), and −0.24 eV (TOP). In comparison, the bilayer adsorption energies (Figure 6e) are −0.16 eV (HCP), −0.21 eV (CCP), and −0.10 eV (TOP). Compared with bare Zr3C2, the adsorption energy of lithium ions at the Zr3C2 functionalized by O and S functional groups is reduced. By contrast, the adsorption energy of Zr3C2F2 and Zr3C2Cl2 on lithium ions increased to a positive value, indicating that the Zr3C2T2 functionalized by fluoride and chlorine could not stably adsorb lithium ions. In addition, the most stable adsorption site of lithium-ion is also affected by the terminals. The most stable adsorption site for lithium ions in the monolayer adsorption of bare Zr3C2 is the HCP site, and the most stable adsorption site in the bilayer adsorption of bare Zr3C2 is the CCP site. When Zr3C2 is functionalized with O and S, the monolayer adsorption of lithium ions starts at the CCP site. For bilayer adsorption in Zr3C2O2, the HCP site is identified as the most stable adsorption site, which is like the switching of adsorption sites in the bare Zr3C2 monolayer adsorption and bilayer adsorption. However, the most stable adsorption site in the bilayer adsorption of Zr3C2S2 is still the CCP site, which is not like the switching of the adsorption site with bare Zr3C2.
To further understand the influence of the functional groups on the lithium storage properties of Zr3C2T2, the relationship between the amount of adsorbed lithium ions (x) and the adsorption energy for both monolayer and bilayer adsorption was investigated. The findings, presented in Figure 6e, show that the curve within the sky blue area represents the variation in adsorption energy during monolayer adsorption (x ≤ 8), while the curve in the light green region corresponds to the bilayer adsorption (9 ≤ x ≤ 16). The results indicate that the adsorption energy increases as the number of adsorbed lithium ions rises. The adsorption energy increases from −0.52 eV to −0.05 eV for bare Zr3C2, from −1.95 eV to −0.49 eV for Zr3C2O2, and from −2.20 eV to −0.44 eV for Zr3C2S2. In addition, the adsorption energy curve of each layer always rises steeply and then gently. Compared with the adsorption energy curve of monolayer adsorption, the front part of the adsorption curve of bilayer adsorption is steeper. This phenomenon is easily inferred that bare Zr3C2 makes it difficult to continue the three-layer adsorption.
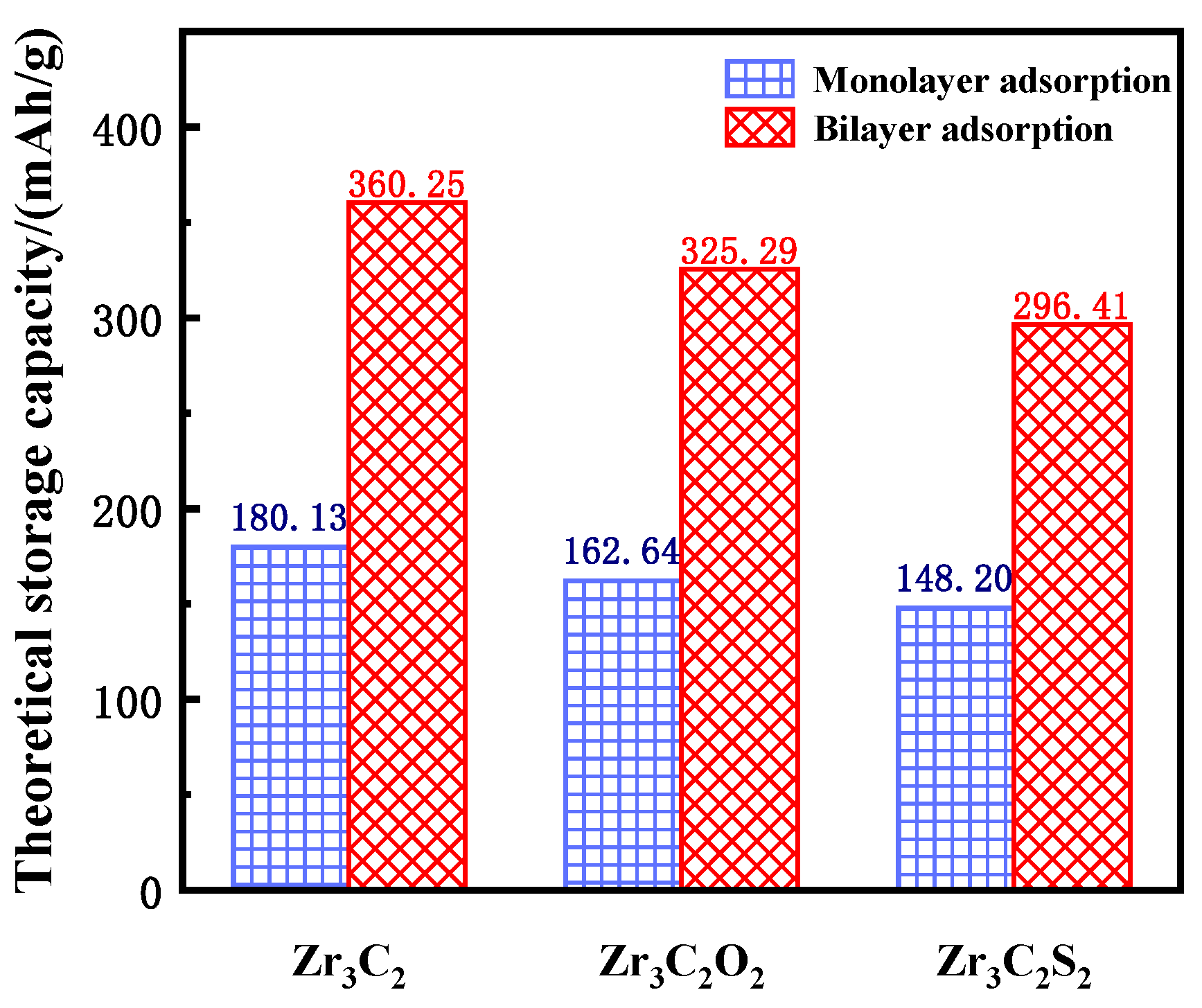
The theoretical lithium storage capacity of bare and functionalized Zr3C2T2 (T = O, S) was calculated using Equation (3) [64]:
where z represents the valence number (z = 1 for Li), F is the Faraday constant (26.810 Ah/mol), and W is the atomic mass (297.68 g/mol for Zr3C2, 329.68 g/mol for Zr3C2O2, and 361.80 g/mol for Zr3C2S2). In Equation (3), 4W is used because a 2 × 2 × 1 supercell was constructed to investigate the lithium storage properties. The theoretical lithium storage capacity of single-layer adsorption and double-layer adsorption was calculated separately, and the results are shown in Figure 7. The sky blue squares represent the theoretical capacity of monolayer adsorption, and the red oblique squares represent the theoretical capacity of bilayer adsorption. The theoretical lithium storage capacities of bare Zr3C2, Zr3C2O2, and Zr3C2S2 are 180.13 mAh/g, 162.64 mAh/g, and 148.20 mAh/g for monolayer adsorption and 360.25 mAh/g, 325.29 mAh/g, and 296.41 mAh/g for bilayer adsorption, respectively. Although the theoretical capacity of the bare Zr3C2 is higher than that of the functionalized Zr3C2T2 (Zr3C2O2 and Zr3C2S2), the application of the functionalized Zr3C2T2 in lithium storage is more promising because it is difficult for the bare Zr3C2 to continue the three-layer adsorption, while Zr3C2O2 and Zr3C2S2 can continue adsorption.
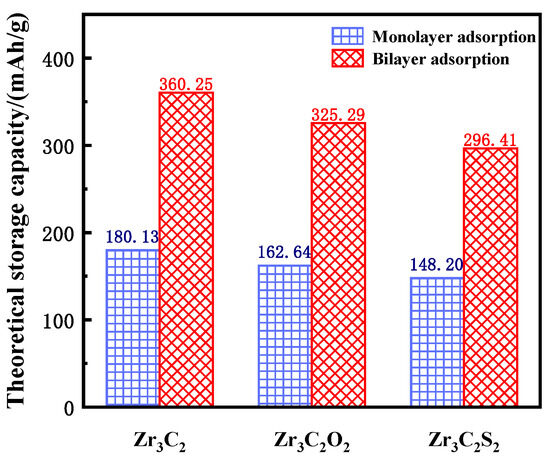
Figure 7.
The theoretical storage capacity of bare Zr3C2 and Zr3C2T2.
4. Conclusions
Using DFT calculations, the effects of surface terminals on the structure, electronic properties, and lithium storage properties of Zr3C2T2 were studied. Functional groups from the oxygen group (T = O, S) and halogen family (T = F, Cl) were considered in this paper. The CCP site was identified as the most stable adsorption site on the Zr3C2T2 surface. The structural properties of Zr3C2T2 were only slightly affected by the surface functional groups, which mainly showed that the presence of functional groups increased the bond length of Zr1-C. The electronic properties of Zr3C2T2 were greatly affected by the O and S functional groups. It was shown that the O and S functional groups had strong hybridization with Zr3C2 in the valence band near the Fermi level and generated a new band, which made the DOS move towards the conduction band. However, the halogen functional groups (F, Cl) had little effect on the electronic properties of Zr3C2. The results of the differential charge density and Mulliken population indicated that the charge transfer between oxygen group atoms and Zr3C2 was more than the counterpart between halogen atoms and Zr3C2. The adsorption energy of lithium ions on the Zr3C2T2 functionalized by oxygen and sulfur functional groups was reduced. By contrast, the Zr3C2T2 functionalized by F and Cl could not stably adsorb lithium ions. Additionally, surface functionalization altered the initial adsorption site of lithium atoms. In monolayer adsorption, the most favorable site for lithium ion adsorption was the HCP site for bare Zr3C2, while it shifted to the CCP site for both Zr3C2O2 and Zr3C2S2. For the bilayer adsorption, the most suitable adsorption sites were CCP for bare Zr3C2, HCP for Zr3C2O2, and CCP for Zr3C2S2. It was revealed that the adsorption energy of both the monolayer and bilayer always rose steeply and then gently. It also showed that bare Zr3C2 made it difficult to continue the three-layer adsorption. The theoretical capacities of the bare Zr3C2 (360.25 mAh/g), Zr3C2O2 (325.29 mAh/g), and Zr3C2S2 (296.41 mAh/g) were predicted.
In future work, the effects of other functional groups and mixed functional groups on the lithium storage performance of the Zr3C2 MXene, the migration of lithium on the MXene surface, and its diffusion energy barrier will be theoretically investigated. In addition, experiments on the effects of Zr3C2 and functional groups on lithium storage will be conducted to validate the theoretical calculations.
Author Contributions
H.L.: methodology, software, investigation, conceptualization, writing—review and editing, visualization, supervision, and funding acquisition; Z.X.: validation, formal analysis, investigation, data curation, writing—original draft preparation; T.G.: investigation, writing—original draft preparation; J.L.: investigation, writing—review and editing; W.L.: investigation, writing—review and editing; Y.L.: investigation, writing—review and editing; S.W.: investigation, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fundamental Research Funds for the Central Universities, CHD (No. 300102312406), and the National College Students Innovation and Entrepreneurship Training Program (No. 202410710100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Acknowledgments
The authors also acknowledge the Northwestern Polytechnical University High Performance Computing Center and Chang’an University High Performance Computing Platform for the allocation of computing time on their machines.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhao, W.; Zhao, C.; Wu, H.; Li, L.; Zhang, C. Progress, challenge and perspective of graphite-based anode materials for lithium batteries: A review. J. Energy Storage 2024, 81, 110409. [Google Scholar] [CrossRef]
- Lin, X.-W.; Li, Y.-B.; Wu, W.-T.; Zhou, Z.-F.; Chen, B. Advances on two-phase heat transfer for lithium-ion battery thermal management. Renew. Sustain. Energy Rev. 2024, 189, 114052. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, L.; Yang, L.; Li, J.; Liu, Y.; Lan, D.; Zhang, S.; Wang, F.; Xu, J.; Liu, D. Ti3C2Tx MXene nanobelts with alkali ion intercalation: Dual-purpose for enhanced lithium-ion batteries and microwave absorption. Carbon 2025, 237, 120148. [Google Scholar] [CrossRef]
- Chen, K.; Yang, H.; Liang, F.; Xue, D. Microwave-irradiation-assisted combustion toward modified graphite as lithium ion battery anode. ACS Appl. Mater. Interfaces 2018, 10, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Huang, J.; Yu, F.; Zhou, Y.; Li, G.; Cheng, W.; Duan, P.; Qi, H.; Xie, H. Chemically bonded MXene/SnSe2 composite with special structural transformation as a high-performance anode for lithium and potassium ions battery. Chem. Eng. J. 2024, 481, 148737. [Google Scholar] [CrossRef]
- Zhu, M.; Han, D.; Cai, C.; Zhong, J.; Weng, J.; Tang, C.; Gao, F.; Zhang, J. Two-dimensional MXene/MOFs heterojunction nanosheets as confinement hosts for dendrite-free lithium metal anodes. Carbon 2024, 227, 119248. [Google Scholar] [CrossRef]
- Toyoura, K.; Koyama, Y.; Kuwabara, A.; Tanaka, I. Effects of off-stoichiometry of LiC6 on the lithium diffusion mechanism and diffusivity by first principles calculations. J. Phys. Chem. C 2010, 114, 2375–2379. [Google Scholar] [CrossRef]
- Zhou, L.-J.; Hou, Z.; Wu, L.-M.; Zhang, Y.-F. First-principles studies of lithium adsorption and diffusion on graphene with grain boundaries. J. Phys. Chem. C 2014, 118, 28055–28062. [Google Scholar] [CrossRef]
- Kong, C.-P.; Hu, Y.-Y.; Bai, F.-Q.; Zhang, H.-X.; Jia, R. F-GDY and F-GDY/Graphene as anodes in lithium-ion batteries: A first-principle investigation. Appl. Surf. Sci. 2022, 595, 153543. [Google Scholar] [CrossRef]
- Thomas, S.; Nam, E.B.; Lee, S.U. Atomistic dynamics investigation of the thermomechanical properties and Li diffusion kinetics in ψ-graphene for LIB anode material. ACS Appl. Mater. Interfaces 2018, 10, 36240–36248. [Google Scholar] [CrossRef]
- Li, L.; Li, Z.; Yoshimura, A.; Sun, C.; Wang, T.; Chen, Y.; Chen, Z.; Littlejohn, A.; Xiang, Y.; Hundekar, P. Vanadium disulfide flakes with nanolayered titanium disulfide coating as cathode materials in lithium-ion batteries. Nat. Commun. 2019, 10, 1764. [Google Scholar] [CrossRef]
- Seh, Z.W.; Yu, J.H.; Li, W.; Hsu, P.-C.; Wang, H.; Sun, Y.; Yao, H.; Zhang, Q.; Cui, Y. Two-dimensional layered transition metal disulphides for effective encapsulation of high-capacity lithium sulphide cathodes. Nat. Commun. 2014, 5, 5017. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Yeom, M.S.; Kim, Y.-T.; Kim, H.; Jung, Y. Polyselenide anchoring using transition-metal disulfides for enhanced lithium–selenium batteries. Inorg. Chem. 2018, 57, 2149–2156. [Google Scholar] [CrossRef]
- Li, W.; Yang, Y.; Zhang, G.; Zhang, Y.-W. Ultrafast and directional diffusion of lithium in phosphorene for high-performance lithium-ion battery. Nano Lett. 2015, 15, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, M.; Anderson, G.; Dharmasena, R.R.; Sumanasekera, G. The prospects of phosphorene as an anode material for high-performance lithium-ion batteries: A fundamental study. Nanotechnology 2017, 28, 075401. [Google Scholar] [CrossRef]
- Yang, Z.; Li, W.; Zhang, J. First-principles study of borophene/phosphorene heterojunction as anode material for lithium-ion batteries. Nanotechnology 2021, 33, 075403. [Google Scholar] [CrossRef]
- Hu, R.; Xu, G.; Yang, Y.; Zhang, J.-M.; Zhong, K.; Huang, Z. Effect of stacking structure on lithium adsorption and diffusion in bilayer black phosphorene. Phys. Rev. B 2019, 100, 085422. [Google Scholar] [CrossRef]
- Tang, Q.; Zhou, Z.; Shen, P. Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2x2 (X = F, OH) monolayer. J. Am. Chem. Soc. 2012, 134, 16909–16916. [Google Scholar] [CrossRef]
- Naguib, M.; Come, J.; Dyatkin, B.; Presser, V.; Taberna, P.-L.; Simon, P.; Barsoum, M.W.; Gogotsi, Y. MXene: A promising transition metal carbide anode for lithium-ion batteries. Electrochem. Commun. 2012, 16, 61–64. [Google Scholar] [CrossRef]
- Nan, J.; Guo, X.; Xiao, J.; Li, X.; Chen, W.; Wu, W.; Liu, H.; Wang, Y.; Wu, M.; Wang, G. Nanoengineering of 2D MXene-based materials for energy storage applications. Small 2021, 17, 1902085. [Google Scholar] [CrossRef]
- Xiong, D.; Li, X.; Bai, Z.; Lu, S. Recent advances in layered Ti3C2Tx MXene for electrochemical energy storage. Small 2018, 14, 1703419. [Google Scholar] [CrossRef]
- Xie, Y.; Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y.; Yu, X.; Nam, K.-W.; Yang, X.-Q.; Kolesnikov, A.I.; Kent, P.R. Role of surface structure on Li-ion energy storage capacity of two-dimensional transition-metal carbides. J. Am. Chem. Soc. 2014, 136, 6385–6394. [Google Scholar] [CrossRef]
- Luo, J.; Tao, X.; Zhang, J.; Xia, Y.; Huang, H.; Zhang, L.; Gan, Y.; Liang, C.; Zhang, W. Sn4+ ion decorated highly conductive Ti3C2 MXene: Promising lithium-ion anodes with enhanced volumetric capacity and cyclic performance. ACS Nano 2016, 10, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lin, S.; Huang, Y.; Tong, P.; Zhao, B.; Zhu, X.; Sun, Y. Synthesis and lithium ion storage performance of two-dimensional V4C3 MXene. Chem. Eng. J. 2019, 373, 203–212. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, K.; Zhu, Y.; Lan, Y.; Hu, L.; Lin, N.; Zhou, J.; Qian, Y. A novel strategy to prepare graphene oxide-wrapped nanocrystals composite for high-performance lithium storage. Mater. Lett. 2016, 175, 32–35. [Google Scholar] [CrossRef]
- Jing, Y.; Zhou, Z.; Cabrera, C.R.; Chen, Z. Graphene, inorganic graphene analogs and their composites for lithium ion batteries. J. Mater. Chem. A 2014, 2, 12104–12122. [Google Scholar] [CrossRef]
- Hao, J.; Zheng, J.; Ling, F.; Chen, Y.; Jing, H.; Zhou, T.; Fang, L.; Zhou, M. Strain-engineered two-dimensional MoS2 as anode material for performance enhancement of Li/Na-ion batteries. Sci. Rep. 2018, 8, 2079. [Google Scholar] [CrossRef]
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2. In MXenes; Jenny Stanford Publishing: Singapore, 2011; pp. 15–29. [Google Scholar]
- Folorunso, O.; Kumar, N.; Hamam, Y.; Sadiku, R.; Ray, S.S. Recent progress on 2D metal carbide/nitride (MXene) nanocomposites for lithium-based batteries. FlatChem 2021, 29, 100281. [Google Scholar] [CrossRef]
- Dong, Y.; Shi, H.; Wu, Z.S. Recent advances and promise of MXene-based nanostructures for high-performance metal ion batteries. Adv. Funct. Mater. 2020, 30, 2000706. [Google Scholar] [CrossRef]
- Shein, I.; Ivanovskii, A. Graphene-like titanium carbides and nitrides Tin+1Cn, Tin+1Nn (n = 1, 2, and 3) from de-intercalated MAX phases: First-principles probing of their structural, electronic properties and relative stability. Comput. Mater. Sci. 2012, 65, 104–114. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Ivanovskii, A.L. Structural and electronic properties and stability of MXenes Ti2C and Ti3C2 functionalized by methoxy groups. J. Phys. Chem. C 2013, 117, 13637–13643. [Google Scholar] [CrossRef]
- Shahpouri, E.; Hassani, S.; Yousefi-Mashhour, H.; Kalantarian, M.M.; Ebadzadeh, T.; Mobasherpour, I. Evaluation of Ti3C2 as electrode material for Li, Na, Mg, Al, K, Ca, and Zn-ion intercalation batteries: A DFT study. Results Chem. 2024, 7, 101385. [Google Scholar] [CrossRef]
- Zhou, H.-Y.; Lin, L.-W.; Sui, Z.-Y.; Wang, H.-Y.; Han, B.-H. Holey Ti3C2 MXene-derived anode enables boosted kinetics in lithium-ion capacitors. ACS Appl. Mater. Interfaces 2023, 15, 12161–12170. [Google Scholar] [CrossRef]
- Chen, Z.-F.; Chen, X.; Chen, C.; Lai, X.; Qin, J.; Chen, C.; Sun, D. A thin, intrinsically stretchable MXene-MWCNTs/polymer current collector for deformable aqueous Li-ion batteries. J. Mater. Chem. A 2024, 12, 2444–2455. [Google Scholar] [CrossRef]
- Bayhan, Z.; El-Demellawi, J.K.; Yin, J.; Khan, Y.; Lei, Y.; Alhajji, E.; Wang, Q.; Hedhili, M.N.; Alshareef, H.N. A laser-induced Mo2CTx MXene hybrid anode for high-performance Li-ion batteries. Small 2023, 19, 2208253. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, D.; Huang, B.; Wang, L.; Xia, Q.; Zhou, A. Effects of surface compositions and interlayer distance on electrochemical performance of Mo2CTx MXene as anode of Li-ion batteries. J. Phys. Chem. Solids 2023, 176, 111238. [Google Scholar] [CrossRef]
- Mashtalir, O.; Lukatskaya, M.R.; Zhao, M.-Q.; Barsoum, M.W.; Gogotsi, Y. Amine-assisted delamination of Nb2C MXene for Li-ion energy storage devices. In MXenes; Jenny Stanford Publishing: Singapore, 2023; pp. 401–414. [Google Scholar]
- Jiang, Y.; Tian, M.; Wang, H.; Wei, C.; Sun, Z.; Rummeli, M.H.; Strasser, P.; Sun, J.; Yang, R. Mildly oxidized MXene (Ti3C2, Nb2C, and V2C) electrocatalyst via a generic strategy enables longevous Li–O2 battery under a high rate. ACS Nano 2021, 15, 19640–19650. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Z.; Yuan, X.; Zhou, N. O-and S-terminated M2C MXenes as anode materials for Na/K-ion batteries. J. Phys. Chem. C 2022, 126, 4267–4275. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Zhang, D.; Liu, X. V2CTx MXene-Encapsulated Liquid Metal Composite as an Anode for Wide-Temperature Li-Ion Batteries. Energy Fuels 2024, 38, 11284–11291. [Google Scholar] [CrossRef]
- Luo, H.; Long, P.; Xiao, J.; Dai, X.; Wang, Z. Mo2CS2 MXene as a promising anode material for metal ion batteries: A first-principles study. Mater. Today Commun. 2024, 38, 108285. [Google Scholar] [CrossRef]
- Liu, J.; Xu, X.; Wang, H.; Wang, P.-F.; Wu, K.; Cheng, Y.; Xiao, B. Achieving high electrical conductivity, energy storage capacity and cycling stability in ammoniated Mo 2 TiC2Tx MXenes as an anode for lithium-ion batteries. J. Mater. Chem. A 2024, 12, 26962–26979. [Google Scholar] [CrossRef]
- Zhou, M.; Shen, Y.; Lv, L.; Zhang, Y.; Meng, X.; Yang, X.; He, Q.; Zhang, B.; Zhou, Z. Bare W-based MXenes (WCrC and MoWC) anode with high specific capacity for Li and Mg-ion batteries. J. Phys. D Appl. Phys. 2023, 57, 015502. [Google Scholar] [CrossRef]
- Wang, D.; Li, F.; Lian, R.; Xu, J.; Kan, D.; Liu, Y.; Chen, G.; Gogotsi, Y.; Wei, Y. A general atomic surface modification strategy for improving anchoring and electrocatalysis behavior of Ti3C2T2 MXene in lithium–sulfur batteries. ACS Nano 2019, 13, 11078–11086. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Zhang, Z.; Wang, L.; Wang, W.; Liu, J.; Hong, Z.; Cho, K.; Wang, W. Origin of theoretical pseudocapacitance of two-dimensional supercapacitor electrodes Ti3C2T2 (T = bare, O, S). J. Mater. Chem. A 2019, 7, 16231–16238. [Google Scholar] [CrossRef]
- Li, H.; Li, A.; Zhang, D.; Wu, Q.; Mao, P.; Qiu, Y.; Zhao, Z.; Yu, P.; Su, X.; Bai, M. First-principles study on the structural, electronic, and lithium storage properties of Ti3C2T2 (T = O, F, H, OH) MXene. ACS Omega 2022, 7, 40578–40585. [Google Scholar] [CrossRef]
- Li, K.; Zeng, J.; Wang, Y.; Zhang, J.; Zhou, Y. A first-principles study of the lithium storage properties of transition metal doped TM-Ti2CO2 (TM = Sc, V, Cr, Mn, Fe, Co, Ni and Cu). Mater. Today Commun. 2024, 40, 109718. [Google Scholar] [CrossRef]
- Abdelsalam, H.; Sakr, M.A.; Teleb, N.H.; Abdelrazek, G.M.; Abd-Elkader, O.H.; Zhang, Q. Unveiling lithium storage potential in Zr2C and Zr2CO2 MXenes: A study of structural stability, electronic properties, and adsorption behavior. Mater. Sci. Eng. B 2025, 314, 118017. [Google Scholar] [CrossRef]
- Zhou, J.; Zha, X.; Chen, F.Y.; Ye, Q.; Eklund, P.; Du, S.; Huang, Q. A two-dimensional zirconium carbide by selective etching of Al3C3 from nanolaminated Zr3Al3C5. Angew. Chem. Int. Ed. 2016, 55, 5008–5013. [Google Scholar] [CrossRef]
- Meng, Q.; Ma, J.; Zhang, Y.; Li, Z.; Hu, A.; Kai, J.-J.; Fan, J. Theoretical investigation of zirconium carbide MXenes as prospective high capacity anode materials for Na-ion batteries. J. Mater. Chem. A 2018, 6, 13652–13660. [Google Scholar] [CrossRef]
- Khan, S.; Kumar, N.; Hussain, T.; Tit, N. Functionalized Hf3C2 and Zr3C2 MXenes for suppression of shuttle effect to enhance the performance of sodium–sulfur batteries. J. Power Sources 2023, 580, 233298. [Google Scholar] [CrossRef]
- Kadangodan Putiyaveettil, A.; Natesan, B. Computational Insights into the Influence of Surface Functionalization Groups on Zirconium Carbide MXenes as Anode Materials for Lithium-Ion Batteries. J. Phys. Chem. C 2024, 128, 12777–12791. [Google Scholar] [CrossRef]
- Khan, K.; Tareen, A.K.; Ahmad, W.; Hussain, I.; Chaudhry, M.U.; Mahmood, A.; Khan, M.F.; Zhang, H.; Xie, Z. Recent Advances in Non-Ti MXenes: Synthesis, Properties, and Novel Applications. Adv. Sci. 2024, 11, 2303998. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Fischer, T.H.; Almlof, J. General methods for geometry and wave function optimization. J. Phys. Chem. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Guo, Y.-L.; Zhao, Y.-Y.; Zeng, G.-L.; Zhang, W.; Ren, C.-L.; Han, H.; Huai, P. A first-principles study on the vibrational and electronic properties of Zr-C MXenes. Commun. Theor. Phys. 2018, 69, 336. [Google Scholar] [CrossRef]
- Li, H.; Qiu, Y.; Gao, D.; Wang, Y.; Zhou, T.; Gao, T.; Xie, Z.; Xu, K.; Yu, P. First-principles study on the adsorption and lithium storage capacities of Hf3C2 and Hf3C2T2 (T = O, F, S) MXenes. FlatChem 2023, 41, 100539. [Google Scholar] [CrossRef]
- Ma, S.; Fan, X.; An, Y.; Yang, D.; Luo, Z.; Hu, Y.; Guo, N. Exploring the catalytic activity of MXenes Mn+1CnO2 for hydrogen evolution. J. Mater. Sci. 2019, 54, 11378–11389. [Google Scholar] [CrossRef]
- Er, D.; Li, J.; Naguib, M.; Gogotsi, Y.; Shenoy, V.B. Ti3C2 MXene as a high capacity electrode material for metal (Li, Na, K, Ca) ion batteries. ACS Appl. Mater. Interfaces 2014, 6, 11173–11179. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).